
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rare earth hydride supported ruthenium catalyst for the hydrogenation of N-heterocycles: boosting the activity via a new hydrogen transfer path and controlling the stereoselectivity†
Yong
Wu
a,
Hongen
Yu
a,
Yanru
Guo
a,
Xiaojing
Jiang
a,
Yue
Qi
a,
Bingxue
Sun
a,
Haiwen
Li
 b,
Jie
Zheng
*a and
Xingguo
Li
*a
b,
Jie
Zheng
*a and
Xingguo
Li
*a
aBeijing National Laboratory for Molecular Science (BNLMS), College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
bPlatform of Inter/Transdisciplinary Energy Research (Q-PIT), International Research Center for Hydrogen Energy, International Institute for Carbon-Neutral Energy Research (I2CNER), Kyushu University, 744 Motooka Nishi-ku, Fukuoka 819-0395, Japan. E-mail: xgli@pku.edu.cn; Fax: +86-10-62765930; Tel: +86-10-62765930
First published on 11th October 2019
Abstract
Hydrogenation of N-heterocycles is of great significance for their wide range of applications such as building blocks in drug and agrochemical syntheses and liquid organic hydrogen carriers (LOHCs). Pursuing a better hydrogenation performance and stereoselectivity, we successfully developed a rare earth hydride supported ruthenium catalyst Ru/YH3 for the hydrogenation of N-heterocycles, especially N-ethylcarbazole (NEC), the most promising LOHC. Full hydrogenation of NEC on Ru/YH3 can be achieved at 363 K and 1 MPa hydrogen pressure, which is currently the lowest compared to previous reported catalysts. Furthermore, Ru/YH3 shows the highest turnover number, namely the highest catalytic activity among the existing catalysts for hydrogenation of NEC. Most importantly, Ru/YH3 shows remarkable stereoselectivity for all-cis products, which is very favorable for the subsequent dehydrogenation. The excellent performance of Ru/YH3 originates from the new hydrogen transfer path from H2 to NEC via YH3. Ru/LaH3 and Ru/GdH3 also reveal good activity for hydrogenation of NEC and Ru/YH3 also possesses good activity for hydrogenation of 2-methylindole, indicating that the use of rare earth hydride supported catalysts is a highly effective strategy for developing better hydrogenation catalysts for N-heterocycles.
Introduction
N-Heterocycles have been attracting much attention because of their great versatility in many pharmaceuticals, alkaloids, agrochemicals, and fine chemicals.1–4 Recently, liquid organic hydrogen carriers (LOHCs) have been proposed as convenient and safe candidates for hydrogen storage and N-heterocycles are very typical LOHCs for their high hydrogen capacities and relatively low operating temperatures.5–8 One of the most well-studied LOHCs is N-ethylcarbazole (NEC), whose hydrogen-rich state dodecahydro-N-ethylcarbazole (12H-NEC) is liquid at room temperature with a hydrogen capacity of 5.8 wt%. Additionally, the operating temperature for hydrogen storage on NEC is below 473 K for the hydrogenation enthalpy is only −50.6 kJ (mol−1 H2).9–13The kinetics of hydrogenation of N-heterocycles is usually sluggish, which is an obstacle for their application as LOHCs. Consequently, effective hydrogenation catalysts are urgently needed. Many homogenous or heterogeneous catalysts were developed for hydrogenation of N-heterocycles, which are mainly quinoline and its derivatives.3,14–21 For the hydrogenation of NEC, the catalysts are mainly Ru based heterogeneous catalysts.22–26 Many factors such as supports and particle sizes will influence the catalytic performance, since the hydrogenation reaction is a stepwise complex reaction (Fig. 1).
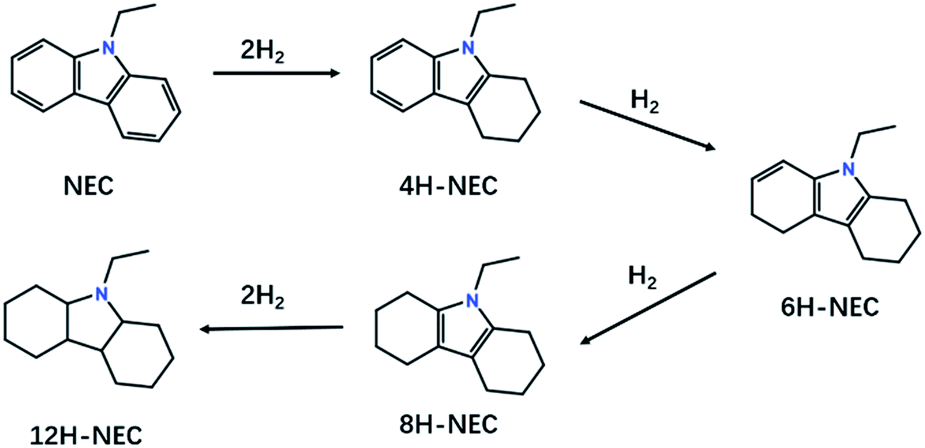 | ||
| Fig. 1 Reaction process of the hydrogenation of NEC. | ||
Recently, we demonstrated that reversible hydrogen absorption/desorption, the very fundamental property of metal hydrides, can be used to promote the hydrogenation of NEC. The Ni/Al2O3-YH3 catalyst we developed is the most efficient noble metal free catalyst for NEC hydrogenation and its catalytic activity is close to that of Ru/Al2O3, the state-of-the-art catalyst for the hydrogenation of NEC.27,28 To further increase catalytic activity and decrease the dosage of the precious metal Ru, we considered that we can directly use YH3 as a support material. Some rare earth hydrides are used as support materials of Ru catalysts for ammonia synthesis.29 Although the original activity of the obtained catalysts is promising, the stability is a problem due to the formation of rare earth nitrides.29 However, this problem will not occur in the hydrogenation of N-heterocycles since there is no nitrogen.
