
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature induced valence phase transition in intermediate-valent YbPd2Al3†
Frank
Stegemann
a,
Juliane
Stahl
b,
Manfred
Bartsch
c,
Helmut
Zacharias
c,
Dirk
Johrendt
b and
Oliver
Janka
 *a
*a
aInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 30, D-48149 Münster, Germany. E-mail: ocjanka@uni-muenster.de
bDepartment Chemie, Ludwig-Maximilians-Universität München, Butenandtstrasse 5-13 (Haus D), D-81377 München, Germany
cCenter for Soft Nanoscience, Westfälische Wilhelms-Universität Münster, Busso-Peus-Straße 10, D-48149 Münster, Germany
First published on 29th October 2019
Abstract
A temperature induced valence phase transition from Yb3+ at higher temperatures to Yb2+ at lower temperatures was observed at T = 110(1) K for intermetallic YbPd2Al3. The title compound has been prepared from the elements in sealed tantalum ampoules. The structure was refined from single-crystal data and the title compound was found to crystallize in the hexagonal YNi2Al3 type structure with space group P6/mmm and lattice parameters of a = 929.56(7) and c = 420.16(3) pm (300 K data). Full ordering of the Pd and Al atoms within the [Pd2Al3]δ− polyanion was observed. Magnetic measurements revealed an anomaly in the dc susceptibility data and intermediate valent Yb at higher temperature, as observed from the effective magnetic moment. The proposed valence phase transition was also observed as a λ-type anomaly in heat capacity measurements (T = 108.4(1) K), however, no systematic shift of the λ-peak was observed in field dependent heat capacity measurements. An antiferromagnetic ordering at this temperature, however, could be excluded, based on field-dependent susceptibility measurements and magnetization isotherms. No dynamic phenomenon was observed in ac susceptibility measurements, excluding e.g. spin-glass behavior. Subsequent temperature dependent single-crystal and powder X-ray diffraction experiments indicated a steep increase in the length of the c axis around T = 110 K upon cooling. However, no structural phase transition was found via single-crystal diffraction experiments conducted at 90 K. The anomaly was also observed in other physical measurements of e.g. the electrical resistivity, indicating a clear change in the electronic structure of the material. X-ray photoelectron spectroscopy conducted at room temperature shows the presence of both, Yb2+ and Yb3+, underlining the mixed-valent state. Members of the solid solution Yb1−xCaxPd2Al3 (x = 0.33, 0.67, 1) were finally used to further study the charge ordering and the present temperature induced valence phase transition.
1 Introduction
In the field of rare-earth element chemistry, the predominant oxidation state of the lanthanoides is the trivalent one. Cerium, praseodymium or terbium exhibit an additional tetravalent oxidation state while for samarium, europium and ytterbium an additional divalent oxidation state exists.1–3 The resulting physical phenomena are numerous, e.g. mixed/intermediate valences in compounds with inequivalent crystallographic cationic sites (Eu3O4,4–6 HP-Tb3O5,7 EuSr2Bi2S4F4 and Eu2SrBi2S4F4,8 Ce2Rh3Sn5 (ref. 9)), inhomogeneous mixed valence states (Sm3S4,10,17 Eu3S4,10 or EuPtP11) as well as intermetallic valence fluctuating materials (CeAl3,12,13 Yb4Pt9Ga24,14 CeMo2Si2C,15 CeRu1−xNixAl16).18 Kondo states or heavy fermion behavior are other prominent physical features, attributed to valence instabilities. Temperature or pressure dependent valence phase transitions in contrast are rather rare. Transitions of this type are found e.g. in metallic Ce,19 in the double perovskites Ba2PrRu1−xIrxO6 (ref. 20) or in the samarium monochalcogenides SmCh (Ch = S, Se, Te).21,22 In EuPtP two new pressure induced valence ordered structures were observed,23 while EuCo2As2 (ref. 24) and YbAl3 (ref. 25) exhibit pressure dependent shifts of their valence. Finally, intermetallic EuPd2Si2 (Ttrans ∼ 200 K)26 and Eu2Pt6Al15 (Ttrans ∼ 45 K)27 as well as the solid solutions CeNi1−xCoxSn (x = 0.4; Ttrans ∼ 95 K)28 and YbxIn1−xCu2 (x = 0.4; Ttrans ∼ 50 K)29–31 exhibit temperature dependent valence phase transitions.Herein, we report on the temperature induced valence phase transition in intermetallic YbPd2Al3 (YNi2Al3 type,32P6/mmm, Z = 3) from intermediate valent Yb at high temperatures to divalent Yb at low temperatures. Upon localization of an electron, a paramagnetic to Pauli-paramagnetic transition occurs at Tanomaly = 110(1) K, which can be observed as a drastic drop in the magnetic susceptibility. The Yb3+ → Yb2+ valence phase transition is furthermore visible in an expansion of the c lattice parameter during cooling, observed in temperature dependent single-crystal and powder X-ray diffraction experiments of YbPd2Al3 and the solid solution Yb1−xCaxPd2Al3 (x = 0.33, 0.67, 1). The (field dependent) specific heat capacity and the electrical resistivity were investigated to underline the proposed valence phase transition, XPS measurements underline the mixed-valent Yb state at room temperature.
2 Experimental section
2.1 Synthesis
Starting materials for the syntheses of YbPd2Al3 and the members of the Yb1−xCaxPd2Al3 series (x = 0.33, 0.67) as well as CaPd2Al3 were ingots of ytterbium (Smart Elements, 99.99%), calcium pieces (Alfa Aesar, 99.5%), palladium sheets (Agosi, 99.99%), and aluminum turnings (Koch chemicals, 99.99%). The calcium ingots were stored under argon; surface impurities were removed mechanically prior to the synthesis. The elements were weighed in the respective atomic ratios and arc-welded33 in tantalum tubes under a dried argon atmosphere of about 800 mbar. The argon gas was purified over titanium sponge (873 K), molecular sieves and silica gel prior to use. The metal tubes were placed in the water-cooled sample chamber of a high-frequency furnace (Hüttinger Elektronik, Freiburg, type TIG 1.5/300 and TIG 5.0/300) in flowing argon.34 The samples were rapidly heated to 1473 K, kept at that temperature for 10 min, annealed for another 5 h at 1023 K and finally rapidly quenched by switching off the high-frequency generator. The temperature was controlled using a radiation pyrometer (Metis MS09, Sensortherm, accuracy: ±50 K). In all cases, the compounds could easily be separated from the ampoules and no reactions with the container material were observed.2.2 X-ray diffraction
