
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging hypervalent iodine and sulfoximine chemistry: a new electrophilic trifluoromethylation reagent†
Jorna
Kalim‡
 a,
Thibaut
Duhail‡
b,
Thanh-Nghi
Le
b,
Nicolas
Vanthuyne
c,
Elsa
Anselmi
b,
Antonio
Togni
*a and
Emmanuel
Magnier
*b
a,
Thibaut
Duhail‡
b,
Thanh-Nghi
Le
b,
Nicolas
Vanthuyne
c,
Elsa
Anselmi
b,
Antonio
Togni
*a and
Emmanuel
Magnier
*b
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 2, 8093 Zürich, Switzerland. E-mail: atogni@ethz.ch
bInstitut Lavoisier de Versailles, UMR CNRS 8180, Université de Versailles-Saint-Quentin, 45 Avenue des Etats-Unis, 78035 Versailles Cedex, France. E-mail: emmanuel.magnier@uvsq.fr
cInstitut des Sciences Moléculaires de Marseille, Centrale Marseille, UMR 7313, Aix-Marseille Université, CNRS, Avenue Escadrille Normandie Niemen, 13013 Marseille Cedex, France
First published on 27th September 2019
Abstract
Electrophilic trifluoromethylation is at the forefront of methodologies available for the installation of the CF3 moiety to organic molecules; research in this field is largely spurred by the availability of stable and accessible trifluoromethylation reagents, of which hypervalent iodine and sulfoximine based compounds have emerged as two prominent reagent classes. Herein, we describe the facile synthesis of an electrophilic trifluoromethylation reagent which merges these two scaffolds in a novel hypervalent iodosulfoximine compound. This presents the first analogue of the well-known Togni reagents which neither compromises stability or reactivity. The electronic and physical properties of this new compound were fully explored by X-ray crystallography, cyclic voltammetry, TGA/DSC and DFT analysis. This solution stable, crystalline reagent was found to be competent in the electrophilic trifluoromethylation of a variety of nucleophiles as well as a source of the trifluoromethyl radical. Furthermore, the possibility of enantioinductive transformations could be probed with the isolation of the first enantiopure hypervalent iodine compound bearing a CF3 group, thus this new reagent scaffold offers the opportunity of structurally diversifying the reagent towards asymmetric synthesis.
Introduction
The unique properties imbued by the trifluoromethyl group to organic molecules has made this motif ubiquitous in compounds of interest in pharmaceutical, agrochemical and materials research.1 Its introduction often leads to changes in the chemical, biological and physical properties of a given compound; for example, lipophilicity and membrane permeability of drug candidates are improved. Furthermore, the energetic cost of breaking the C–F bond compared to other C-heteroatom bonds means that fluorinated compounds display enhanced chemical stability. The accessibility of this group is largely dependent on the development of CF3 transfer reagents, which can be classified according to their nucleophilic, electrophilic or radical character. The utility of nucleophilic trifluoromethylation reagents, exemplified by trifluoromethyltrimethylsilane (TMSCF3), has been expansively studied, however the need for differential functional group tolerance has spurred extensive research in the development of electrophilic trifluoromethylation reagents.2One prominent class of electrophilic trifluoromethylating reagents are hypervalent iodine compounds 1 and 2 (Fig. 1), first reported by one of our groups in 2006.3 Reagents 1 and 2, have been used to transform a wide variety of nucleophiles, for the generation of new C–, N–, O–, P–, or S–CF3 bonds. In many cases this process corresponds to a substitution of a hydrogen atom by the CF3 group. Synthetically and commercially, reagents 1 and 2 are readily accessible as crystalline shelf-stable compounds, resulting in vigorous research efforts by one of our groups and others towards utilizing them in disparate applications. This research has received particular attention, given the increasing interest of the synthetic community for new methodologies in organofluorine chemistry. The activation of 1 and 2 can be achieved using Brønsted or Lewis acids which increases their electrophilic or iodonium-like character, thus matching the electrophilic character of the reagent to the nucleophile.3c,4 One of our groups recently reported the activation of reagent 2 with TiCl4 which enabled the trifluoromethylation of sulfonate salts, a transformation which is otherwise difficult to achieve.4b
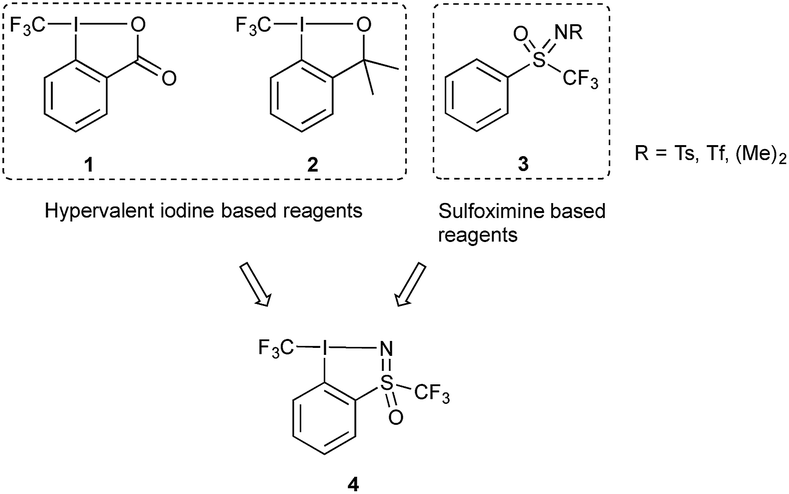 | ||
| Fig. 1 Known electrophilic fluoroalkylation reagents, 1, 2 and 3 and newly designed hypervalent iodosulfoximine reagent 4. | ||
Another class of electrophilic fluoroalkylating reagents are sulfoximine based compounds of type 3 (Fig. 1).5 They have been successfully employed for the introduction of trifluoro- and monofluoromethyl groups by Shibata and coworkers,6 difluoromethyl by the groups of Hu7 as well as Olah and Prakash,8 and for bromodifluoro- and dichlorofluoromethyl by one of our groups.9 In 2012, Hu and coworkers greatly expanded the field of application of perfluoroalkylated sulfoximine reagents by showing that mono- and difluoromethyl derivatives can also be efficient reagents for the nucleophilic transfer of perfluorinated entities.10 More recently, Akita and coworkers have published the use of the same reagent as a source for generating difluoromethyl radicals.11 One year later, one of our groups extended this methodology to other perfluoroalkyl radicals including the trifluoromethyl group.12 One important property of these compounds is the ease with which they can be structurally diversified and their properties finely tuned for targeted applications.