Here we report a robust Ru/YH3 catalyst for the hydrogenation of N-heterocycles. The temperature for complete NEC hydrogenation on Ru/YH3 can be as low as 363 K. For sufficient reaction rate, we choose 403 K as the optimized temperature. At 403 K, the hydrogen pressure can be below 1 MPa. Besides the lowest hydrogenation temperature and hydrogen pressure among the reported studies, the hydrogenation rate is also the fastest while using the same amount of Ru. Additionally, Ru/YH3 exhibits extraordinary stereoselectivity for the all-cis products.
Based on our experimental results, we believe that there is a new hydrogen transport path in our system during the reaction, which contributes to the high catalytic activity. More specifically, there is hydrogen transfer between gaseous hydrogen and hydrogen in YH3 as YH3 is also a hydrogen storage material which can absorb and desorb hydrogen reversibly rapidly, so that the hydrogen in YH3 can spill-over to Ru, enabling the hydrogenation of NEC molecules absorbed on Ru.
Results and discussion
The properties of catalysts
Ru/YH3 was obtained by an improved chemical vapor deposition method and Ru/Al2O3 was prepared by the same method.29–31 Ru/Al2O3 is a reference catalyst. Fig. 2 shows the XRD patterns of Ru/YH3 and Ru/Al2O3. There are only signals of crystalline YH3 (ICSD 98-015-4809)32 in the XRD patterns of Ru/YH3. The XRD patterns of Ru/Al2O3 are almost the same as the XRD patterns of pristine Al2O3 (ICSD 98-008-8027)33 (Fig. S1†). The signal peaks of Al2O3 are broad, as the size is about 20 nm according to the supplier. There are no signals of Ru in the XRD patterns, indicating that the size of Ru is very small.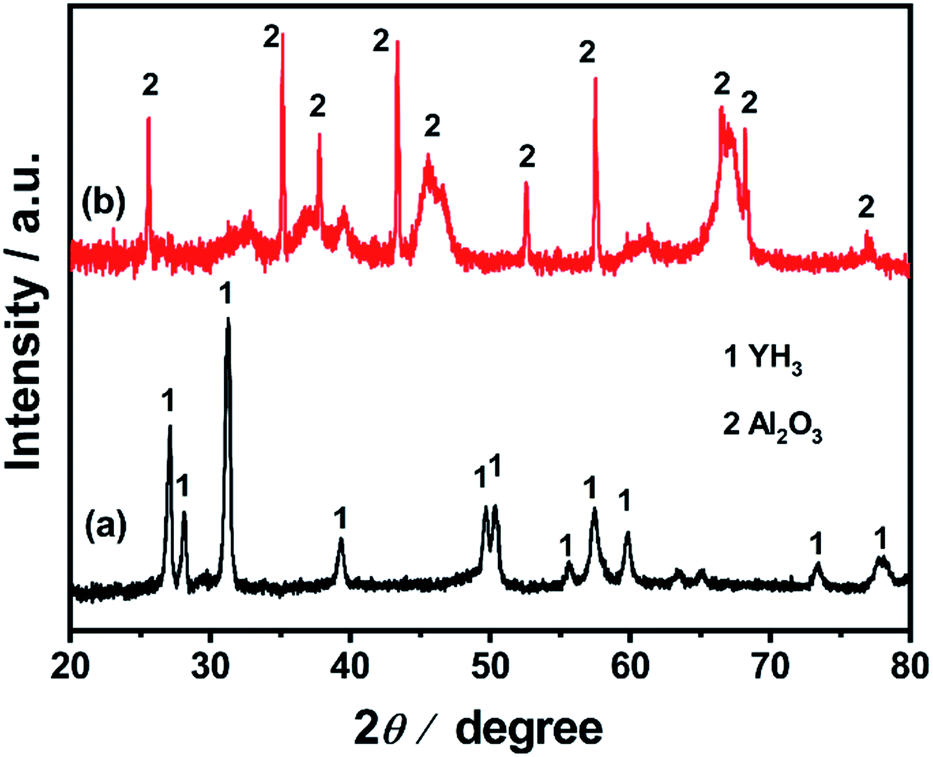 | ||
| Fig. 2 XRD patterns of (a) Ru/YH3 and (b) Ru/Al2O3. | ||
The TEM images of YH3 and Al2O3 (Fig. S2†) and the HRTEM images of Ru/YH3 and Ru/Al2O3 (Fig. 3) well prove the point. The size of YH3 ranges from 50 nm to 500 nm, while the size of Al2O3 is around 20 nm as expected. The size of Ru is approximately 5 nm, and Ru nanoparticles are uniformly dispersed on the supports according to the elemental mapping for both Ru/YH3 and Ru/Al2O3. The lattice fringes of Ru (010), Ru (002), Ru (011) and YH3 (111) can be seen in the HRTEM images of Ru/YH3, implying that YH3 has not been oxidized during the catalyst preparation process, which corresponds with the results of XRD patterns of Ru/YH3.
 | ||
| Fig. 3 HRTEM images and elemental mapping of (a–f) Ru/YH3 and (g–k) Ru/Al2O3. | ||
This can also be demonstrated from the XPS spectra of Ru/YH3 (Fig. S3†). The binding energies that can be ascribed to Y 3d5/2 and Y 3d3/2 are 157.8 eV and 159.8 eV, while the previously reported results for YH3 are 157.7 eV and 159.6 eV.34,35 In the case of Y2O3, they are 156.8 eV and 158.9 eV. In the case of YH2, they are 156.5 eV and 158.5 eV.34,35 These results indicate that there is no surface oxidation in Ru/YH3. The binding energy of Ru 3d5/2 appears at 280.4 eV, exhibiting a positive chemical shift of ca. 0.4 eV from the nominal value of 280.0 eV in Ru nanoparticles.36 However, the binding energy of Ru 3d5/2 in Ru/Al2O3 appears at the same position as that in Ru/YH3, indicating the same electronic effect of support materials on Ru/Al2O3 and Ru/YH3.