The polycrystalline Yb1−xCaxPd2Al3 (x = 0, 0.33, 0.67, 1) samples were characterized by Guinier patterns (imaging plate detector, Fujifilm BAS-1800) with Cu Kα1 radiation and α-quartz (a = 491.30 and c = 540.46 pm) as an internal standard. The lattice parameters were deduced from least-squares fits of the Guinier data35 using the structural models determined by single-crystal X-ray diffraction experiments. For the solid solution Yb1−xCaxPd2Al3 (x = 0, 0.33, 0.67, 1) Vegard's law-like behavior was observed for the unit cell volume along with a linear trend of the hexagonal lattice parameters (Table 1). Temperature dependent powder X-ray diffraction experiments between 10 and 300 K were conducted on a Huber G670 Guinier Imaging Plate diffractometer (Co Kα1 radiation, Ge-111 monochromator, silicon as external standard) with Low Temperature Device 670.4 and closed cycle He cryostat (Lakeshore temperature controller, model 331). The lattice parameters of the temperature dependent measurements are listed in Table S1.†| Compound | T (K) | a (pm) | c (pm) | V (nm3) |
|---|---|---|---|---|
| a Single-crystal data. b Guinier powder X-ray data. | ||||
| YbPd2Al3a | 90(2) | 928.02(6) | 422.22(3) | 0.3149 |
| YbPd2Al3a | 140(2) | 928.02(6) | 419.56(3) | 0.3129 |
| YbPd2Al3a | 300(2) | 929.56(7) | 420.16(3) | 0.3144 |
| YbPd2Al3b | RT | 928.9(2) | 420.25(9) | 0.3140 |
| Yb0.68(1)Ca0.32(1)Pd2Al3a | RT | 930.98(7) | 421.40(3) | 0.3163 |
| Yb0.67Ca0.33Pd2Al3b | RT | 932.3(1) | 421.73(4) | 0.3174 |
| Yb0.34(1)Ca0.66(1)Pd2Al3a | RT | 933.53(7) | 422.66(3) | 0.3190 |
| Yb0.33Ca0.67Pd2Al3b | RT | 934.1(2) | 423.0(1) | 0.3197 |
| CaPd2Al3a | RT | 936.28(6) | 423.96(3) | 0.3219 |
| CaPd2Al3b | RT | 936.3(1) | 423.86(5) | 0.3218 |
Small single-crystals of the Yb1−xCaxPd2Al3 (x = 0, 0.33, 0.67, 1) series were selected from the obtained samples. The crystals were glued to thin quartz fibers using beeswax and investigated by Laue photographs taken on a Buerger camera (white molybdenum radiation; imaging plate technique, Fujifilm, BAS-1800) in order to check their quality. Temperature dependent intensity data sets of YbPd2Al3 were collected with a Stoe StadiVari four-circle diffractometer (Mo Kα radiation; λ = 71.073 pm; μ-source; oscillation mode; hybrid-pixel-sensor, Dectris Pilatus 100 K (ref. 36)) with an open Eulerian cradle setup. Additional measurements at room temperature were conducted on a Stoe IPDS-II image plate system (graphite monochromatized Mo Kα radiation; λ = 71.073 pm; oscillation mode). Numerical absorption corrections along with scaling (for the StadiVari data sets) were applied. To obtain further information about possible site preferences (Yb:Ca) within the solid solution, four independent single-crystals of the samples with the nominal compositions Yb0.33Ca0.67Pd2Al3 and Yb0.33Ca0.67Pd2Al3 were collected each (Table S2†). Details about the data collection and the crystallographic parameters are summarized in Tables S3–S6.†
CCDCs 1868211, 1868849, 1875396, 1880784–1880786, 1892594, 1892597–1892598, 1895369–1895370 and 1910744 contain the supplementary crystallographic data for this paper.†
2.3 Physical property investigations
2.4. X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectra were measured using an Axis-Ultra spectrometer (Kratos, Manchester, UK) in ultrahigh vacuum (pressure < 10−7 Pa). Monochromatic Al Kα radiation (hν = 1486.6 eV) was used with a 12 kV accelerating voltage and 10 mA emission current. The charge neutralizer was run with a filament current of 1.82 A, a charge balance of 2.25 V and a filament bias of 1.0 V. A pass energy filter of 40 eV was applied to the hemispherical analyzer (electrostatic lens mode), enabling high-energy resolution for narrow scans with a step size of 0.1 eV at a dwell time of 4 s. Cold-pressed pellets (ø = 4.5 mm, d = 1.0–1.5 mm) of polycrystalline YbPd2Al3, Yb0.67Ca0.33Pd2Al3, Yb0.33Ca0.67Pd2Al3, and CaPd2Al3 along with a piece of elemental palladium were attached to the XPS sample holder using conductive carbon tape. An area of about 1 × 2 mm2 on the samples surfaces were irradiated at an X-ray incidence angle of 30° relative to the sample normal. Due to different static charge phenomena of the samples, the C 1s signal, caused by organic surface contaminations, was used as an internal reference and its binding energy was set to 285.0 eV (referenced to the Fermi level) for all samples. The peak positions were determined by Gaussian fits of the spectra and are listed in Table 2.| Yb0 4d | Yb2+ 4d | Yb3+ 4d | Pd 3d | Ca 2p | |
|---|---|---|---|---|---|
| a This work. | |||||
| YbPd2Al3a | — | 182.5 | 185.6 | 335.1 | — |
| Yb0.67Ca0.33Pd2Al3a | — | 182.4 | 185.7 | 335.3 | 347.3 |
| Yb0.33Ca0.67Pd2Al3a | — | 182.8 | 185.9 | 335.3 | 347.3 |
| CaPd2Al3a | — | — | — | 335.4 | 347.3 |
| Yb71 | 181.2 | — | — | — | — |
| Yb2O3 (ref. 71) | — | — | 185.4 | — | — |
| Pda | — | — | — | 335.5 | — |
| Pd72 | — | — | — | 335.2 | — |
| Pd73 | — | — | — | 335.3 | — |
| PdAl3 (ref. 72) | — | — | — | 337.7 | — |
| CaO65 | — | — | — | — | 347.1 |
| CaCO3 (ref. 66) | — | — | — | — | 347.1 |
| CaMoO4 (ref. 67) | — | — | — | — | 347.4 |