Given the growing body of research in new trifluoromethylating reagents based on compounds of the type shown in Fig. 1, we were interested in merging these two scaffolds to generate a new reagent, resulting in the design of compound 4. Vastly altering the scaffold of 1 and 2 has been investigated in our labs and others, however in most cases these modifications either resulted in diminished reactivity, product instability, or proved difficult to synthetically access.13 One such example saw the introduction of a para-NO2 group on compound 1, which resulted in reduced solubility and instability under acidic reaction conditions.13d In 2018 an acyclic chloro(phenyl)trifluoromethyliodane reagent was reported, which showed similar reactivity to 1 and 2.13e Previous attempts at synthesising this compound by Yagupolskii and Umemoto proved challenging, presumably due to the lack of stability of the acyclic structure.14 The reported compound was found to be stable as a solid, but substantial degradation was observed in commonly used solvents, for example 65% decomposition in MeCN-d3 after 1 day. This finding suggests cyclic hypervalent iodine compounds such as 1 and 2 are more stable in solution compared to acyclic analogues. A further challenge in trifluoromethylation chemistry that has yet to be addressed is the introduction of chirality onto the reagent scaffold,13b in order to enable the synthesis of enantioenriched trifluoromethylated substrates. Given the chirality of the sulfoximine group in 3, we anticipated that reagent 4 could be used to investigate enantioinductive transformations.
In this paper we describe the synthesis and characterization of an enantiopure, hypervalent iodosulfoximine compound 4 which is also an efficient trifluoromethylating reagent. To the best of our knowledge, this is the first example of a hypervalent iodine bonded to the nitrogen of a sulfoximine moiety incorporated into the 5-membered heterocyclic ring. The properties of compound 4 were investigated by X-ray crystallography, cyclic voltammetry, TGA/DSC and DFT. We conducted a comparative reaction screening which shows that in most cases reagent 4 reacts in a very similar fashion to reagent 1 and 2, a very gratifying finding as this is the first successful modification of the original iodine(III) reagents which does not significantly impact reactivity or reduce stability.
Results and discussion
Reagent design and synthesis
A sulfoximine substituent on the aromatic ring ortho to the iodine center was expected to have a strong enough electron withdrawing character to stabilize the 3-centre 4-electron hypervalent iodine bond. Hypervalent iodine compounds bearing a sulfoximine group have previously been reported by Bolm and co-workers, however the reported compounds were designed for the transfer of the sulfoximine moiety, and hence not a part of the 5-membered heterocycle.15Synthetic accessibility to iodine(I) reagent precursor 5 has previously been reported by the Versailles lab, starting from phenyl trifluoromethyl sulfoxide 6 (Scheme 1).16 This three-step procedure involves the preparation of a sulfilimine 7 (via a Ritter-like process), its subsequent oxidation and final N-deprotection sequence to deliver the sulfoximine 8. This protocol can be safely followed on a multi-gram scale, allowing for the preparation of 33 g of 8.17 In the next step, the trifluoromethylsulfoximine group acts as an ortho-directing substituent and various electrophiles can be introduced. Thus, ortho-lithiation followed by quenching with excess iodine gave the iodoarene 5 in excellent yields on a 15 g scale.18 In a more recent report, sulfoximine 8 could be accessed via a one-pot protocol starting from commercially available phenyl trifluoromethyl sulfide, using phenyliodine diacetate and ammonium carbamate followed by acid hydrolysis. Application of this method gives compound 5 in two steps as opposed to three, however large scale synthesis via this route has not yet been established.19
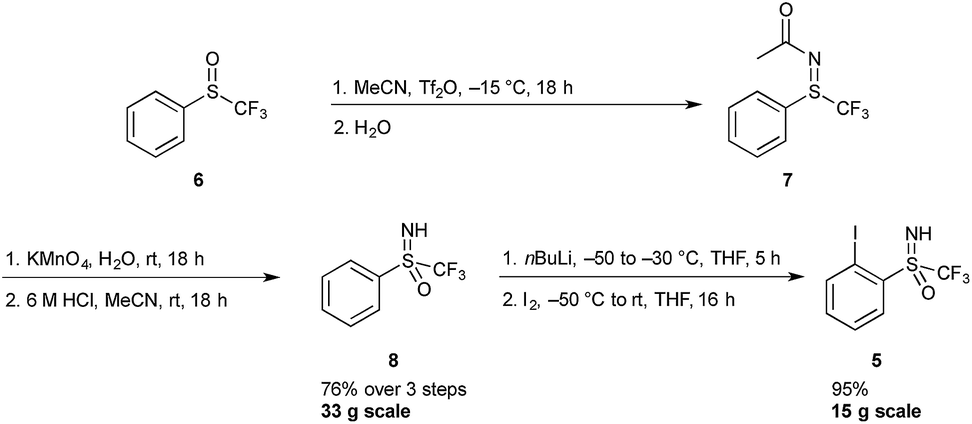 | ||
| Scheme 1 Synthesis of iodosulfoximine compound 5. Tf2O = trifluoromethanesulfonic anhydride. | ||
The improved one-pot procedure for the synthesis of reagent 1 was then successfully applied to iodosulfoximine 5 (Scheme 2), allowing for the isolation of reagent 4.20 Oxidation is first achieved using trichloroisocyanuric acid (TCICA) and 5 mol% trifluoroacetic acid (TFA) as a Brønsted acid catalyst,21 which facilitated the reaction at room temperature. After one hour, complete conversion to the chloroiodane 9 was confirmed by 19F NMR, and a subsequent ligand exchange at the iodine atom with potassium acetate was also found to be facile at room temperature with full conversion in 30 minutes. This is in contrast to the conditions reported for the synthesis of 1, which involved heating at reflux for both the oxidation and ligand exchange steps. Finally, a second ligand exchange with TMSCF3 gave 4 in excellent yield (74%) over the three steps. Reagent 4 could be purified by recrystallization from a mixture of pentane/MeCN/Et2O (see ESI for details†), a simplified protocol compared to that reported previously for 1 and 2.20 The one-pot protocol could also be applied on a 10 g scale to give compound 4 in 75% (see ESI for details†). This yield compares well with that of the synthesis of 1, which can be obtained in 72% over three-steps from 2-iodobenzoic acid, and is improved compared to the four-step protocol for the synthesis of 2 which can be obtained in 67% yield from methyl 2-iodobenzoate.20
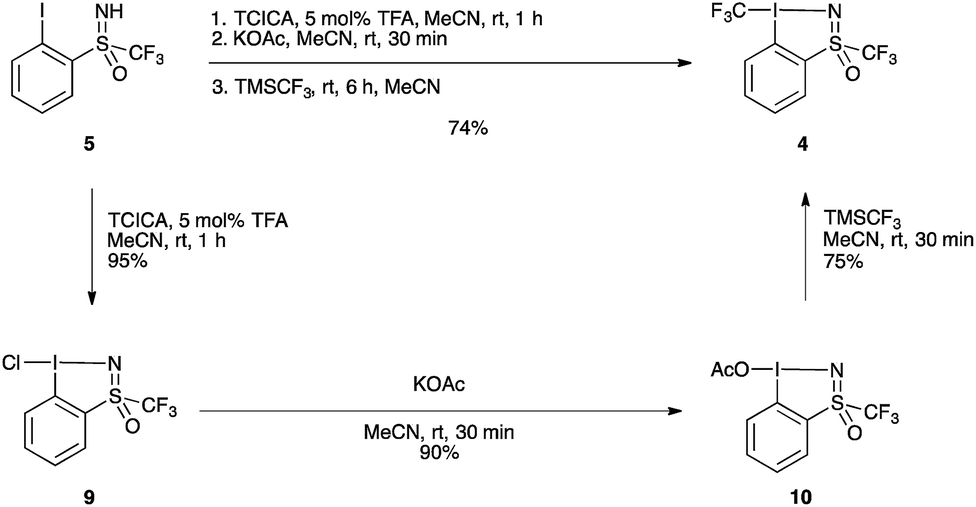 | ||
| Scheme 2 Synthesis of hypervalent iodosulfoximine reagent 4. TCICA = trichloroisocyanuric acid, TFA = trifluoroacetic acid. | ||
Additionally, the intermediate chlorinated and acetylated iodine(III) precursors could be isolated in excellent yields, 95% for chloroiodane 9 and 90% for 10 (starting from 9) following the standard procedures (see ESI for details†).20 Reagent 4 could be accessed from isolated compound 10, albeit with no improvement of yield compared to the one-pot protocol starting from 5. It is worth noting the presence of a trifluoromethyl group linked to the sulfur atom is crucial as the same transformation with the S–methyl sulfoximine failed (see ESI, Scheme S1†).