Since the particle sizes of YH3 and Al2O3 are much different and Al2O3 is porous, the surface areas of Ru/YH3 and Ru/Al2O3 must be different. Indeed, the BET surface areas of Ru/YH3 and Ru/Al2O3 are 11.3 and 103.2 m2 g−1 from the nitrogen adsorption/desorption isotherms (Fig. S4†), respectively. Due to the bigger surface area of Ru/Al2O3, which signifies a smaller loss of Ru during the chemical vapor deposition process, the Ru loading amount of Ru/Al2O3 is 4.6 wt% while that of Ru/YH3 is 1.3 wt% based on the results of ICP-OES, even though the expected loading amounts are 5 wt% for all catalysts.
The catalytic performances
It seems that the catalytic performance of Ru/YH3 will be terrible because of the low surface area and low Ru loading amount. Unexpectedly, the hydrogenation rates are almost the same when adding the same amount of Ru/YH3 and Ru/Al2O3 (Fig. 4a). The 1H NMR spectra of the corresponding hydrogenation products (Fig. 4b) confirm that the products are both complete hydrogenated products (12H-NEC) as the chemical shifts (>3.6 ppm) ascribed to H which bonds to C (sp2) disappear.9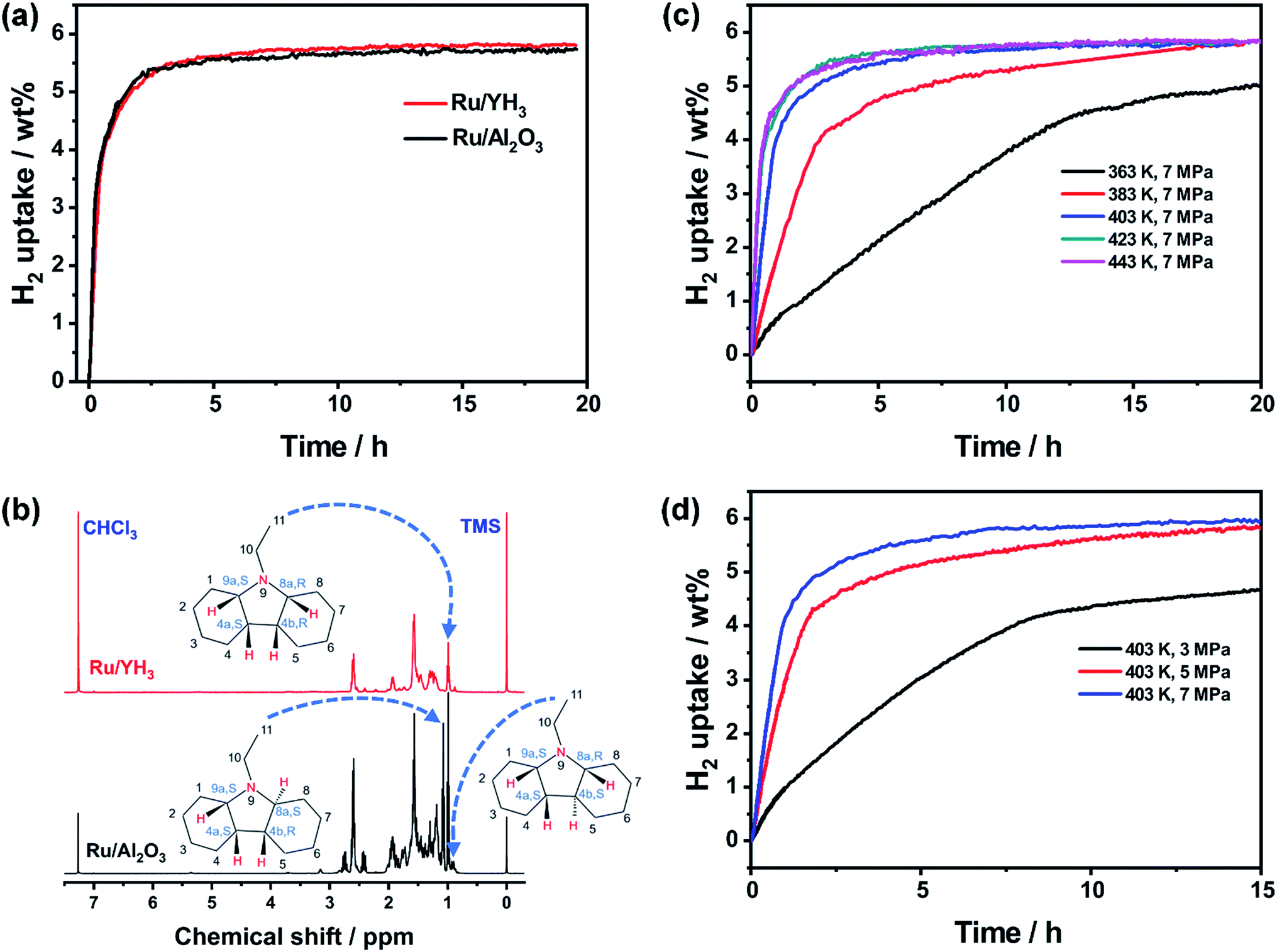 | ||
| Fig. 4 (a) Hydrogen absorption kinetics and (b) 1H NMR spectra of corresponding hydrogenation products of NEC using Ru/YH3 and Ru/Al2O3 as catalysts (403 K, 7 MPa H2, 1.00 g NEC, 50 mg catalysts). Hydrogen absorption kinetics of NEC using Ru/YH3 as the catalyst (c) at different temperatures and (d) under different hydrogen pressures (1.00 g NEC, 50 mg catalyst). | ||
As the Ru loading amount of Ru/Al2O3 is higher than that of Ru/YH3, the TON (turnover number, the amount of substance transformed per mole active metal per hour) of Ru/YH3 is higher than the TON of Ru/Al2O3 (Table 1). That is, the catalytic performance of Ru/YH3 is better. Compared to the previously reported catalysts for NEC hydrogenation, the TON of Ru/YH3 is also the highest (Table 1).