3 Results and discussion
3.1 Structure refinement
A selected single-crystal of YbPd2Al3 was first measured at 300 K and a careful analysis of the obtained data set indicated a hexagonal primitive lattice. During the refinement, space group P6/mmm was found to be correct; isotypism with the YNi2Al3 type structure32 was already evident from the Guinier powder pattern and from a systematic check of the Pearson database.38 The structure solution was conducted using the SuperFlip algorithm.39 All structures were refined on F2 with anisotropic displacement parameters for all atoms using Jana2006.40,41 As a check for the correct composition, the occupancy parameters were refined in a separate series of least-squares cycles. All sites were fully occupied within three standard deviations with exception of the solid solution (vide infra). No significant residual peaks were evident in the final difference Fourier syntheses. The final positional parameters and interatomic distances are listed in Tables S5 and S6.† The Yb:Ca site occupancies of the single-crystals obtained for members of the solid solutions are additionally listed in Table S2.† Due to the unusual anomaly visible in the susceptibility measurement (vide infra) additional data sets of YbPd2Al3 were recorded at 140 and 90 K. Data sets of the single-crystals obtained from the samples with nominal compositions Yb0.67Ca0.33Pd2Al3 and Yb0.33Ca0.67Pd2Al3 as well as CaPd2Al3 were recorded at room temperature. The obtained data sets of all measurements were handled as described above; the results will be discussed in the crystal chemistry section below.3.2 Temperature dependent powder X-ray diffraction
Guinier powder patterns were recorded of all prepared samples to check for phase purity and refine the lattice parameters (Table 1). Additionally, temperature dependent powder X-ray diffraction patterns of YbPd2Al3 were recorded between 10–300 K upon cooling with intervals of 10 K from 10–90 and 120–300 K, between 90 and 120 K intervals of 5 K were used. In order to obtain the powder lattice parameters, the unit cell dimensions of the 90 K single-crystal measurement were used as starting point and refined using WinPLOTR42 of the FullProf Suite.43 The lattice parameters and the unit cell volumes obtained from these measurements are listed in Table S2† and depicted in Fig. 1, along with the unit cell parameters obtained from the single-crystal X-ray diffraction experiments. With decreasing temperature, both a and c axis initially contract until ∼120 K. At lower temperatures, however, a quite drastic expansion of ∼3 pm for the c axis is visible, starting around 105 K. The a axis in contrast does not show this effect.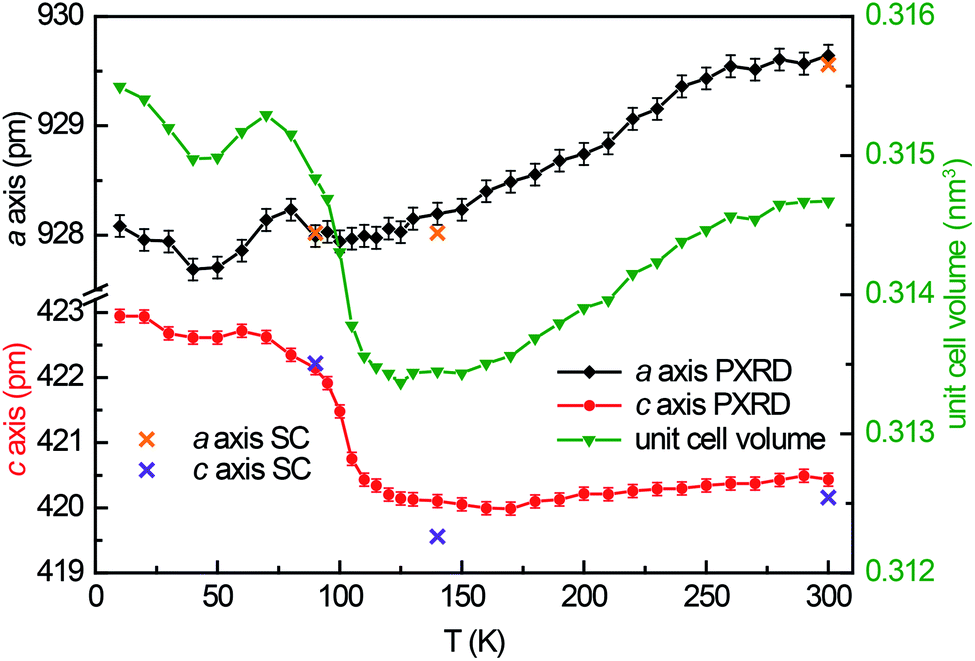 | ||
| Fig. 1 Temperature dependent lattice parameters of YbPd2Al3 obtained from temperature dependent powder X-ray diffraction experiments upon cooling from 300 to 10 K and from single-crystal X-ray diffraction experiments. | ||
3.3 Crystal chemistry
YbPd2Al3 and the other representatives of the solid solution Yb1−xCaxPd2Al3 (x = 0.33, 0.67, 1) all crystallize in the hexagonal crystal system with space group P6/mmm in the YNi2Al3 type structure. No superstructure reflections as in ErPd2Al3 (ref. 44) were observed. The structure description and discussion in the following paragraphs is based on the 300 K measurement of YbPd2Al3. The crystal structure can be described by a polyanionic [Pd2Al3]δ− framework with two crystallographically independent Yb atoms residing in the cavities of the polyanion (Fig. 2). Within the polyanionic network short heteroatomic Pd–Al distances of 254 (Pd–Al2) and 259 pm (Pd–Al1) are found along with longer homoatomic Al–Al (278 and 283 pm) and Pd–Pd distances (299 pm). The observed interatomic Pd–Al distances are in line with those found in binary PdAl45 (264 pm, CsCl type), PdAl2 (ref. 46) (255 pm, Co2Si type), Pd5Al3 (ref. 46) (250–287 pm, Rh5Ge3 type) or ternary YbPdAl47,48 (256–267 pm, TiNiSi type) and Yb8Pd24Al49 (253 pm, Ce8Pd24Sb type). The homoatomic distances are in line with those reported e.g. for elemental Al50 (286 pm), binary Pd5Al3 (ref. 46) (Pd–Pd = 271–306 pm) or ternary Yb8Pd24Al49 (Pd–Pd = 266–307 pm). The two independent Yb atoms both reside in a sixfold capped hexagonal prism, depicted in Fig. 3. The interatomic Yb–Pd distances range from 300–317 pm, the Yb–Al contacts from 324–352 pm. Both are in the same range as those in e.g. YbPdAl47,48 (Yb–Pd = 286–303 pm, Yb–Al = 306–326 pm).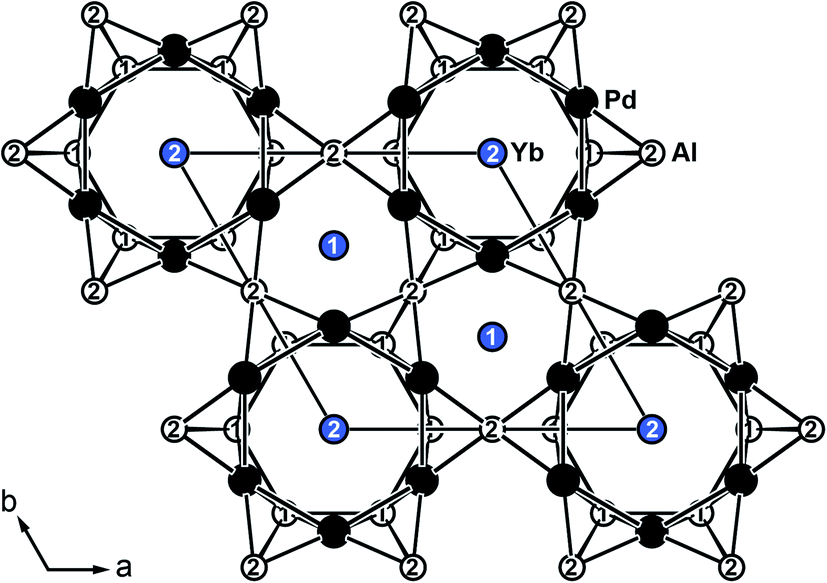 | ||
| Fig. 2 Extended unit cell of hexagonal YbPd2Al3 (YNi2Al3 type) depicted along [001]. Yb atoms are shown in blue, Pd atoms in black and Al atoms in white. | ||
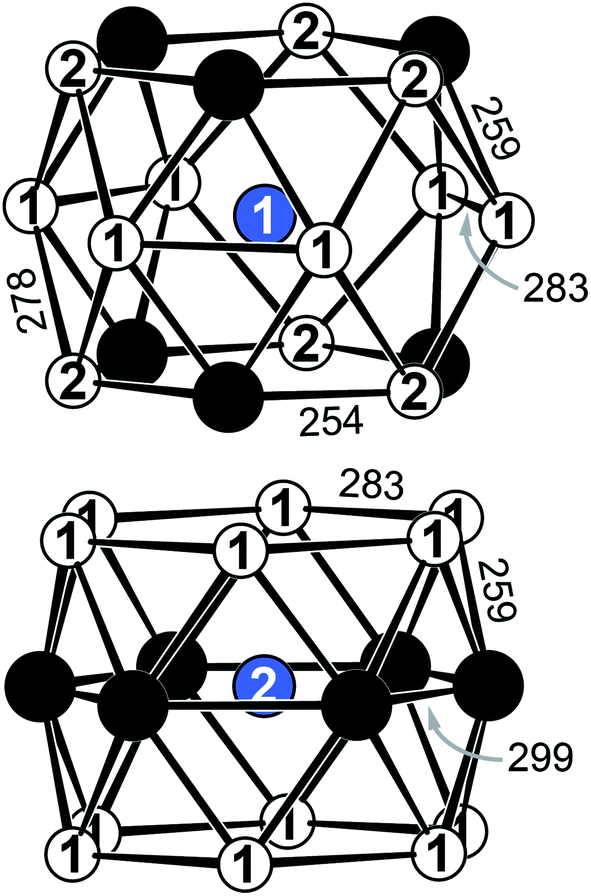 | ||
| Fig. 3 Sixfold capped hexagonal prisms surrounding the two crystallographically independent Yb atoms in YbPd2Al3. Interatomic distances are given in pm. Yb atoms are shown in blue, Pd atoms in black and Al atoms in white. | ||
Upon cooling, the unit cell contracts (vide supra), therefore the respective interatomic distances contract accordingly when cooling from 300 to 140 K. When decreasing the temperature further to 90 K, the Yb1–Al1, Yb1–Al2, Yb2–Al1 (Fig. S1,† top) and Yb1–Pd (Fig. S1,† bottom) distances increase, also in line with the observed unit cell changes. The interatomic Yb2–Pd and Pd–Pd distances in contrast slightly decrease (Fig. S1,† bottom). This, however, is not surprising since they form the caps of the hexagonal prism. Upon expansion of the prism along [001] the caps move towards the center of the prisms, shortening the respective Yb2–Pd and Pd–Pd distances. It is important to note again, that no structural transition was observed, the hexagonal space group symmetry remains at 90 K.