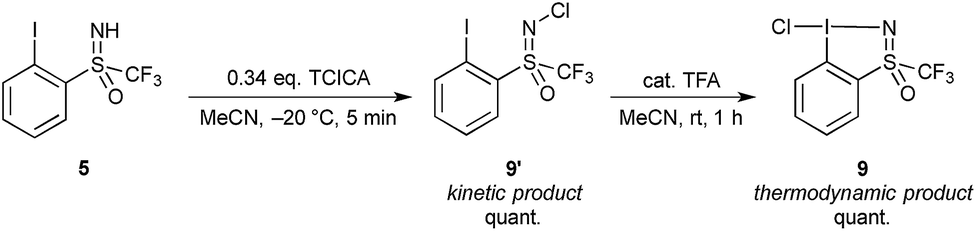
Given the chirality of the sulfoximine group on the reagent scaffold, we envisaged taking advantage of this property for the preparation of an enantiopure reagent. We were pleased to find that the iodosulfoximine 5 could be separated by preparative chiral HPLC using a Chiralpak AS-H column and hexane/isopropanol (80/20) as mobile phase on a 2 g scale. The separated enantiomers were configurationally stable on the bench as confirmed by analytical chiral HPLC after several weeks. With the stable enantiomeric form of (+) and (−)-5 in hand we proceeded to synthesize the enantiopure form of (−) and (+)-4 respectively (see ESI for optical rotation values†). Gratifyingly, the one-pot protocol for the preparation of reagent 4 preserved the absolute configuration of the sulfur atom as confirmed by analytical chiral HPLC, which gave a single peak suggesting no epimerization had occurred. Furthermore, good quality single crystals that were used for an X-ray diffraction study of the structure of chloroiodane (−)-9 could be obtained by slow evaporation of a dichloromethane solution, thereby determining the absolute configuration to be (S)-(−)-9 (Fig. 2).
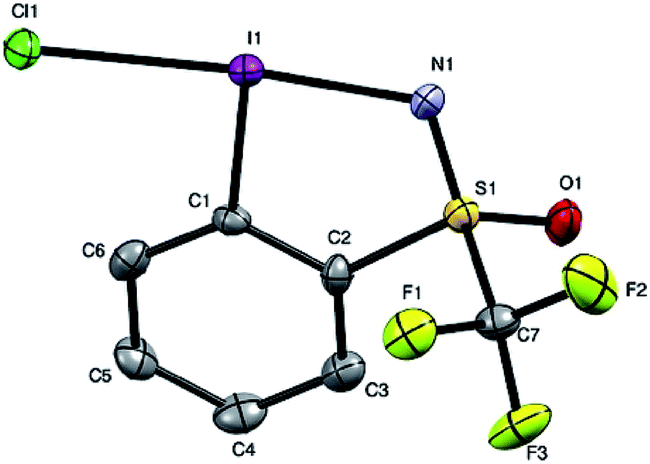 | ||
| Fig. 2 Structure of (S)-(−)-chloroiodane, 9, as determined by X-ray diffraction. Selected bond lengths and angles: I1–Cl1: 2.576(2) Å; I1–N1: 2.102(6) Å; N1–S1: 1.498(8) Å; angle: Cl1–I1–N1: 174.9(2)°; C1–I1–Cl1: 89.5(2)°; C1–I1–N1: 85.4(3)°; torsion: C6–C1–I1–Cl1: 5.1(7)°; C2–C1–I1–N1: 3.3(6)°. H atoms are omitted for clarity; thermal ellipsoids given at 50% probability. | ||
Tautomerization of chlorinated 5
Interestingly, during our first attempts at oxidizing the iodine center with TCICA using the original conditions reported for the synthesis of 1 and 2 without use of a Brønsted acid catalyst, an unexpected product was obtained. We found that conducting the reaction at room temperature with 0.34 eq. of TCICA gave a mixture of products, the desired cyclic structure 9 and another product characterized by a new peak in the 19F NMR spectrum at −63.89 ppm in MeCN-d3. We assigned this new compound to an acyclic N-chlorinated structure 9′. Furthermore, we found that conducting the reaction at −20 °C gave a mixture of 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of 9′ to 9, and slow irreversible transformation of 9′ into 9 could be observed at room temperature over 12 hours. Hence, 9′ is the kinetic product for the chlorination with TCICA, with 9 being the thermodynamically favoured tautomer (Scheme 3).
:1 of 9′ to 9, and slow irreversible transformation of 9′ into 9 could be observed at room temperature over 12 hours. Hence, 9′ is the kinetic product for the chlorination with TCICA, with 9 being the thermodynamically favoured tautomer (Scheme 3).
 | ||
| Scheme 3 Oxidative chlorination of 5 with TCICA giving N-chlorinated compound 9′ which tautomerizes to cyclic chloroiodane 9. | ||
The structure of N-chlorinated compound 9′ could be confirmed by NMR and HRMS analysis, crystal structural analysis was inaccessible as compound 9′ tautomerizes to 9 in solution at low temperatures as well as in the solid state. To further corroborate the acyclic structure of 9′ we compared the obtained NMR data with that of the newly synthesized and previously reported compounds (see ESI, Table S1†). The Versailles lab has recently reported the synthesis of an N-chlorinated sulfoximine analogue of 8via the same method,22 and the 19F NMR signal of this compound was reported to be at −68.19 ppm in MeCN-d3, which is a 4 ppm difference to that of 9′. Analysis of the 19F NMR chemical shifts of the synthesized cyclic reagents 4, 9 and 10 showed a 13 ppm difference between the signal of acyclic 9′ and that of the cyclic structures (which are in −77 ppm region). Additionally, to confirm this acyclic structure we examined the 13C NMR shifts for the carbon atom bound to iodine on the aromatic ring, Cipso (see ESI, Table S1†) which gives a distinct chemical shift depending on the oxidation state of the iodine atom. In iodine(I) compounds the ipso-carbon is strongly shielded as a result of relativistic effects, which is reduced in iodine(III) and iodine(V) species.23 This trend is observed in both cyclic and acyclic variants of oxidized iodine species and is not impacted strongly by the nature of the ligands at the iodine atom (see Table S1 in ESI†).23 When comparing the 13C NMR shifts of the new and previously synthesized compounds we found that derivatives of iodine(I) gave a signal in the 95 ppm region, whilst iodine(III) species had a 13C NMR chemical shift of 110–122 ppm for Cipso, a significant difference of 15–27 ppm.