| Catalyst | Condition | Yield/% | TONa/h−1 | Reference |
|---|---|---|---|---|
| a TON is calculated as the amount of substance completely transformed per mole active metal per hour based on the hydrogenation curves. | ||||
| 5 wt% Ru/Al2O3 | 5 wt% Cat., 403 K, 7 MPaH2 | 99.5 | 68.7 | 9 |
| 5 wt% Ru/TiO2 | 5 wt% Cat., 403 K, 7 MPaH2 | 95.0 | 17.8 | 24 |
| Ru black | 5 wt% Cat., 403 K, 7 MPaH2 | 85.0 | 0.5 | 24 |
| 5 wt% Ru/AC | 10 wt% Cat., 403 K, 7 MPaH2 | 100 | 23.2 | 22 |
| 5 wt% Rh/AC | 20 wt% Cat., 403 K, 7 MPaH2 | 100 | 17.0 | 22 |
| 5 wt% Pd/AC | 20 wt% Cat., 403 K, 7 MPaH2 | 67.0 | 7.7 | 22 |
| F–LaNi5 | 10 wt% Cat., 443 K, 6 MPaH2 | 87.9 | 0.5 | 37 |
| RANEY® Ni | 10 wt% Cat., 453 K, 5 MPaH2 | 86.2 | 0.4 | 38 |
| 65 wt% Ni/Al2O3–SiO2 | 20 wt% Cat., 403 K, 7 MPaH2 | 50.0 | 0.3 | 23 |
| 5.2 wt% Ru/Al2O3 | 10 wt% Cat., 413 K, 6 MPaH2 | 96.6 | 24.2 | 28 |
| 0.52 mol% Pd2Ru/SiCN | 20 wt% Cat., 383 K, 2 MPaH2 | 97.9 | 7.2 | 39 |
| 4.6 wt% Ru/Al2O3 | 5 wt% Cat., 403 K, 7 MPaH2 | 100 | 90.0 | This work |
| 1.3 wt% Ru/YH3 | 5 wt% Cat., 403 K, 7 MPaH2 | 100 | 318.6 | This work |
Additionally, the hydrogenation on the Ru/YH3 catalyst is stereoselective. There are six stereoisomers in full hydrogenation products of NEC.9 When the catalyst is Ru/Al2O3, the products are (4a S, 4b R, 8a S, 9a S)–12H-NEC (47 mol%), (4a S, 4b R, 8a R, 9a S)–12H-NEC (41 mol%) and (4a S, 4b S, 8a R, 9a S)–12H-NEC (12 mol%) (Fig. 4b), which is in agreement with the previous reports.9 When the catalyst is Ru/YH3, the products are (4a S, 4b R, 8a R, 9a S)–12H-NEC (91 mol%) and (4a S, 4b S, 8a R, 9a S)–12H-NEC (9 mol%) (Fig. 4b). The main product on the Ru/YH3 catalyst is an all-cis product, which is easier to dehydrogenate on the catalytic surface with less steric hindrance. The formation all-cis 12H-NEC is favourable in dynamics but unfavourable in thermodynamics; therefore faster rates may result in a better stereoselectivity. Stereoselective hydrogenation of NEC was only reported on Ru black before; however only 70 mol% all-cis 12H-NEC was obtained.22,23 The authors proved that the stereoselectivity originated from the flatter surface of Ru black, which is unfavourable for stereoisomerization from all-cis 12H-NEC to other more thermodynamically stable products.22,23
The surface structure of Ru in Ru/YH3 is similar to that of Ru black according to the HRTEM images (Fig. 3a–c), resulting from the low surface area of YH3. However, the hydrogenation rate on Ru/YH3 is much faster than that on Ru black; hence Ru/YH3 exhibits a better stereoselectivity for all-cis 12H-NEC.
To further investigate the influences of temperature and hydrogen pressure, we performed a series of experiments under different conditions. The results (Fig. 4c and d) show that full hydrogenation can be accomplished at only 363 K (Fig. S5†). The higher the temperature, the faster the reaction rate. However, the increase is unremarkable when the temperature is over 403 K, which is limited by mass transfer of substrates.9 As a result, we choose 403 K as the optimized temperature. An increment of the reaction rate is also observed with increased hydrogen pressure. When the pressure is 3 MPa, the time to full hydrogenation is as long as 60 h (Fig. S6†). Therefore, high pressure is necessary to obtain a high enough rate in this case.