For the members of the solid solutions Yb1−xCaxPd2Al3 with x = 0.33 and 0.67, mixing of Yb and Ca on the two crystallographic Yb sites (1a and 2d) was observed. For both compositions, four single-crystals each were selected and investigated, showing the same Yb/Ca distribution. For nominal Yb0.67Ca0.33Pd2Al3 a distribution of 80(1)% Yb and 20(1)% Ca on the 2d site and 46(1)% Yb and 54(1)% Ca on the 1a site was observed resulting to a refined composition of Yb0.68(1)Ca0.32(1)Pd2Al3 for all compounds. For nominal Yb0.33Ca0.67Pd2Al3 a distribution of 44(1)% Yb and 56(1)% Ca on the 2d site and 15(1)% Yb and 85(1)% Ca on the 1a site was found leading to Yb0.34(1)Ca0.66(1)Pd2Al3 (Table S2†). This narrow distribution is very intriguing and points towards a potential charge ordering.
3.4 Physical properties
A powdered sample of YbPd2Al3 was first investigated by dc-magnetic susceptibility measurements between 3–300 K in zero-field-cooled mode (ZFC) at 10 kOe. The susceptibility χ and its inverse χ−1 are depicted in Fig. 4 (top). With decreasing temperature the susceptibility starts to rise, followed by a sharp decrease at Tanomaly = 110(1) K. The temperature of the anomaly was determined from the maximum of dχ/dT, plotted in blue. Below 25 K the susceptibility rises again reaching χ = 0.0079(1) emu mol−1 at 3 K. This feature is likely to be caused by traces of paramagnetic impurities such as Yb2O3 that orders antiferromagnetically at 2.3 K.51 In order to determine the effective magnetic moment, a fit of the inverse magnetic susceptibility between 175 and 300 K using the modified Curie–Weiss law with χ0 = −1.28(1) × 10−4 emu mol−1 (obtained from CaPd2Al3 at 300 K) was conducted. The effective magnetic moment is μeff = 2.75(1) μB, in line with the observations of the temperature dependence of the effective magnetic moment (vide infra). The Weiss constant is θP = 0.9(2) K, suggesting no dominant magnetic interaction in the paramagnetic temperature regime. In order to understand the behavior around T = 110 K, the temperature dependent effective magnetic moment per Yb atom was calculated according toand is plotted in Fig. 4 (middle, top). The effective magnetic moment initially increases almost linear with increasing temperature starting at μeff = 0.43(1) μB at 3 K before again a rapid increase around Tanomaly = 110(1) K is observed. Above 150 K the moment saturates with μeff = 2.72(1) K at 300 K measured at 10 kOe (μeff = 2.59(5) K at 300 K measured at 50 Oe, see Fig. S6†). The magnetization isotherms recorded at 3 and 10 K are slightly curved, the isotherm at 50 K is linear, typical for a paramagnetic material, while the isotherm recorded at 150 K finally exhibits a much stronger increase, in line with a higher contribution of paramagnetic Yb3+ at this temperature (Fig. 4, middle, bottom). The saturation magnetization at 3 K and 80 kOe is μsat = 0.05(2) μB, which is drastically lower compared to the expected saturation magnetization of 4 μB according to gJ × J for Yb3+. The reported magnetic behavior (reduced effective magnetic moment and low saturation magnetization), along with the structural investigations, can only be explained if YbPd2Al3 contains partially paramagnetic Yb3+ cations (4f13) at higher temperatures, which are reduced to diamagnetic Yb2+ cations (4f14) below Tanomaly = 110(1) K by a spontaneous electron localization from the conduction band.
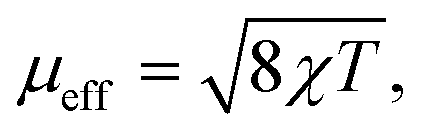
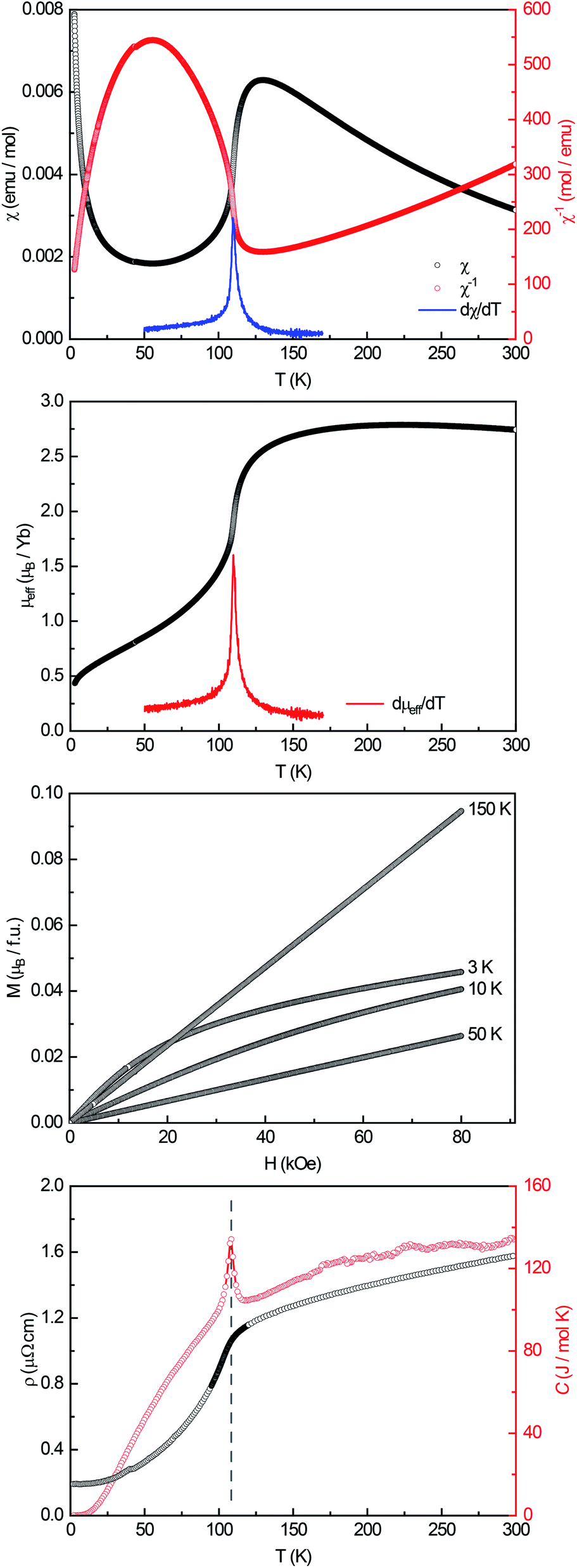 | ||
| Fig. 4 Magnetic properties of YbPd2Al3. Top: temperature dependence of the magnetic susceptibility (χ and χ−1 data) measured in ZFC mode at 10 kOe; dχ/dT is shown in blue. Middle, top: temperature dependence of the effective magnetic moment; dμeff/dT is plotted in red. Middle, bottom: magnetization isotherms recorded at 3, 10, 50 and 150 K. Bottom: temperature dependence of the electrical resistivity (black) and heat capacity (red). | ||
A similar behavior, also featuring a sharp rise of the magnetic susceptibility, was observed in the solid solution YbxIn1−xCu2 with the strongest effect for x = 0.4.29–31 The sharp rise was attributed to a spontaneous valence change from Yb2+ at low temperatures to Yb3+ at high temperatures with Tanomaly ∼ 50 K. The observed magnetic behavior is furthermore significantly different compared to intermediate valence or valence fluctuating ytterbium compounds like YbCu2Si2,52 Yb1−xScxAl2,53 Yb2PdGe6,54 or Yb4Pt9Ga24,14 which can be described by the interconfiguration fluctuation (ICF) model introduced by Sales and Wohlleben.52 The explanation of the susceptibility data via a valence phase transition is also in line with the observed structural effects (vide supra), since Yb2+ is significantly larger than Yb3+ (113 vs. 86 pm2). An only partial Yb3+ character above Tanomaly can be derived from the calculated magnetic moment of μeff = 2.72(1) μB at 300 K measured at 10 kOe, which is significantly lower compared to the calculated moment of μcalc = 4.536 μB of a free Yb3+ ion. When looking back at the crystal structure, the two Yb sites have different site multiplicities (Yb1 on 2d, Yb2 on 1a). Therefore, a charge separation into Yb2+ and Yb3+ is possible. When calculating the net magnetic moment with the paramagnetic Yb3+ solely on the 2d site an effective moment of μeff,calc = 3.70 μB is obtained, while Yb3+ solely on the 1a site results in μeff,calc = 2.62 μB. The latter matches the observed effective moment quite well. This assumption is additionally underlined by the fact that the Yb1–Pd and Yb1–Al distances enlarge upon the valence change from Yb3+ → Yb2+ as seen from the temperature dependent single crystal X-ray diffraction experiments. In order to check for a structural segregation of Yb2+ and Yb3+, members of the solid solution Yb1−xCaxPd2Al3 with x = 0.33 and 0.67 have been synthesized. The results of these investigations will be discussed below.