The instability of N-chloroamines and their use as oxidants in organic synthesis is well described; N-chloroamides such as TCICA and N-chlorosuccinimide are known to undergo reactions with amines via Cl+ oxidation, hence it is unsurprising to find that TCICA reacts preferentially with the imine moiety.24 To probe the mechanism of the tautomerization process we monitored the reaction profile by 19F NMR spectroscopy (see ESI for details†). The rate of decay of 9′ and the rate of formation of 9 was found to be the same. Increasing the concentration or the temperature accelerates the tautomerization process, with the fastest reaction rates observed with the addition of TFA at room temperature. Furthermore, an induction period was seen at various temperatures and concentrations, with no intermediate observed and only traces (<1%) of by-products identified in the 19F NMR spectrum. Given the sigmoidal curve for the reaction profile, which is characteristic of autocatalytic processes, we could rule out such a mechanism by addition of 10 mol% of product 9 to a mixture of 5, which was found to have the same reaction profile as the same reaction mixture without the addition of compound 9 (see ESI for details†). Since many chlorination reactions using N-haloamines have been proposed to occur via formation of radical species such as variations of the Hofmann–Löffler–Freytag reaction,25 we explored this possibility by conducting the reaction in the absence of light, but found the reaction to proceed; EPR studies of the reaction mixture also gave no signal for radical intermediates. Kinetic studies conducted thus far have proved inconclusive. However, from these qualitative findings we could rule out covalent isomerization processes where the Cl atom migrates from nitrogen to iodine or bimolecular transition states involving two 9′ molecules, where Cl atom transfer occurs in a concerted fashion, as these would show first or second order reaction profiles respectively. One suggestion for the mechanism by which this occurs would be that the reaction proceeds via an ionic mechanism involving an iodonium [8-I-2] species, these intermediates have been previously reported to form via a dissociative mechanism from [10-I-3] compounds.26 Further investigation of this unusual reaction mechanism is underway in our laboratories.
Structure and properties
In order to get a full understanding of the stability and reactivity of 4, we performed several analytical measurements. TGA/DSC analysis revealed no melting event to occur before the onset of rapid thermal decomposition at 146.3 °C within a short temperature window. A similar observation was made in previous reports for compound 1.27 The solution stability of the reagent was checked in several solvents. After 1 day traces of decomposition products (<5%) were observed in methanol-d4 and 15% after 4 days, similarly 8% decomposition was observed after 4 days in CDCl3, but the compound was stable in MeCN-d3 for 1 week, and only traces (<5%) of decomposition products were observed in acetone-d6 after 1 week. Thus, the reagent is stable in commonly used solvents for trifluoromethylation reactions for extended periods, and the minor decomposition observed is indicative of its reactivity.Good quality single crystals of compound 4 for X-ray diffraction could be obtained by slow evaporation of a dichloromethane solution (Fig. 3). The structural features of this new reagent were compared with that of compounds 1 and 2 (Table 1).3a,b The I1–N1 bond of compound 4 is elongated relative to the I1–O1 bond of reagent 2 (2.284(2) in 4vs. 2.1176(14) Å in 2) and is the same as that of compound 1 (2.283(2) Å), indicating that the carboxylate and sulfoximine moieties have similar electron withdrawing properties. This trend is reversed when looking at the I1–CF3 bond lengths, which is shortest in compound 1 (2.219 Å), with similar bond lengths observed for compounds 2 and 4 (2.267(2) and 2.250(3) Å respectively). Interestingly, the lengthening of the I1–N1 bond in the sulfoximine reagent does not coincide with a significant shortening of the I1–CF3 bond length which is typically observed in cyclic hypervalent iodine structures of this type. The CF3–I1–N1 bond angle in the sulfoximine reagent (173.35(9)°) is closer to 180°, compared to the I1–C8–O1 bond angles of reagents 1 and 2 which are around 170°, a consequence of the large N1–S1 bond length (1.482(2) Å). Furthermore, structural overlap of the three compounds (Table 1) highlights the distortion of the torsion angles N1–I1–C1–C2 (4.6(2)°) and C8–I1–C1–C6 (10.1(2)°) in 4 out of plane of the phenyl group, which is not as pronounced as the corresponding angles in compound 2 (11.56(15)° and 13.21(2)°), with 1 being essentially co-planar.