The H transfer mechanism
The catalytic activity of Ru/YH3 is superior to that of Ru/Al2O3, while the surface area of Ru/YH3 is much smaller than the surface area of Ru/Al2O3. The XPS spectra of Ru/Al2O3 and Ru/YH3 indicate that the electron effect of Al2O3 and YH3 is the same (Fig. S4†). The size of Ru is similar in the two catalysts and the flat surface of Ru in Ru/YH3 is unhelpful to surpass Ru/Al2O3. As YH3 has no activity (Fig. S7†), it must have some other positive influence on the catalytic hydrogenation of NEC.Although rare earth hydrides can reversibly adsorb and desorb hydrogen, they are not good hydrogen storage materials due to their low hydrogen capacities and high dehydrogenation temperatures. However, this property may be helpful in other application domains.27,40,41 The conversion temperature between YH3 and YH2 is as high as 642 K at 101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 Pa hydrogen pressure.42 In spite of this, some hydrogen can be released under the conditions of hydrogenation reaction according to the pressure-composition isotherm (PCI) curves of Y–H systems.42 In order to illustrate this, the TPD/MS curves of YH3 and Ru/YH3 are measured. The onset hydrogenation temperatures are around 373 K for both YH3 and Ru/YH3 (Fig. S8a†), suggesting that YH3 can desorb some hydrogen during the hydrogenation reaction. In contrast, the transformation of YH3−x into YH3 is easy at the hydrogenation temperature of NEC according to the PCI curve of YH3.42 In this situation, the hydrogenation of NEC on the Ru/YH3 catalyst can be schematically described as in eqn (1) and (2):
325 Pa hydrogen pressure.42 In spite of this, some hydrogen can be released under the conditions of hydrogenation reaction according to the pressure-composition isotherm (PCI) curves of Y–H systems.42 In order to illustrate this, the TPD/MS curves of YH3 and Ru/YH3 are measured. The onset hydrogenation temperatures are around 373 K for both YH3 and Ru/YH3 (Fig. S8a†), suggesting that YH3 can desorb some hydrogen during the hydrogenation reaction. In contrast, the transformation of YH3−x into YH3 is easy at the hydrogenation temperature of NEC according to the PCI curve of YH3.42 In this situation, the hydrogenation of NEC on the Ru/YH3 catalyst can be schematically described as in eqn (1) and (2):
| YH3 + NEC-Ru → YH3−x + 12H-NEC-Ru | (1) |
| YH3−x + H2 → YH3 | (2) |
Here YH3−x (0 < x < 1) denotes partially H deficient YH3. NEC-Ru and 12H-NEC-Ru mean NEC and 12H-NEC molecules adsorbed on the Ru surface. We have stated that eqn (1) and (2) are thermodynamically feasible by introducing the concept of hydrogen chemical potential in our previous report.27
To verify the assumption experimentally, we conduct the hydrogenation reaction using stoichiometric Ru/YH3 without H2. To avoid the influence of the gaseous H2 from the pyrolysis of YH3, we first set the temperature as 353 K, which is lower than the onset decomposition temperature of YH3. The NMR spectra of the product (Fig. S8b†) clearly show that a partial hydrogenation product tetrahydro-N-ethylcarbazole (4H-NEC) is obtained, and the pressure does not increase during the reaction. When the temperature is up to 403 K, the result (Fig. S8b†) is similar except that the amount of 4H-NEC increases. Although YH3 can release H2 at 403 K, the rate is very slow (Fig. S6†), consequently the pressure still does not increase. These results illustrate that there is indeed a new hydrogen transfer path in the hydrogenation reaction as eqn (1) and (2) show, which is the main cause of the high activity of Ru/YH3.
More general applicability
Although the catalytic performance of Ru/YH3 is excellent, the hydrogen pressure is very high to guarantee the selectivity for full hydrogenation products, which is unfavourable for the practical application. The plateau pressure of YH3 is lower than 1.4 kPa when the temperature is lower than 523 K, indicating that the reversible hydrogen absorption and desorption of YH3 can occur under much lower hydrogen pressure at reaction temperature.42 As the new hydrogen transfer path plays a key role in the reaction, the hydrogen pressure may be decreased by using more Ru/YH3 catalyst.To test the hypothesis, we use 100 wt% catalyst and 1 MPa H2 to run the hydrogenation reaction. Indeed, the hydrogenation rate is still fast, especially in the initial stage (Fig. 5a) when using Ru/YH3. Oppositely, the performance of Ru/Al2O3 is undesirable under this condition (Fig. 5a). If we use a flow reactor and maintain the hydrogen pressure, which is closer to the practical situation in industrial production, the ratio of Ru/YH3 to NEC will be higher and the reaction rate will be faster.