In addition to the dc-magnetization experiments, heat capacity and electrical resistivity measurements have been performed. The heat capacity (Fig. 4, bottom, red) clearly shows a sharp and intense λ-shaped anomaly at Tanomaly = 108.4(1) K, in line with the other observed temperatures. Moreover, field dependent heat capacity measurements were conducted with applied magnetic fields of H = 0, 10, 50 and 90 kOe (Fig. S2†) within a temperature range of ±20 K around the anomaly. The observed temperature shifts are negligible, since all peak temperature are the same within three standard deviations. The change in the integrated areas can be explained by the stronger magnetic fields. Field dependent susceptibility measurements also indicate no temperature dependence of the anomaly (Fig. S3†). As both, the heat capacity and the susceptibility measurements show no significant temperature dependent shifts, no magnetic ordering takes place at the anomaly, in line with the M(H) investigations. Additional ac-magnetic susceptibility measurements conducted on YbPd2Al3 revealed no frequency dependency of the zero-field as well as field dependent in-phase χ′(ω,T) and out-of-phase χ′′(ω,T) susceptibilities, therefore excluding dynamic processes or spin-glass behavior55 (Fig. S4†). In the resistivity measurements (Fig. 4, bottom, black) a nearly linear decrease is visible from 300 to 150 K before again an abrupt decrease is observed, coinciding with the anomaly visible in the heat capacity. This decrease can be explained by the localization of an electron into the open-shell 4f13 configuration of Yb3+ to form the close-shell Yb2+ (4f14) ion. The present behavior is comparable to the Kondo/impurity scattering observed for trivalent intermetallic cerium compounds (4f1), e.g. Ce3Al56 or Ce3Al11,57 in which the resistivity drops significantly below the Kondo temperature. Trivalent Yb compounds act as ‘hole’ equivalents to the cerium compounds.
As mentioned before, the calculated magnetic moment of YbPd2Al3 is nearly in line with one Yb3+ and two Yb2+ ions per unit cell. Therefore, the divalent Yb2+ ions were replaced by the also diamagnetic Ca2+ ions, which are similar in size (Ca2+: 106 pm; Yb2+: 113 pm2) leading to an ideal formula of Yb0.33Ca0.67Pd2Al3. Additionally, also Yb0.67Ca0.33Pd2Al3 and the diamagnetic CaPd2Al3 were synthesized. All compounds were structurally as well as magnetically characterized. They exhibit a linear trend in the unit cell volume, known as Vegard behavior as well as a linear trend in the hexagonal lattice parameters (Table 1). The magnetic behavior is plotted in Fig. S5.† In comparison with YbPd2Al3, the Ca-containing members of the solid solutions Yb0.33Ca0.67Pd2Al3 and Yb0.67Ca0.33Pd2Al3 also exhibit a paramagnetic behavior, in line with Yb3+ contributions. From the magnitude of the magnetic susceptibility and the effective magnetic moment, it is evident that the amount of trivalent Yb atoms is decreasing (Fig. S6†). As expected, CaPd2Al3 finally exhibits a temperature independent diamagnetic behavior (χdia = −1.28(1) × 10−4 emu mol−1 at 300 K). When looking at the data of Yb0.33Ca0.67Pd2Al3 and Yb0.67Ca0.33Pd2Al3, the “bump-like” feature is smeared and stretched over a larger temperature region; hence measurements up to 395 K were conducted. This behavior can be explained by the disorder in the solid solutions and the distribution of Ca and Yb on the two available crystallographic sites (1a and 2d), which has been proven by single-crystal structure investigations (Table S2†). However, due to the mixing of Yb and Ca, no strict charge separation is present.
3.5 X-ray photoelectron spectroscopy (XPS)
When looking at intermetallic compounds with the general composition RExTyXz (RE: rare-earth element, T: transition metal/group 6–12, X: element of group 13–15) the transition metal, especially when being a 5d element, is the most electronegative element involved. We have shown for a number of gold (e.g. Na2Au3Al,58 AEAuX (AE = Ca–Ba, X = Al–In),59 AEAu2Al2 (AE = Ca, Sr)60 or Ba3Au5Al6 (ref. 60)) or platinum intermetallics (e.g. Ba3Pt4Al4 (ref. 61) and REPt2Al3 (RE = Y, Dy–Tm) series62) that a significant electron transfer onto the 5d metal takes places. For the presented compounds, electronegativities of 1.00 (Ca), 2.20 (Pd) and 1.61 (Al), according to the Pauling scale, are found.2 For Yb no value is listed on this scale, however, for the neighboring elements values of 1.25 (Tm) and 1.27 (Lu) are reported, making Pd again the most electronegative element.2 Since the most electronegative elements acquire additional electron density, a shift towards lower binding energies can be observed in the respective XPS measurements.58–61
Fig. 5 (top) depicts the XPS spectra of the Pd 3d and Ca 2p lines of elemental Pd (black), YbPd2Al3 (red), Yb0.67Ca0.33Pd2Al3 (green), Yb0.33Ca0.67Pd2Al3 (blue), and CaPd2Al3 (orange). As can be seen from the plot and the data in Table 2, only minor shift of the 3d5/2 and 3d3/2 lines of the Pd reference and the investigated compounds can be observed. This is at the first glimpse slightly surprising, but can be explained in a straightforward manner. First, the electronegativity difference between Pd and Al is smaller compared to e.g. Pt/Al or Au/Al. Also all investigated compounds exhibit more Al than Pd atoms (atomic ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Hence, electrons are needed for additional bonds in the [Pd2Al3]δ− polyanion, reducing the electron density at the Pd atoms. In total, the electron density is close to metallic palladium with a small shift to lower binding energies, indicating a slightly negative charge on the Pd atoms. Additionally, a rather pronounced ‘two-peak’ structure is observed for YbPd2Al3. For the other investigated compounds, a tailing of the peaks towards higher binding energies is visible. This can be explained by so-called shake-up related effects.63,64 After photoionization, ions can be left in an excited energy state, which lies a few eV above the ground state. In these cases, the kinetic energy of the emitted photoelectron is reduced and can be seen as an additional peak at a higher binding energy compared to the main line. For metals, conduction electrons also can undergo shake-up events that follow the basic core electron photoemission. In these cases, instead of a discrete structure also only a tail on the higher binding energy side of the main can be evident. Similar effects, also causing satellite transitions are shake-down and shake-off events. For Yb0.67Ca0.33Pd2Al3, Yb0.33Ca0.67Pd2Al3 and CaPd2Al3, additionally the Ca 2p lines are observed in this region. These are in line with typical inorganic calcium compounds, e.g. CaO,65 CaCO3,66 or CaMoO4,67 proving the electron transfer from the alkaline-earth atoms and the formal divalent oxidation state for calcium.