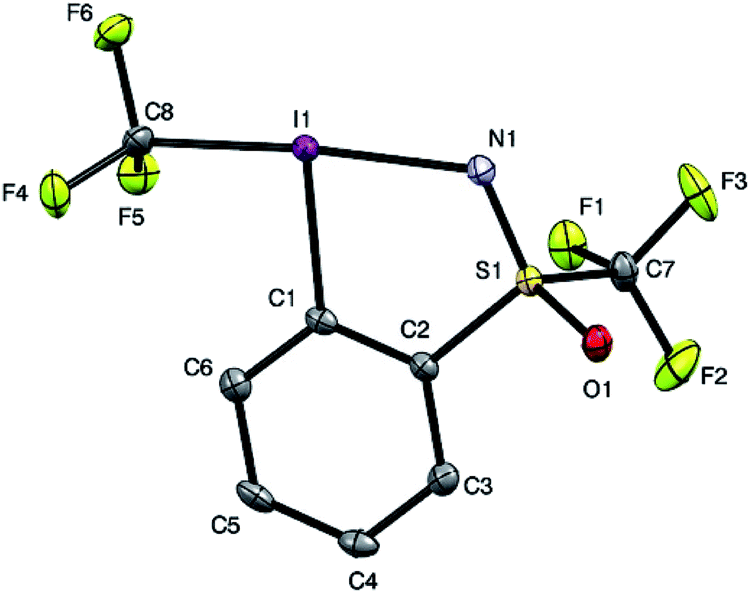 | ||
| Fig. 3 Structure of hypervalent iodine compound 4 (see Table 1 for structural properties). H atoms are omitted for clarity; thermal ellipsoids given at 50% probability. | ||
|
|
|||
|---|---|---|---|
| Reagent 1a | Reagent 2a | Reagent 4 | |
| a Data for reagent 1 and 2 taken from the literature.3a,b X = O1 or N1. | |||
| I1–X1 | 2.283(2) | 2.1176(14) | 2.284(2) |
| I1–CF3 | 2.219(4) | 2.267(2) | 2.250(3) |
| I1–C1 | 2.113(3) | 2.1211(19) | 2.132(2) |
| X1–I1–CF3 | 170.49(12) | 169.78(7) | 173.35(9) |
| C1–I1–X1 | 76.79(11) | 78.71(6) | 82.73(8) |
| C1–I1–CF3 | 93.74(14) | 91.11(8) | 91.76(9) |
| X1–I1–C1–C2 | 0.3(2) | 11.56(15) | 4.6(2) |
| F3C–I1–C1–C6 | 0.7(3) | 13.21(18) | 10.1(2) |
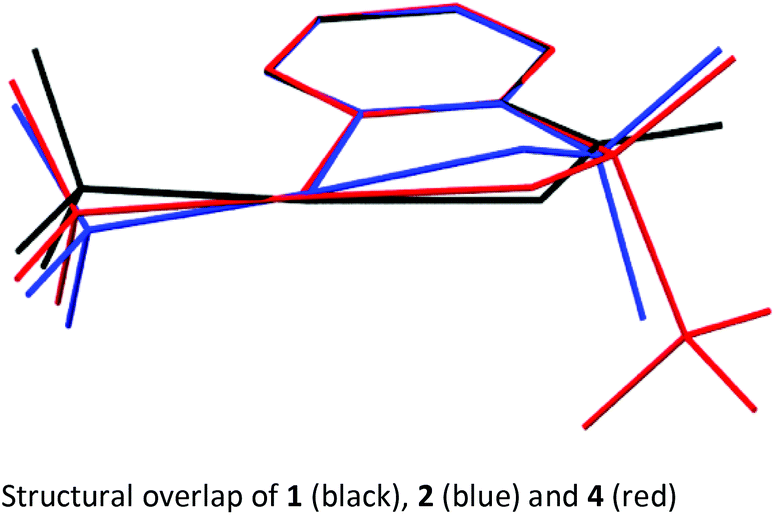
The structural properties of these hypervalent iodine reagents can be loosely related to their reactivity in that a lengthening of the I1–X1 bond length (usually coinciding with the shortening of the I1–CF3 bond length) results in faster initial rates of reaction when toluene sulfonic acid is used as the model substrate, with 1 showing much faster reactivity than 2.13b However, this correlation only loosely holds as solid-state bond lengths may not truly reflect solution bond lengths due to crystal packing effects.
Given the novel structure of 4, we aimed at gaining a deeper understanding of the electronic effects of the sulfoximine moiety on the hypervalent iodine scaffold and how this may compare to the existing reagents. Molecular electrostatic potential (MEP) surfaces were generated for the visualisation of electron density on all three compounds for comparison using the previously reported methods (Fig. 4, see ESI for details†).28 Comparing the MEP electron density of the three reagents, a strong polarization towards the ligands is observed in all three compounds, with the calculated dipole moments following the trend: 5.2 D in 1; 5.1 D in 4; and 3.0 D in 2. The electron density on the heteroatom directly bound to iodine is similar in all three compounds at around −0.05 au. Larger differences are seen in the expression of the “σ-hole” – a region of positive charge at the iodine atom,28 the most strongly expressed region of positive potential is seen in compound 1 followed by that of compound 4. These electronic properties have an impact on the strength and directionality of reagent–substrate or reagent–solvent interaction, wherein a nucleophile or polar solvent would be much more strongly coordinated to compounds 1 and 4 compared to 2.
Finally, cyclic voltammetry experiments were conducted to compare the redox potentials of the new reagent 4 with that of 1 and 2 (see ESI for details†). The first cathodic peak potential for reagent 4vs. [FeCp2]/[FeCp2]+ was found to be −2.02 V which is similar to that of 1 (−1.91 V vs. [FeCp2]/[FeCp2]+), compound 2 was found to be the most difficult to reduce with a first cathodic peak potential of −2.42 V under the same conditions. Hence, the cyclic voltammetry data further corroborates the similarity of compound 4 to 1, both of which are more easily reduced compared to compound 2.