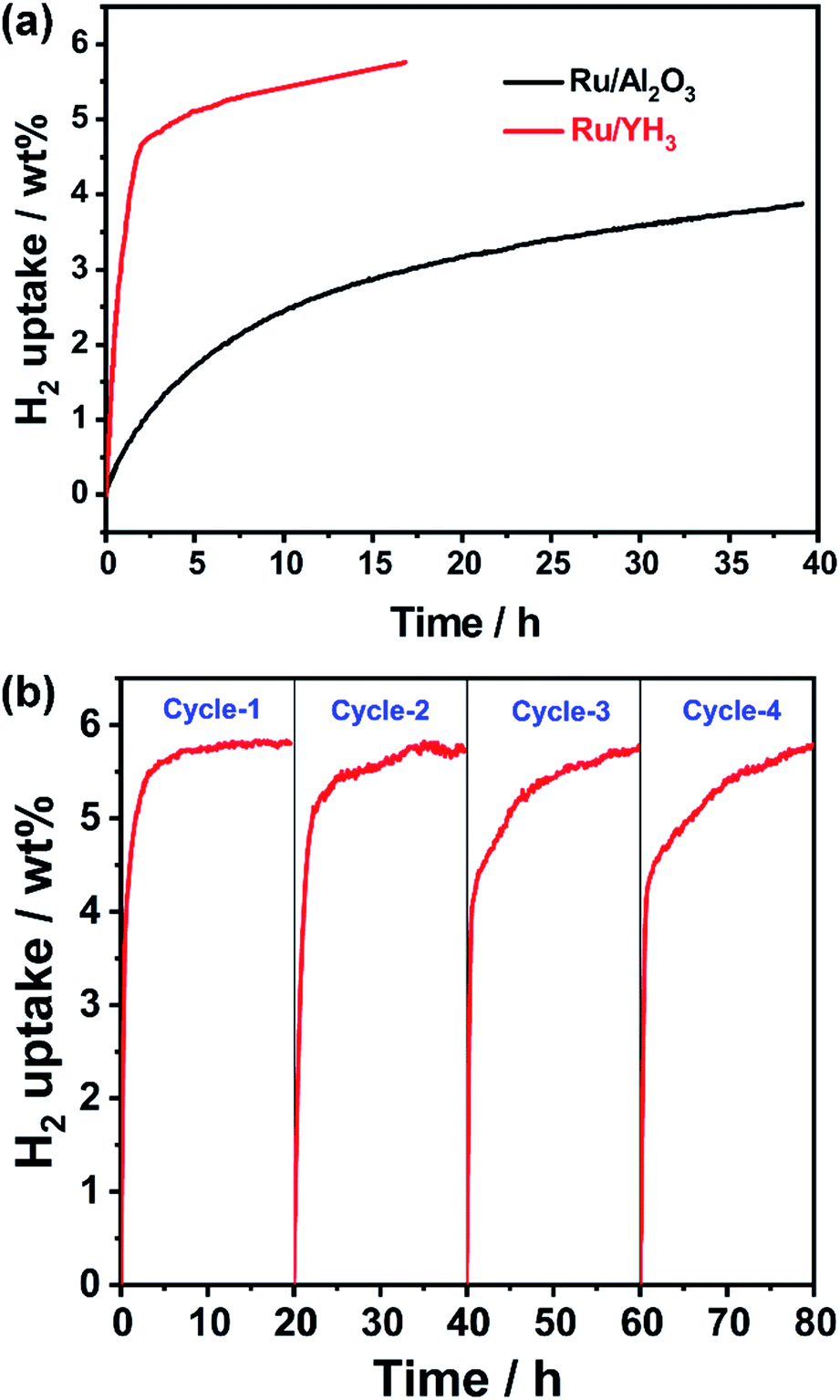 | ||
| Fig. 5 (a) Hydrogen absorption kinetics of NEC using Ru/YH3 and Ru/Al2O3 as catalysts (403 K, 1 MPa H2, 1.00 g NEC, 1.00 g catalyst); (b) hydrogenation kinetics on Ru/YH3 for four repeated cycles (403 K, 7 MPa H2, 1.00 g NEC per experiment, 50 mg catalyst). | ||
If we want to use Ru/YH3 in a continuous flow reactor, the catalytic stability is significant. The results of stability test (Fig. 5b) show that the rates are relatively stable, while the slight decrease of rates can be attributed to the unseparated products. In other words, the stability of the Ru/YH3 catalyst is satisfactory.
To investigate the range of applicability, we use Ru/YH3 to catalyse the hydrogenation reactions of another N-heterocycle, 2-methylindole (MID), under similar conditions. The results (Fig. 6a and S9†) show that MID can be hydrogenated completely in 10 h, implying that Ru/YH3 is a versatile catalyst for hydrogenation of N-heterocycles to some extent.
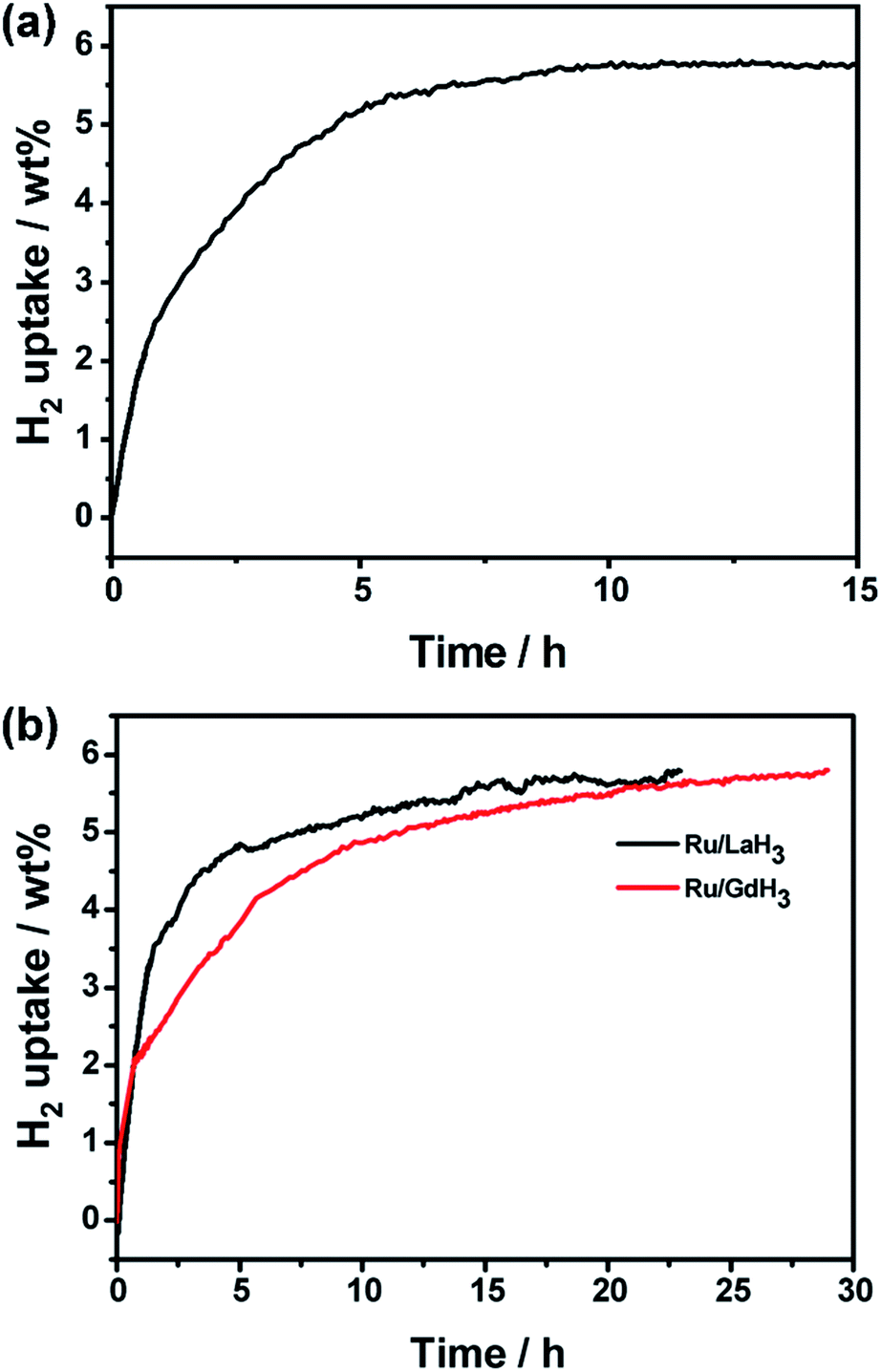 | ||
| Fig. 6 (a) Hydrogen absorption kinetics of 2-methylindole using Ru/YH3 as the catalyst; (b) hydrogen absorption kinetics of NEC using Ru/LaH3 and Ru/GdH3 as the catalyst. (403 K, 7 MPa H2, 1.00 g substrates, 50 mg catalyst). | ||
Considering the fact that the properties of rare earth hydrides are similar, we use LaH3 and GdH3 to replace YH3 in our experiments. As expected, the hydrogenation reaction can also be completed with high activity (Fig. 6b). This implies that rare earth hydrides are generic in this system. The overall rate of Ru/LaH3 is faster than that of Ru/GaH3, but slower than that of Ru/YH3. As the density becomes higher from Y to La to Gd, the volume and the surface area will be smaller when the weight is the same. Thus, the amount of Ru loading will decrease, resulting in worse catalytic performance. As a consequence, YH3 is a better choice for this system.
Conclusions