:2). Hence, electrons are needed for additional bonds in the [Pd2Al3]δ− polyanion, reducing the electron density at the Pd atoms. In total, the electron density is close to metallic palladium with a small shift to lower binding energies, indicating a slightly negative charge on the Pd atoms. Additionally, a rather pronounced ‘two-peak’ structure is observed for YbPd2Al3. For the other investigated compounds, a tailing of the peaks towards higher binding energies is visible. This can be explained by so-called shake-up related effects.63,64 After photoionization, ions can be left in an excited energy state, which lies a few eV above the ground state. In these cases, the kinetic energy of the emitted photoelectron is reduced and can be seen as an additional peak at a higher binding energy compared to the main line. For metals, conduction electrons also can undergo shake-up events that follow the basic core electron photoemission. In these cases, instead of a discrete structure also only a tail on the higher binding energy side of the main can be evident. Similar effects, also causing satellite transitions are shake-down and shake-off events. For Yb0.67Ca0.33Pd2Al3, Yb0.33Ca0.67Pd2Al3 and CaPd2Al3, additionally the Ca 2p lines are observed in this region. These are in line with typical inorganic calcium compounds, e.g. CaO,65 CaCO3,66 or CaMoO4,67 proving the electron transfer from the alkaline-earth atoms and the formal divalent oxidation state for calcium.
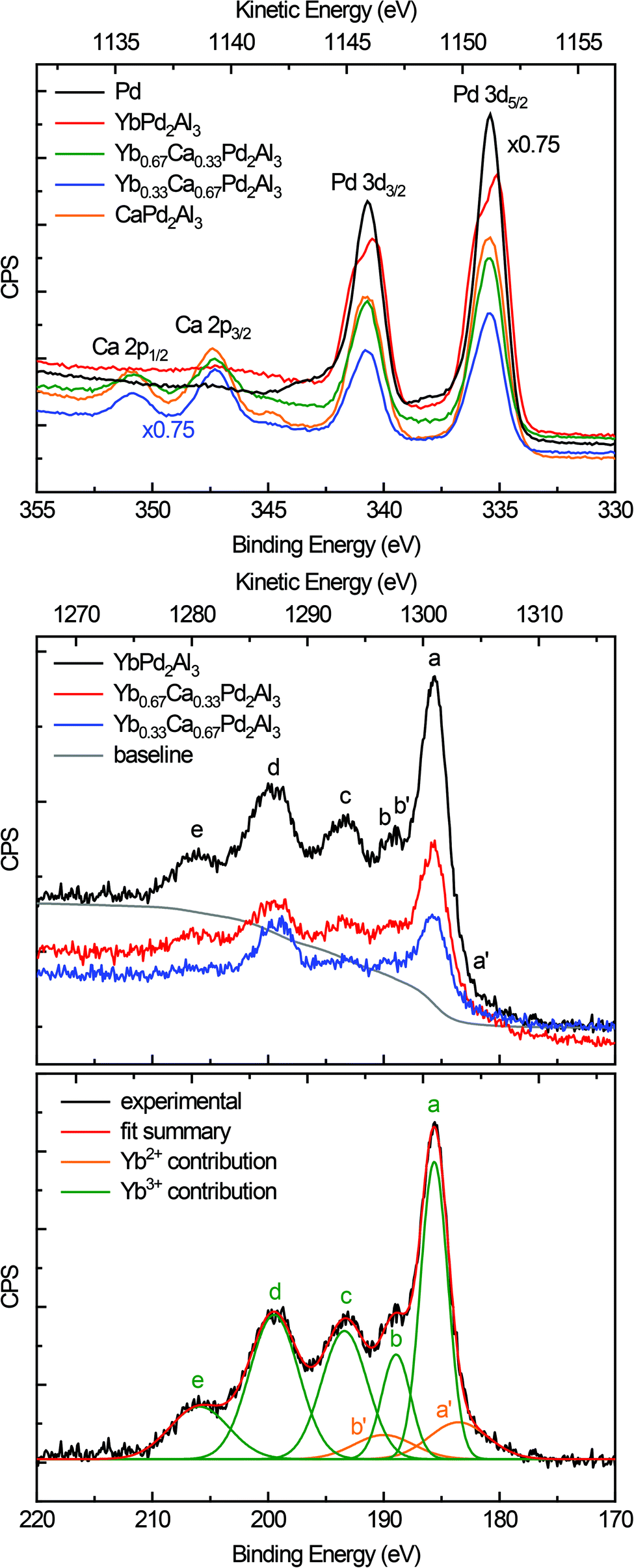 | ||
| Fig. 5 (Top) X-ray photoemission spectra of the Pd 3d and Ca 2p lines of elemental Pd (black), YbPd2Al3 (red), Yb0.67Ca0.33Pd2Al3 (green), Yb0.33Ca0.67Pd2Al3 (blue), and CaPd2Al3 (orange). (Middle) X-ray photoemission spectra of the Yb 4d lines of YbPd2Al3 (black), Yb0.67Ca0.33Pd2Al3 (red), and Yb0.33Ca0.67Pd2Al3 (blue). The background of YbPd2Al3 is depicted as solid grey line. (bottom) Fit of the X-ray photoemission spectra of the Yb 4d lines of YbPd2Al3. The sum is depicted in red. (CPS = counts per second, linear scale). | ||
Fig. 5 (middle) depicts the XPS spectra of the Yb 4d lines of YbPd2Al3 (black), Yb0.67Ca0.33Pd2Al3 (red) and Yb0.33Ca0.67Pd2Al3 (blue). The spectrum consists of five peaks that are easy visible (labeled a to e), which are caused by Coulomb and exchange interactions of the Yb3+ (4f13) open shell ions. According to the literature,68,69 the peak a is attributed to 3H6 and 1G4 terms, the small peak c corresponds to the 3G6 and 1F3 terms, while finally the broad peaks d and e can be assigned to the 3G4, 3D3 and 3P2 as well as the 1H5 terms, respectively. Peak b originates from the 1D2 and 3H5 terms, which overlaps the 4d3/2 peak (b′) of the divalent Yb atoms. These Yb2+ ions show a rather simple spectrum due to the completely filled 4f states. Hence, the exchange interaction between a core hole and the 4f electrons is negligible. The spectrum consists of a doublet of the 4d5/2 (a′) and 4d3/2 (b′) peaks with a relative intensity ratio of 3:2, which has been fixed during fitting. The observed fit of the spectrum of YbPd2Al3 is depicted in Fig. 5 (bottom), the fits of the spectra of Yb0.67Ca0.33Pd2Al3 and Yb0.33Ca0.67Pd2Al3 are shown in the ESI† in Fig. S7 and S8.† Since no cleaving or Ar+ sputtering of the specimen was possible, surface oxidation due to sample handling in air or oxidation under X-ray radiation 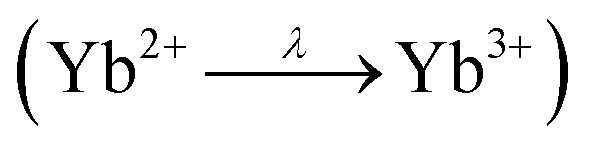 occurred. Similar X-ray induced oxidation processes have been observed for e.g. EuCl2,70 where besides Eu2+ also significant amounts of Eu3+ are visible in the XPS investigations. Subsequently, the relative Yb3+/Yb2+ ratio is significantly biased and cannot be used for the determination of the amount of Yb2+ present in the sample. However, the presence of both lines still proves the presence of both, di- and trivalent Yb atoms. A better quantification might be achievable using Hard X-ray Photoelectron Spectroscopy (HAXPES) investigations which can monitor the Yb 3d3/2 and 3d5/2 or XANES measurements.
occurred. Similar X-ray induced oxidation processes have been observed for e.g. EuCl2,70 where besides Eu2+ also significant amounts of Eu3+ are visible in the XPS investigations. Subsequently, the relative Yb3+/Yb2+ ratio is significantly biased and cannot be used for the determination of the amount of Yb2+ present in the sample. However, the presence of both lines still proves the presence of both, di- and trivalent Yb atoms. A better quantification might be achievable using Hard X-ray Photoelectron Spectroscopy (HAXPES) investigations which can monitor the Yb 3d3/2 and 3d5/2 or XANES measurements.