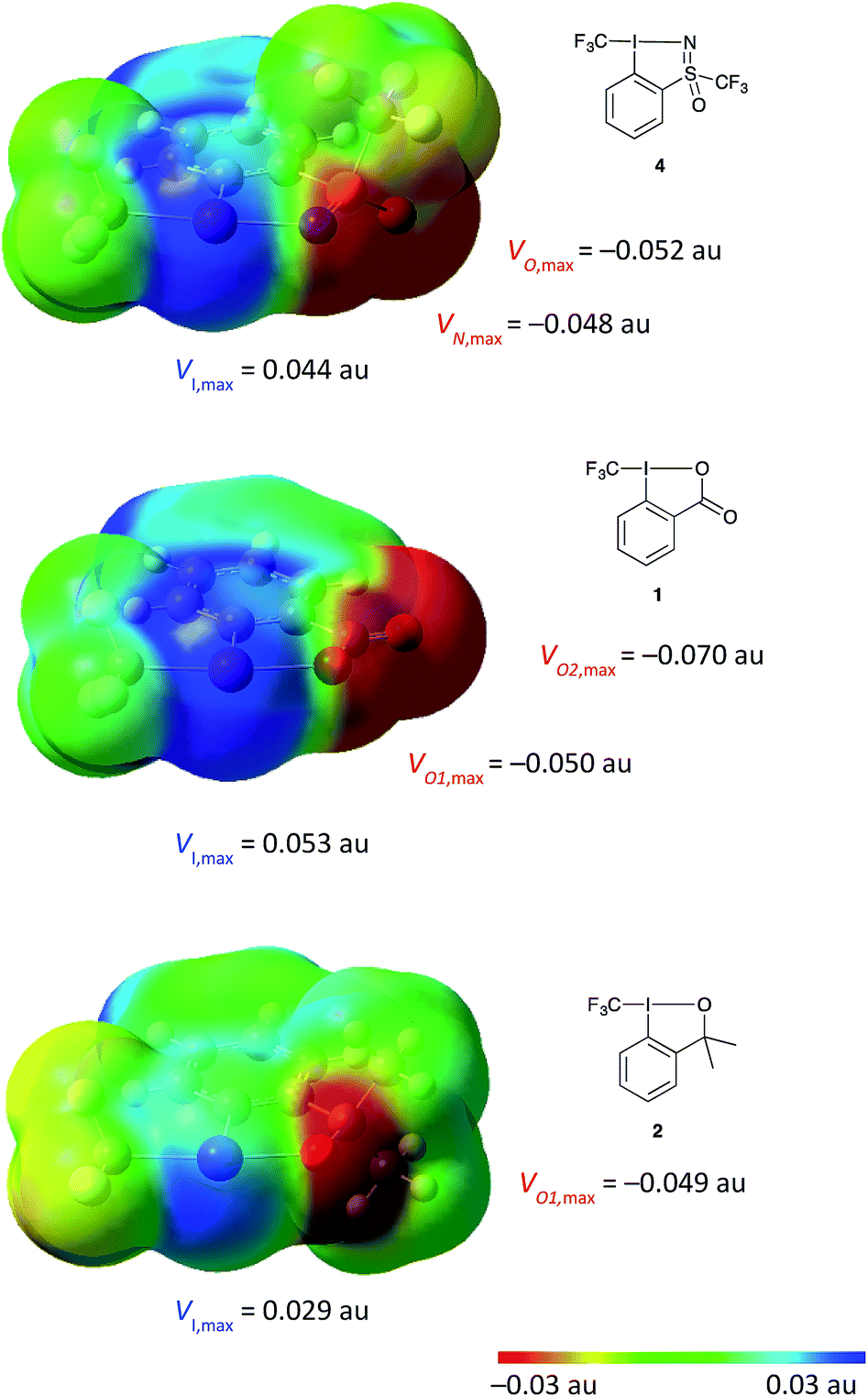 | ||
| Fig. 4 DFT calculation done at the B3LYP, aug-cc-pVTZ (for H, C, O, N), aug-cc-pV(T+d)Z (for S) and aug-cc-pVTZ-PP (for I). MEP mapped onto the isodensity surface at 0.001 au, within the vdW contact distances containing 96% of molecular electron density (see ESI for details†).28 | ||
Reactivity studies
After establishing the structural similarity of this new reagent 4 to the existing trifluoromethylation reagents 1 and 2, we proceeded to investigate its reactivity. To this aim, we attempted the trifluoromethylation of various S-, C-, O-, and P-centered nucleophiles (Scheme 4), and compared the yields to the previously reported results for 1 and 2. Trifluoromethylation of sulfonic acids was investigated first (entry 1), as this has been established as a clean and fast reference reaction in one of our labs for testing newly designed reagents.13b To our satisfaction, we found that compound 4 performed comparatively to 1 when camphorsulfonic acid 11 is used as a substrate, giving 92% 19F NMR yield after 90 minutes for 12.29 Additionally, iodoarene 5 could be recovered from the reaction mixture, with 70% recovery after column chromatography in the case of entry 1 (Scheme 4), providing the opportunity to potentially re-use the material. Trifluoromethylation of chlorothiophenol 13 gave comparable results, and 14 was obtained in 73% 19F NMR yield (entry 2); typical yields for the trifluoromethylation of aromatic thiols with reagent 2 have been reported in 72–91% yield.3b Diphenylphosphine 15 (entry 3) also proved to be a viable substrate giving the trifluoromethylated product 16 in 69% yield.30 Oxytrifluoromethylation of styrene 17 (entry 4) gave satisfactory yields of 66% respectively, compared to 93% for the same transformation using reagent 1.31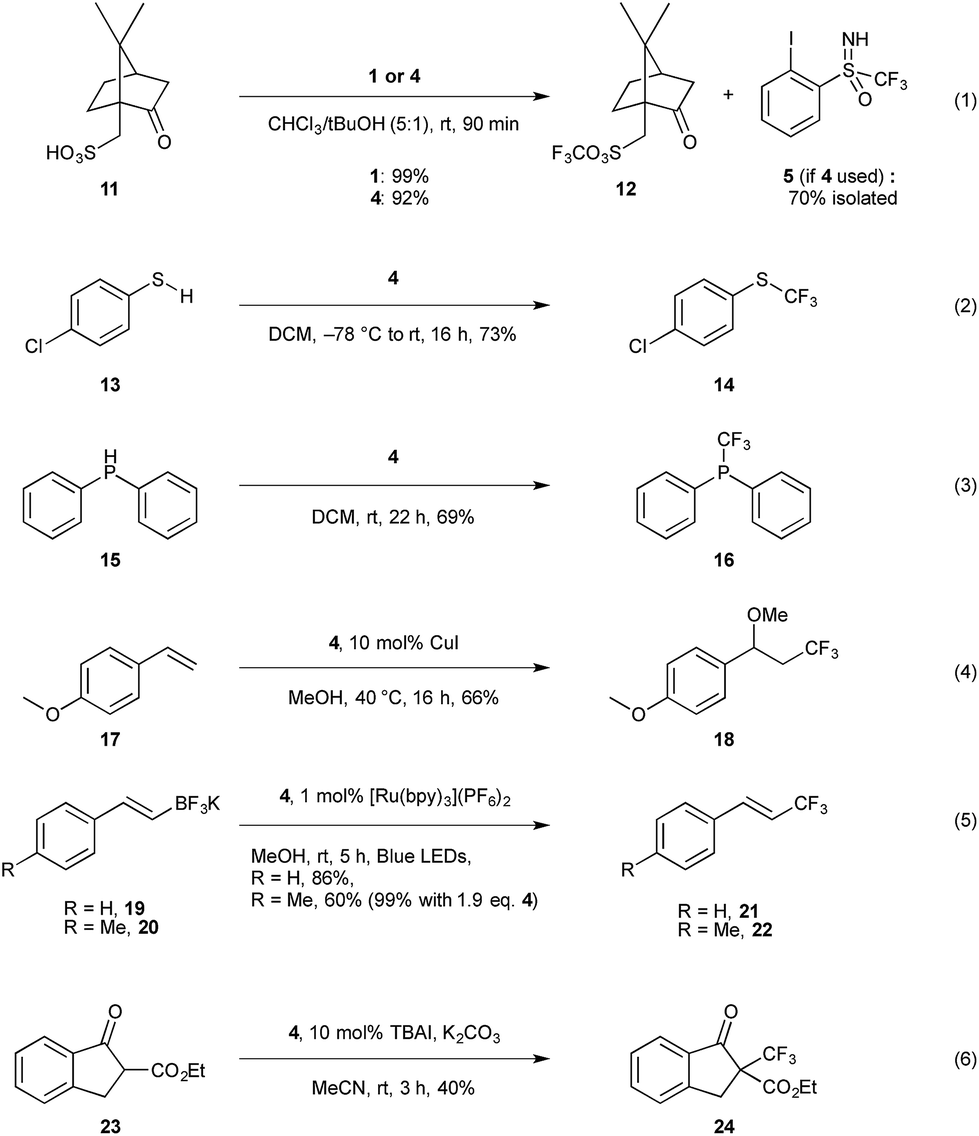 | ||
| Scheme 4 Reactivity screening of compound 4 with various nucleophiles. Yields determined by 19F NMR using α,α,α-trifluorotoluene as an internal standard. TBAI = tetrabutylammonium iodide. | ||
Inspired by the work of Akita and co-workers, we investigated the visible light induced electrophilic trifluoromethylation of trifluoroborates, 19 and 20.32 We obtained very favourable results using 1 mol% of [Ru(bpy)3](PF6)2 photocatalyst giving the trifluoromethylated alkene 21 in 86% yield when using 1.2 eq. of 4 for the potassium salt of styryltrifluoroborate (entry 5). The equivalent reaction with 1 is reported in the literature to give a yield of 81% with 5 mol% of the same photocatalyst, however in our hands the 19F NMR yield for this reaction was 35% after 5 h, and 56% after 15 h. We observed a similar trend using para-methyl styryltrifluoroborate which, under the literature reported conditions gave only 45% yield for 22 when reproduced in our lab vs. 81% reported in the literature with compound 1. Under the same conditions compound 4 gave 60% yield, which could be further improved to 99% when 1.9 eq. of 4 is used.