In summary, we have developed a rare earth hydride supported ruthenium catalyst Ru/YH3 for hydrogenation of N-heterocycles. For hydrogenation of a well-studied and promising liquid organic hydrogen carrier, ethylcarbazole (NEC), Ru/YH3 shows the highest catalytic activity as it has the highest turnover number. This means that the amount of precious metal Ru can be decreased, which is essential for the application in hydrogen storage. The outstanding activity is due to the new hydrogen transfer path from H2 to NEC provided by reversible hydrogen absorption and desorption of YH3. Additionally, complete hydrogen adsorption can be accomplished at the lowest temperature (363 K) and the lowest hydrogen pressure (1 MPa). Moreover, Ru/YH3 is also robust in terms of stability, low-pressure hydrogenation and stereoselectivity for all-cis products, which is desirable from the point of view of the application of NEC in hydrogen storage. The Ru/YH3 catalyst is also active for hydrogenation of 2-methylindole, another N-heterocycle. Other rare earth hydrides are also effective supports. The idea of rare earth hydride supported catalysts is an excellent strategy for developing N-heterocycle hydrogenation catalysts and new applications of rare earth hydrides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the MOST of China (No. 2018YFB1502102) and NSFC (No. 51771002, 51431001, 21771006, U1607126 and 21621061). The authors are also grateful for support from the Progress 100 program of Kyushu University.Notes and references
- V. Sridharan, P. A. Suryavanshi and J. Carlos Menendez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed.
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed.
- Y. Wu, Z. Chen, W.-C. Cheong, C. Zhang, L. Zheng, W. Yan, R. Yu, C. Chen and Y. Li, Chem. Sci., 2019, 10, 5345–5352 RSC.
- P. Ryabchuk, A. Agapova, C. Kreyenschulte, H. Lund, H. Junge, K. Junge and M. Beller, Chem. Commun., 2019, 55, 4969–4972 RSC.
- R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134–138 RSC.
- T. He, Q. Pei and P. Chen, J. Energy Chem., 2015, 24, 587–594 CrossRef.
- P. M. Modisha, C. N. M. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778–2796 CrossRef CAS.
- M. Niermann, A. Beckendorff, M. Kaltschmitt and K. Bonhoff, Int. J. Hydrogen Energy, 2019, 44, 6631–6654 CrossRef CAS.
- K. M. Eblagon, D. Rentsch, O. Friedrichs, A. Remhof, A. Zuettel, A. J. Ramirez-Cuesta and S. C. Tsang, Int. J. Hydrogen Energy, 2010, 35, 11609–11621 CrossRef CAS.