4. Conclusion
YbPd2Al3 and its solid solution Yb1−xCaxPd2Al3 (x = 0.33, 0.67, 1) have been synthesized from the elements using sealed tantalum ampoules. The compounds crystallize in the hexagonal YNi2Al3 type structure (P6/mmm) with lattice parameters of a = 930–936 and c = 420–424 pm. During dc magnetic susceptibility measurements, an anomaly at Tanomaly ∼ 110 K was observed. Antiferromagnetic ordering was discarded due to the recorded magnetization isotherms, the observed expansion of the unit cell below Tanomaly and the absence of a significant shift in field dependent heat capacity and dc susceptibility measurements. The observed feature is also visible in measurements of the electrical resistivity and the heat capacity, in contrast, no dynamic features were observed in ac magnetization experiments. Therefore, this feature can only be attributed to a temperature induced ytterbium valence change. While above 110 K a mixed-valent Yb state (as seen from the effective magnetic moment) is present, an intrinsic one electron reduction upon formation of a nearly diamagnetic compound at low temperatures takes place. The temperature induced valence change is not only characterized by the drastically expansion of the c axis, observed in temperature dependent powder and single-crystal X-ray measurements, but also by a significant distortion of the Yb1 polyhedra. XPS investigation proved the presence of Yb2+ and Yb3+, however, overestimating the amounts of trivalent Yb due to surface oxidation of the specimen. A possible explanation for this valence change is the lower overall energy of the purely divalent system at low temperatures, since the open shell 4f13 system of Yb3+ transitions into a fully closed shell Yb2+ (4f14) system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dipl.-Chem. Simon Gausebeck for the help with the sample preparation, Dr R.-D. Hoffmann, Dipl.-Ing. U. Ch. Rodelwald and Dipl.-Ing. J. Kösters for the collection of the single-crystal diffractometer data.References
- A. Szytuła and J. Leciejewicz, Handbook of Crystal Structures and Magnetic Properties of Rare Earth Intermetallics, CRC Press, Boca Raton, 1994 Search PubMed.
- J. Emsley, The Elements, Clarendon Press, Oxford University Press, Oxford, New York, 1998 Search PubMed.
- S. Cotton, Lanthanides & Actinides, Macmillan Education LTD, Houndsmillo, Basingstoke, Hampsire, London, 1991 Search PubMed.
- H. Bärnighausen and G. Brauer, Acta Crystallogr., 1962, 15, 1059 CrossRef.
- R. Rau, Acta Crystallogr., 1966, 20, 716–723 CrossRef CAS.
- F. F. Y. Wang, Phys. Status Solidi B, 1966, 14, 189–192 CrossRef CAS.
- M. Glätzle, O. Janka, V. Svitlyk, D. Chernyshov, M. Bartsch, H. Zacharias, R. Pöttgen and H. Huppertz, Chem.–Eur. J., 2018, 24, 15236–15245 CrossRef PubMed.
- Z. Haque, G. S. Thakur, R. Parthasarathy, B. Gerke, T. Block, L. Heletta, R. Pöttgen, A. G. Joshi, G. K. Selvan, S. Arumugam, L. C. Gupta and A. K. Ganguli, Inorg. Chem., 2017, 56, 3182–3189 CrossRef CAS PubMed.
- M. B. Gamża, R. Gumeniuk, U. Burkhardt, W. Schnelle, H. Rosner, A. Leithe-Jasper and A. Ślebarski, Phys. Rev. B, 2017, 95, 165142 CrossRef.
- F. L. Carter, J. Solid State Chem., 1972, 5, 300–313 CrossRef CAS.
- N. Lossau, H. Kierspel, J. Langen, W. Schlabitz, D. Wohlleben, A. Mewis and C. Sauer, Z. Phys. B: Condens. Matter, 1989, 74, 227–232 CrossRef CAS.
- K. H. J. Buschow and H. J. Vandall, Solid State Commun., 1970, 8, 363–365 CrossRef CAS.
- K. H. Mader and W. M. Swift, J. Phys. Chem. Solids, 1968, 29, 1759–1764 CrossRef CAS.
- O. Sichevych, Y. Prots, Y. Utsumi, L. Akselrud, M. Schmidt, U. Burkhardt, M. Coduri, W. Schnelle, M. Bobnar, Y.-T. Wang, Y.-H. Wu, K.-D. Tsuei, L. H. Tjeng and Y. Grin, Inorg. Chem., 2017, 56, 9343–9352 CrossRef CAS PubMed.
- U. B. Paramanik, U. Anupam, U. Burkhardt, R. Prasad, C. Geibel and Z. Hossain, J. Alloys Compd., 2013, 580, 435–441 CrossRef CAS.
- O. Niehaus, U. C. Rodewald, P. M. Abdala, R. S. Touzani, B. P. T. Fokwa and O. Janka, Inorg. Chem., 2014, 53, 2471–2480 CrossRef CAS PubMed.
- B. Batlogg, E. Kaldis, A. Schlegel, G. von Schulthess and P. Wachter, Solid State Commun., 1976, 19, 673–676 CrossRef CAS.
- B. Batlogg, E. Kaldis and P. Wachter, J. Magn. Magn. Mater., 1976, 3, 96–98 CrossRef CAS.
- D. C. Koskenmaki and K. A. Gschneidner Jr, Handbook on the Physics and Chemistry of Rare Earths, 1978, vol. 1, p. 337 Search PubMed.
- M. Wakeshima, Y. Izumiyama, Y. Doi and Y. Hinatsu, Solid State Commun., 2001, 120, 273–278 CrossRef CAS.
- A. Jayaraman, V. Narayanamurti, E. Bucher and R. G. Maines, Phys. Rev. Lett., 1970, 25, 1430–1433 CrossRef CAS.
- E. Kaldis and P. Wachter, Solid State Commun., 1972, 11, 907–912 CrossRef CAS.
- M. Akihiro, M. Shigeki, U. Masafumi, W. Hirofumi, M. Kazuyuki, U. Yoshiya, M. Masaichiro, K. Naomi, N. Kiyofumi, H. Naohisa, O. Yasuo and I. Naoki, J. Phys.: Condens. Matter, 2018, 30, 105603 CrossRef PubMed.
- A. P. Menushenkov, A. A. Yaroslavtsev, A. Y. Geondzhian, R. V. Chernikov, L. Nataf, X. Tan and M. Shatruk, J. Supercond. Novel Magn., 2017, 30, 75–78 CrossRef CAS.
- R. S. Kumar, A. Svane, G. Vaitheeswaran, V. Kanchana, E. D. Bauer, M. Hu, M. F. Nicol and A. L. Cornelius, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075117 CrossRef.