Considering reagent 4 is reminiscent of Shibata's fluorinated Johnson reagent on the sulfoximine moiety, we were curious to explore whether this could be a second reactive site on the scaffold.5 β-Keto ester 23 was chosen as a model substrate as this is known to react with Shibata's reagent to give the trifluoromethylated product 24 in 74% yield.5a Using the literature reported conditions, with 1.5 eq. of 4, DBU (1,8-diazabicyclo[5.4.0]undec-7) as base in CH2Cl2 and stirring for 2 h, only 29% 19F NMR yield of 24 was obtained. The 19F NMR of the reaction mixture showed 73% of unreacted 4 as well as 30% iodoarene 5 and 42% sulfoximine 8. Thus, the CF3 moiety at the iodine was transferred leaving the sulfoximine moiety intact. Stirring the reaction mixture longer did not lead to higher yields, however the yield could be improved by conducting the reaction under phase transfer conditions (entry 6), which gave moderate yields of 40%, whereas use of reagent 2 gives the desired product in 67% yield.3b
Finally, we were curious to explore the possibility of using the enantiopure sulfoximine reagent for asymmetric transformations. We first turned our attention to trifluoromethylation of chiral sulfonic acids, as the mechanism for this is well established to occur via protonation of the reagent by the substrate and coordination of the sulfonate to the iodine center, followed by reductive elimination to give the corresponding trifluoromethylated sulfonic acid.31 In order to enable kinetic resolution, we took 2 eq. of racemic 4 and reacted it under the standard conditions with 1 eq. of (1S)-(+)-10-camphor sulfonic acid (entry 1, Scheme 4). Once the reaction was complete, the remaining reagent was recovered after recrystallization of the reaction mixture, however no preferential reaction was observed, with 0% ee in recovered 4 observed by analytical chiral HPLC. Although this result was disappointing, we speculated that the stereogenic centre in camphorsulfonic acid was not close enough to the reactive centre to facilitate enantiodiscrimination. We next attempted the trifluoromethylation of β-keto ester 23 under phase transfer conditions (entry 6). However, chiral analytical HPLC analysis of the purified reaction mixture showed only racemic 24. Although these results have so far proven unsuccessful, we aim to further investigate the possibility of asymmetric transformations by increasing the steric bulk on the sulfoximine group through substitution of the CF3 moiety or using Brønsted or Lewis acid catalysts.
Conclusions
In conclusion we have described the facile synthesis of the new hypervalent iodine trifluoromethylation reagent, 4, which to our delight shows stability and reactivity comparable to those of the parent reagents 1 and 2. Compound 4 could also be isolated in its enantiopure form and is, to the best of our knowledge, the first described enantiopure hypervalent iodine compound bearing a transferable CF3 moiety. This is a promising start to explore the possibility of enantioselective trifluoromethylation of prochiral substrates, a longstanding challenge in electrophilic trifluoromethylation chemistry. Investigations towards this objective are currently underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Agence Nationale de la Recherche (ANR) is warmly acknowledged for PRCI funding (ANR-17-CE07-0048-01). Financial support by the Swiss National Science Foundation (Grant 200021E_175680/1) is acknowledged. Thanh-Nghi Le is grateful to the Vietnamese Government, the National Institute of Medicinal Materials in Hanoi for a doctoral fellowship. Drs Giang Vo-Thanh, Bruce Pégot and Patrick Diter are acknowledged for fruitful discussions. Jorna Kalim would like to thank Dr Reinhard Kissner, Dr René Verel and Grégoire Le Corre for help with measurements and discussions.Notes and references
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) T. Hiyama, Organofluorine Compounds: Chemistry and Applications, Springer-Verlag, Berlin, Heidelberg, New York, 2000 CrossRef; (d) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem.–Eur. J., 2019, 25, 11797 CrossRef CAS PubMed.
- (a) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806 CrossRef CAS PubMed; (b) N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6, 65 Search PubMed.
- (a) P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef CAS PubMed; (b) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS PubMed; (c) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed.
- (a) P. Liebing, E. Pietrasiak, E. Otth, J. Kalim, D. Bornemann and A. Togni, Eur. J. Org. Chem., 2018, 27–28, 3771 CrossRef; (b) P. Liebing, J. Kalim, N. Arefyeva, F. Oehler, M. Wickleder and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 8585 CrossRef CAS PubMed.
- (a) V. Bizet, R. Kowalczyk and C. Bolm, Chem. Soc. Rev., 2014, 43, 2426 RSC; (b) X. Shen and J. Hu, Eur. J. Org. Chem., 2014, 4437 CrossRef CAS; (c) A.-L. Barthelemy and E. Magnier, C. R. Chim., 2018, 21, 711 CrossRef CAS.
- (a) S. Noritake, N. Shibata, S. Nakamura, T. Toru and M. Shiro, Eur. J. Org. Chem., 2008, 3465 CrossRef CAS; (b) Y. Nomura, E. Tokunaga and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 1885 CrossRef CAS PubMed.
- (a) W. Zhang, F. Wang and J. Hu, Org. Lett., 2009, 11, 2109 CrossRef CAS PubMed; (b) W. Zhang, W. Huang and J. Hu, Angew. Chem., Int. Ed., 2009, 48, 9858 CrossRef CAS PubMed.
- G. K. S. Prakash, Z. Zhang, F. Wang, C. Ni and G. A. Olah, J. Fluorine Chem., 2011, 132, 792 CrossRef CAS.
- C. Urban, F. Cadoret, J.-C. Blazejewski and E. Magnier, Eur. J. Org. Chem., 2011, 4862 CAS.