- M. Sobota, I. Nikiforidis, M. Amende, B. S. Zanon, T. Staudt, O. Hoefert, Y. Lykhach, C. Papp, W. Hieringer, M. Laurin, D. Assenbaum, P. Wasserscheid, H.-P. Steinrueck, A. Goerling and J. Libuda, Chem.–Eur. J., 2011, 17, 11542–11552 CrossRef CAS PubMed.
- Y. Dong, M. Yang, P. Mei, C. Li and L. Li, Int. J. Hydrogen Energy, 2016, 41, 8498–8505 CrossRef CAS.
- B. Wang, T.-y. Chang, Z. Jiang, J.-j. Wei, Y.-h. Zhang, S. Yang and T. Fang, Int. J. Hydrogen Energy, 2018, 43, 7317–7325 CrossRef CAS.
- B. Wang, T.-y. Chang, X. Gong, Z. Jiang, S. Yang, Y.-s. Chen and T. Fang, ACS Sustainable Chem. Eng., 2019, 7, 1760–1768 CrossRef CAS.
- I. Sorribes, L. Liu, A. Domenech-Carbo and A. Corma, ACS Catal., 2018, 8, 4545–4557 CrossRef CAS.
- A. Vivancos, M. Beller and M. Albrecht, ACS Catal., 2018, 8, 17–21 CrossRef CAS.
- X. Xue, M. Zeng and Y. Wang, Appl. Catal., A, 2018, 560, 37–41 CrossRef CAS.
- J.-W. Zhang, D.-D. Li, G.-P. Lu, T. Deng and C. Cai, ChemCatChem, 2018, 10, 4980–4986 CAS.
- S. Zhang, Z. Xia, T. Ni, Z. Zhang, Y. Ma and Y. Qu, J. Catal., 2018, 359, 101–111 CrossRef CAS.
- G. Jaiswal, M. Subaramanian, M. K. Sahoo and E. Balaraman, ChemCatChem, 2019, 11, 2449–2457 CrossRef CAS.
- S. Wang, H. Huang, C. Bruneau and C. Fischmeister, ChemSusChem, 2019, 12, 2350–2354 CAS.
- Z. Wei, F. Shao and J. Wang, Chin. J. Catal., 2019, 40, 980–1002 CrossRef CAS.
- K. M. Eblagon, K. Tam and S. C. E. Tsang, Energy Environ. Sci., 2012, 5, 8621–8630 RSC.
- K. M. Eblagon, K. Tam, K. M. K. Yu and S. C. E. Tsang, J. Phys. Chem. C, 2012, 116, 7421–7429 CrossRef CAS.
- K. M. Eblagon, K. Tam, K. M. K. Yu, S.-L. Zhao, X.-Q. Gong, H. He, L. Ye, L.-C. Wang, A. J. Ramirez-Cuesta and S. C. Tsang, J. Phys. Chem. C, 2010, 114, 9720–9730 CrossRef.
- K. M. Eblagon and S. C. E. Tsang, Appl. Catal., B, 2014, 160, 22–34 CrossRef.
- K. M. Eblagon and S. C. E. Tsang, Appl. Catal., B, 2015, 163, 599–610 CrossRef CAS.
- Y. Wu, H. Yu, Y. Guo, Y. Zhang, X. Jiang, B. Sun, K. Fu, J. Chen, Y. Qi, J. Zheng and X. Li, J. Mater. Chem. A, 2019, 7, 16677–16684 RSC.
- C. Wan, Y. An, G. Xu and W. Kong, Int. J. Hydrogen Energy, 2012, 37, 13092–13096 CrossRef CAS.
- H. Mizoguchi, M. Okunaka, M. Kitano, S. Matsuishi, T. Yokoyama and H. Hosono, Inorg. Chem., 2016, 55, 8833–8838 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- F. Hayashi, Y. Toda, Y. Kanie, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2013, 4, 3124–3130 RSC.
- V. K. Fedotov, V. E. Antonov, I. O. Bashkin, T. Hansen and I. Natkaniec, J. Phys.: Condens. Matter, 2006, 18, 1593–1599 CrossRef CAS.
- M. Oetzel and G. Heger, J. Appl. Crystallogr., 1999, 32, 799–807 CrossRef CAS.
- A. Fujimori and L. Schlapbach, J. Phys. C: Solid State Phys., 1984, 17, 341–351 CrossRef CAS.
- T. Mongstad, A. Thogersen, A. Subrahmanyam and S. Karazhanov, Sol. Energy Mater. Sol. Cells, 2014, 128, 270–274 CrossRef CAS.
- D. Zhu, H. Jiang, L. Zhang, X. Zheng, H. Fu, M. Yuan, H. Chen and R. Li, ChemCatChem, 2014, 6, 2954–2960 CrossRef CAS.
- F. Wu, Y. An, L. Song, G. Xu and L. Xia, Chem. React. Eng. Technol., 2015, 31, 407–411 CAS.
- X. Ye, Y. An and G. Xu, J. Alloys Compd., 2011, 509, 152–156 CrossRef CAS.
- D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima and R. Kempe, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- J. Wang, K. Fu, X. Li and G. Li, Vacuum, 2019, 162, 67–71 CrossRef CAS.
- J. Wang, G. Li, K. Fu and X. Li, J. Mater. Sci., 2019, 54, 13334–13343 CrossRef CAS.
- K. Fu, X. Jiang, Y. Guo, S. Li, J. Zheng, W. Tian and X. Li, Prog. Nat. Sci.: Mater. Int., 2018, 28, 332–336 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures. See DOI: 10.1039/c9sc04365a |
| This journal is © The Royal Society of Chemistry 2019 |