- E. V. Sampathkumaran, L. C. Gupta, R. Vijayaraghavan, K. V. Gopalakrishnan, R. G. Pillay and H. G. Devare, J. Phys. C: Solid State Phys., 1981, 14, L237 CrossRef CAS.
- M. Radzieowski, F. Stegemann, T. Block, J. Stahl, D. Johrendt and O. Janka, J. Am. Chem. Soc., 2018, 140, 8950–8957 CrossRef CAS PubMed.
- D. T. Adroja, B. D. Rainford, J. M. de Teresa, A. del Moral, M. R. Ibarra and K. S. Knight, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 12790–12797 CrossRef PubMed.
- I. Felner and I. Nowik, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 617–619 CrossRef CAS PubMed.
- I. Felner, I. Nowik, D. Vaknin, U. Potzel, J. Moser, G. M. Kalvius, G. Wortmann, G. Schmiester, G. Hilscher, E. Gratz, C. Schmitzer, N. Pillmayr, K. G. Prasad, H. de Waard and H. Pinto, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 6956–6963 CrossRef CAS PubMed.
- H. Müller, E. Bauer, E. Gratz, K. Yoshimura, T. Nitta and M. Mekata, J. Magn. Magn. Mater., 1988, 76, 159–160 CrossRef.
- O. S. Zarechnyuk and R. M. Rykhal', Visnyk Lvivskoho Derzhavnoho Universytetu Serija Khimichna, 1981, 23, 45–47 Search PubMed.
- R. Pöttgen, T. Gulden and A. Simon, GIT Labor-Fachz., 1999, 43, 133–136 Search PubMed.
- D. Kußmann, R.-D. Hoffmann and R. Pöttgen, Z. Anorg. Allg. Chem., 1998, 624, 1727–1735 CrossRef.
- K. Yvon, W. Jeitschko and E. Parthé, J. Appl. Crystallogr., 1977, 10, 73–74 CrossRef.
- Dectris, Pilatus 100K-S Detector System, Technical Specification and Operating Procedure (version 1.7), Baden, 2011 Search PubMed.
- L. J. van der Pauw, Philips research reports, 1958, 13, 1–9 Search PubMed.
- P. Villars and K. Cenzual, Pearson's Crystal Data: Crystal Structure Database for Inorganic Compounds (on DVD), ASM International®, Materials Park, Ohio (USA), release 2018/2019 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Jana2006, The crystallographic computing system, Institute of Physics, Praha, Czech Republic, 2006 Search PubMed.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr., 2014, 229, 345–352 Search PubMed.
- T. Roisnel and R. Rodríguez-Carvajal, Materials Science Forum, Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), 2000, pp. 118–123 Search PubMed.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- F. Eustermann, R.-D. Hoffmann and O. Janka, Z. Kristallogr., 2017, 232, 573–581 CAS.
- H. Pfisterer and K. Schubert, Naturwissenschaften, 1950, 37, 112–113 CrossRef CAS.
- K. Schubert, H. Breimer, W. Burkhardt, E. Günzel, R. Haufler, H. L. Lukas, H. Vetter, J. Wegst and M. Wilkens, Naturwissenschaften, 1957, 44, 229–230 CrossRef CAS.
- G. Cordier, T. Friedrich, R. Henseleit, A. Grauel, U. Tegel, C. Schank and C. Geibel, J. Alloys Compd., 1993, 201, 197–201 CrossRef CAS.
- G. Cordier and R. Henseleit, Z. Kristallogr., 1993, 203, 144–145 CAS.
- S. Surjeet and S. K. Dhar, J. Phys.: Condens. Matter, 2002, 14, 11795 CrossRef.
- A. W. Hull, Phys. Rev., 1917, 10, 661–696 CrossRef CAS.
- R. M. Moon, W. C. Koehler, H. R. Child and L. J. Raubenheimer, Phys. Rev., 1968, 176, 722–731 CrossRef CAS.
- B. C. Sales and D. K. Wohlleben, Phys. Rev. Lett., 1975, 35, 1240–1244 CrossRef CAS.
- G. J. Lehr, D. T. Morelli, H. Jin and J. P. Heremans, J. Appl. Phys., 2013, 114, 223712 CrossRef.
- D. Kaczorowski, A. V. Gribanov, P. Rogl and S. F. Dunaev, J. Alloys Compd., 2016, 685, 957–961 CrossRef CAS.
- J. A. Mydosh, J. Magn. Magn. Mater., 1996, 157–158, 606–610 CrossRef.
- M. Sera, T. Satoh and T. Kasuya, J. Magn. Magn. Mater., 1987, 63–64, 82–84 CrossRef CAS.
- H. J. Van Daal and K. H. J. Buschow, Phys. Lett. A, 1970, 31, 103–104 CrossRef CAS.
- F. Stegemann, C. Benndorf, Y. Zhang, M. Bartsch, H. Zacharias, B. P. T. Fokwa, H. Eckert and O. Janka, Inorg. Chem., 2017, 56, 1919–1931 CrossRef CAS PubMed.
- C. Benndorf, F. Stegemann, S. Seidel, L. Schubert, M. Bartsch, H. Zacharias, B. Mausolf, F. Haarmann, H. Eckert, R. Pöttgen and O. Janka, Chem.–Eur. J., 2017, 23, 4187–4196 CrossRef CAS PubMed.
- F. Stegemann, C. Benndorf, Y. Zhang, M. Bartsch, H. Zacharias, B. P. T. Fokwa, H. Eckert and O. Janka, Z. Anorg. Allg. Chem., 2017, 643, 1379–1390 CrossRef CAS.
- F. Stegemann, C. Benndorf, T. Bartsch, R. S. Touzani, M. Bartsch, H. Zacharias, B. P. T. Fokwa, H. Eckert and O. Janka, Inorg. Chem., 2015, 54, 10785–10793 CrossRef CAS PubMed.
- F. Eustermann, S. Gausebeck, C. Dosche, M. Haensch, G. Wittstock and O. Janka, Crystals, 2018, 8, 169 CrossRef.
- D. Briggs, Surface analysis by Auger and X-ray photoelectron spectroscopy SurfaceSpectra, Manchester, 2003 Search PubMed.
- S. Hüfner, Photoelectron spectroscopySpringer, Berlin, 2010 Search PubMed.
- H. Van Doveren and J. A. T. H. Verhoeven, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 265–273 CrossRef CAS.
- Q. Liu, J. S. Laskowski, Y. Li and D. Wang, Int. J. Miner. Process., 1994, 42, 251–266 CrossRef CAS.
- V. I. Nefedov, M. N. Firsov and I. S. Shaplygin, J. Electron Spectrosc. Relat. Phenom., 1982, 26, 65–78 CrossRef CAS.
- Y. Ohno, J. Electron Spectrosc. Relat. Phenom., 2008, 165, 1–4 CrossRef CAS.
- I. Chorkendorff, J. Onsgaard, J. Schmidt-May and R. Nyholm, Surf. Sci., 1985, 160, 587–598 CrossRef CAS.
- D. Kim, Y.-H. Jin, K.-W. Jeon, S. Kim, S.-J. Kim, O. H. Han, D.-K. Seo and J.-C. Park, RSC Adv., 2015, 5, 74790–74801 RSC.
- S. B. M. Hagström, P. O. Hedén and H. Löfgren, Solid State Commun., 1970, 8, 1245–1248 CrossRef.
- F. U. Hillebrecht, J. C. Fuggle, P. A. Bennett, Z. Zołnierek and C. Freiburg, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 2179–2193 CrossRef CAS.
- K. Kishi, Y. Motoyoshi and S. Ikeda, Surf. Sci., 1981, 105, 313–324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868211, 1868849, 1875396, 1880784–1880786, 1892594, 1892597–1892598, 1895369–1895370 and 1910744. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04437j |
| This journal is © The Royal Society of Chemistry 2019 |