- (a) X. Shen, W. Zhang, T. Luo, X. Wan, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 6966 CrossRef CAS PubMed; (b) X. Shen, W. Zhang, C. Ni, Y. Gu and J. Hu, J. Am. Chem. Soc., 2012, 134, 16999 CrossRef CAS PubMed; (c) X. Shen, W. Miao, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2014, 53, 775 CrossRef CAS PubMed; (d) X. Shen, Q. Liu, T. Luo and J. Hu, Chem.–Eur. J., 2014, 20, 6795 CrossRef CAS PubMed; (e) X. Shen, M. Zhou, C. Ni, W. Zhang and J. Hu, Chem. Sci., 2014, 5, 117 RSC; (f) T. Luo, R. Zhang, W. Zhang, X. Shen, T. Umemoto and J. Hu, Org. Lett., 2014, 16, 888 CrossRef CAS PubMed; (g) T. Luo, R. Zhang, X. Shen, W. Zhang, C. Ni and J. Hu, Dalton Trans., 2015, 44, 19636 RSC.
- (a) Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Chem.–Eur. J., 2016, 22, 1262 CrossRef CAS PubMed; (b) N. Noto, T. Koike and M. Akita, J. Org. Chem., 2016, 81, 7064 CrossRef CAS.
- M. Daniel, G. Dagousset, P. Diter, P.-A. Klein, B. Tuccio, A.-M. Goncalves, G. Masson and E. Magnier, Angew. Chem., Int. Ed., 2017, 56, 3997 CrossRef CAS PubMed.
- Chiral variations of Togni-type reagents have been previously reported in ref. 13b, however the racemates were not seperated and their use in asymmetric synthesis was not explored. (a) E. Pietrasiak and A. Togni, Organometallics, 2017, 36, 3750 CrossRef CAS; (b) K. Niedermann, J. M. Welch, R. Koller, J. Cvengroš, N. Santschi, P. Battaglia and A. Togni, Tetrahedron, 2010, 66, 5753 CrossRef CAS; (c) N. Santschi, C. Matthey, R. Schwenk, E. Otth and A. Togni, Eur. J. Org. Chem., 2015, 9, 1925 CrossRef; (d) N. Santschi, R. C. Sarott, E. Otth, R. Kissner and A. Togni, Beilstein J. Org. Chem., 2014, 10, 1 CrossRef PubMed; (e) C. Xu, X. Song, J. Guo, S. Chen, J. Gao, J. Jiang, F. Gao, Y. Li and M. Wang, Org. Lett., 2018, 20, 3933 CrossRef CAS PubMed.
- (a) L. M. Yagupolskii, I. I. Maletina, N. V. Kondratenko and V. V. Orda, Synthesis, 1978, 835 CrossRef CAS; (b) T. Umemoto, Y. Kuriu, H. Shuyama, O. Miyano and S.-I. Nakayama, J. Fluorine Chem., 1982, 20, 695 CrossRef CAS; (c) T. Umemoto, Y. Kuriu, H. Shuyama, O. Miyano and S.-I. Nakayama, J. Fluorine Chem., 1986, 31, 37 CrossRef CAS.
- (a) H. Wang, Y. Cheng, G. Raabe and C. Bolm, Angew. Chem., Int. Ed., 2016, 55, 12655 CrossRef CAS PubMed; (b) H. Wang, D. Zhang and C. Bolm, Chem.–Eur. J., 2018, 24, 14942 CrossRef CAS PubMed; (c) H. Wang, D. Zhang, H. Sheng and C. Bolm, J. Org. Chem., 2017, 82, 11854 CrossRef CAS PubMed.
- Y. Macé, C. Urban, C. Pradet, J. Marrot, J.-C. Blazejewski and E. Magnier, Eur. J. Org. Chem., 2009, 52, 3150 CrossRef.
- Unpublished results under submission.
- T.-N. Le, P. Diter, B. Pegot, C. Bournaud, M. Toffano, R. Guillot, G. Vo-Thanh and E. Magnier, Org. Lett., 2016, 18, 5102 CrossRef CAS PubMed.
- S. Chaabouni, J.-F. Lohier, A. -L. Barthelemy, T. Glachet, E. Anselmi, G. Dagousset, P. Diter, B. Pégot, E. Magnier and V. Reboul, Chem.–Eur. J., 2018, 24, 17006 CrossRef CAS PubMed . For the preparation of non-fluorinated sulfoximines using phenyl diacetate and ammonium carbamate see: A. Tota, M. Zenzola, S. J. Chawner, S. S. John-Campbell, C. Carlucci, G. Romanazzi, L. Degennaro, J. A. Bull and R. Luisi, Chem. Commun., 2017, 53, 348 RSC.
- V. Matoušek, E. Pietrasiak, R. Schwenk and A. Togni, J. Org. Chem., 2013, 78, 6763 CrossRef PubMed.
- (a) E. E. van Tamelen and K. B. Sharpless, Tetrahedron Lett., 1967, 8, 2655 CrossRef; (b) C. R. Pitts, D. Bornemann, P. Liebing, N. Santschi and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 1950 CrossRef CAS PubMed.
- A. Prieto, P. Diter, M. Toffano, J. Hannedouche and E. Magnier, Adv. Synth. Catal., 2019, 361, 436 CrossRef CAS.
- A. R. Katritzky, J. K. Gallos and H. D. Durst, Magn. Reson. Chem., 1989, 27, 815 CrossRef CAS.
- (a) U. Tilstam and H. Weinmann, Org. Process Res. Dev., 2002, 6, 384 CrossRef CAS; (b) C. Pastoriza, J. M. Antelo, J. Crugeiras and A. Peña-Gallego, J. Phys. Org. Chem., 2014, 27, 407 CrossRef CAS; (c) L. D. Luca and G. Giacomello, Synlett, 2004, 12, 2180 CrossRef; (d) S. Gaspa, M. Carraro, L. Pisano, A. Poecheddu and L. D. Luca, Eur. J. Org. Chem., 2019, 3544 CrossRef CAS.
- (a) S. Wawzonek and P. J. Thelen, J. Am. Chem. Soc., 1950, 72, 2118 CrossRef CAS; (b) H. Togo, Y. Hoshina, T. Muraki, H. Nakayama and M. Yokoyama, J. Org. Chem., 1998, 63, 5193 CrossRef CAS; (c) E. J. Corey and W. R. Hertler, J. Am. Chem. Soc., 1960, 82, 1657 CrossRef CAS.
- V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds, John Wiley & Sons, West Sussex, 2014 Search PubMed.
- N. Santschi, B. J. Jelier and T. Nauser, Phys. Chem. Chem. Phys., 2017, 19, 18172 RSC.
- H. P. Magalhães, A. Togni and H. P. Lüthi, J. Org. Chem., 2017, 82, 11799 CrossRef.
- R. Koller, Q. Huchet, P. Battaglia, J. M. Welch and A. Togni, Chem. Commun., 2009, 5993 RSC.
- P. Eisenberger, I. Kieltsch, N. Armanino and A. Togni, Chem. Commun., 2008, 1575 RSC.
- H. Egami, R. Shimizu and M. Sodeoka, Tetrahedron, 2012, 53, 5503 CrossRef CAS.
- Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 2037 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, full characterisation and spectroscopic data. CCDC 1947035 and 1947034 for 4 and 9 respectively. See DOI: 10.1039/c9sc04289j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |