
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciplex emissions derived from exceptionally long-distance donor and acceptor molecules†
Yong-Jin
Pu
 *ab,
Yuki
Koyama
ab,
Daisuke
Otsuki
b,
Minjun
Kim
a,
Hiroya
Chubachi
b,
Yuki
Seino
a,
Kazushi
Enomoto
a and
Naoya
Aizawa
ac
*ab,
Yuki
Koyama
ab,
Daisuke
Otsuki
b,
Minjun
Kim
a,
Hiroya
Chubachi
b,
Yuki
Seino
a,
Kazushi
Enomoto
a and
Naoya
Aizawa
ac
aRIKEN Center for Emergent Matter Science (CEMS), Wako, Saitama 351-0198, Japan. E-mail: yongjin.pu@riken.jp
bGraduate School of Organic Materials Science, Yamagata University, Yonezawa, Yamagata 992-8510, Japan
cJST-PRESTO, Kawaguchi, Saitama 332-0012, Japan
First published on 23rd September 2019
Abstract
Intermolecular electron–hole coupling in organic semiconductor excited states plays important roles in organic light-emitting diodes and organic photovoltaics, and the distance of the coupling is typically only on the order of a few nanometers. Here, we report exceptionally long-distance coupled exciplex emissions between electron-donor and electron-acceptor molecules even with a 70 nm-thick spacer layer. Donor/spacer (∼70 nm)/acceptor-type stacked films showed a low-energy band emission, which is not ascribed to the emission of the donor, spacer, and acceptor themselves, but well corresponds to the energy difference between the highest occupied molecular orbital of the donor and the lowest unoccupied molecular orbital of the acceptor. Delayed transient photoluminescence (PL) and electroluminescence (EL) decays and PL quenching by oxygen at the low-energy band were observed and are consistent with the characteristics of the exciplex species.
Introduction
An exciplex is an intermolecular complex in an excited state, formed from electron-donor and electron-acceptor molecules. These have been widely studied especially in the context of photosynthesis chemistry, in an effort to understand photo-induced electron transfer and to create long-lived charge-separated states with donor–spacer–acceptor system frameworks, including electron-donating porphyrin derivatives, electron-accepting fullerenes, carbon nanotubes, and π-conjugated molecules.1–3 The energy level of an exciplex state is lower than that of each independent electron-donor and electron-acceptor molecule, and thus its emission wavelength becomes bathochromically-shifted from the original emission of each molecule. Photoluminescence (PL) quantum yields of exciplex emissions tend to be low, and therefore the exciplex emissions have not been widely utilized in organic light-emitting diode (OLED) applications.4,5 However, Goushi et al. recently used the exciplex state between a hole-transporting layer (HTL) and an electron-transporting layer (ETL), in which the energy difference between the excited singlet state and triplet state becomes small and favourable to achieve thermally activated delayed fluorescence (TADF), enhancing the internal quantum efficiency of fluorescent OLEDs.6 Park and Sun et al. reported an exciplex co-host for phosphorescent and fluorescent emitters in OLEDs having a high external quantum efficiency (EQE) of approximately 30%.7,8 An exciplex can be considered as a partially charge-transfer (CT) excited state, and it is also important to understand and enhance charge separation of bound hole–electron pairs at the p–n interface for organic photovoltaics.9,10 In organic molecules with low dielectric constants in particular, hole–electron pairs are tightly bound within small exciton Bohr radii.For exciplex formation, the distance between electron-donor and electron-acceptor molecules has to be very small, on the order of a few nanometers at most.11–13 However, Nakanotani et al. observed exciplex emissions derived from long-distance coupled donor and acceptor molecules with a 10 nm thick spacer layer,14 and then, Chapran et al., Li et al., and Sych et al. also observed similar exciplex emissions with a several nanometer spacer layer, using different types of donor and acceptor molecules.15–17 Colella et al. reported that donor and acceptor molecules diluted in a host matrix (i.e. a spacer molecule) showed efficient exciplex emission.18 Supporting those reports, Deotare et al. found that bound electron–hole pairs moved to a distance of approximately 10 nm at the p–n interface.19 These extraordinarily long-distance electron–hole couplings between the donor and acceptor molecules are still not well understood, and developing and understanding such exotic exciplex states (i.e. long-distance coupled excited CT states) are both a fascinating and challenging direction of study.
Here we report abnormally long-distance exciplex emissions in OLEDs even with an ∼70 nm-thick spacer layer between electron-donor molecules and electron-acceptor molecules. Various types of devices and stacked films were fabricated and investigated to prove that the emergent low-energy band emission originates from the long-distance coupled exciplex species and to exclude other possibilities. Electroluminescence (EL) and transient EL of the devices with different spacer layers were measured to compare with PL and transient PL of the donor:acceptor mixed or stacked films. A device exclusively showing electromer emission was fabricated to judge the relevance of the electromer to the low-energy band emission. Various donor molecules in the OLEDs were systematically used, while the types of spacer and acceptor molecules were fixed, to investigate the influence of the energy difference between the donor and acceptor molecules on the low-energy band emission. Oxygen dependence and temperature dependence of the PL were measured to obtain more confident evidence that the excited triplet state is involved in the low-energy band emission as a TADF-type exciplex. The type of spacer molecule was also changed to help understand the role of the interfaces between the donor and the spacer and between the spacer and the acceptor in the emission mechanism.
Results and discussion
1,1′-Bis(di-4-tolyl-aminophenyl)cyclohexane (TAPC), 9,10-bis(3,5-dimethoxyphenyl)anthracene (DMA), and 9,10-bis(3,5-dicyanophenyl)anthracene (DCA) were used as the donor molecule (D), spacer molecule (S), and acceptor molecule (A), respectively. Chemical structures of the compounds, PL spectra of their neat films and co-evaporated mixed films, and transient PL decays are shown in Fig. 1 and PL quantum yields of the films are summarized in Table S1.† Co-evaporated films with each combination of TAPC:DMA (D:S), DMA:DCA (S:A), and TAPC:DCA (D:A) showed bathochromically shifted PL compared to the PL of their respective neat films (D, S, and A), and these red-shifted emissions are ascribed to exciplex emissions from the D:S, S:A, and D:A complexes. PL decays of the D:S, S:A, and D:A complexes were on the submicrosecond scale (125–299 ns), although the PL decays from the neat films of D, S, and A themselves were on the subnanosecond scale (0.4–0.98 ns), supporting the exciplex formation of D:S, S:A, and D:A (Tables S2 and S3†).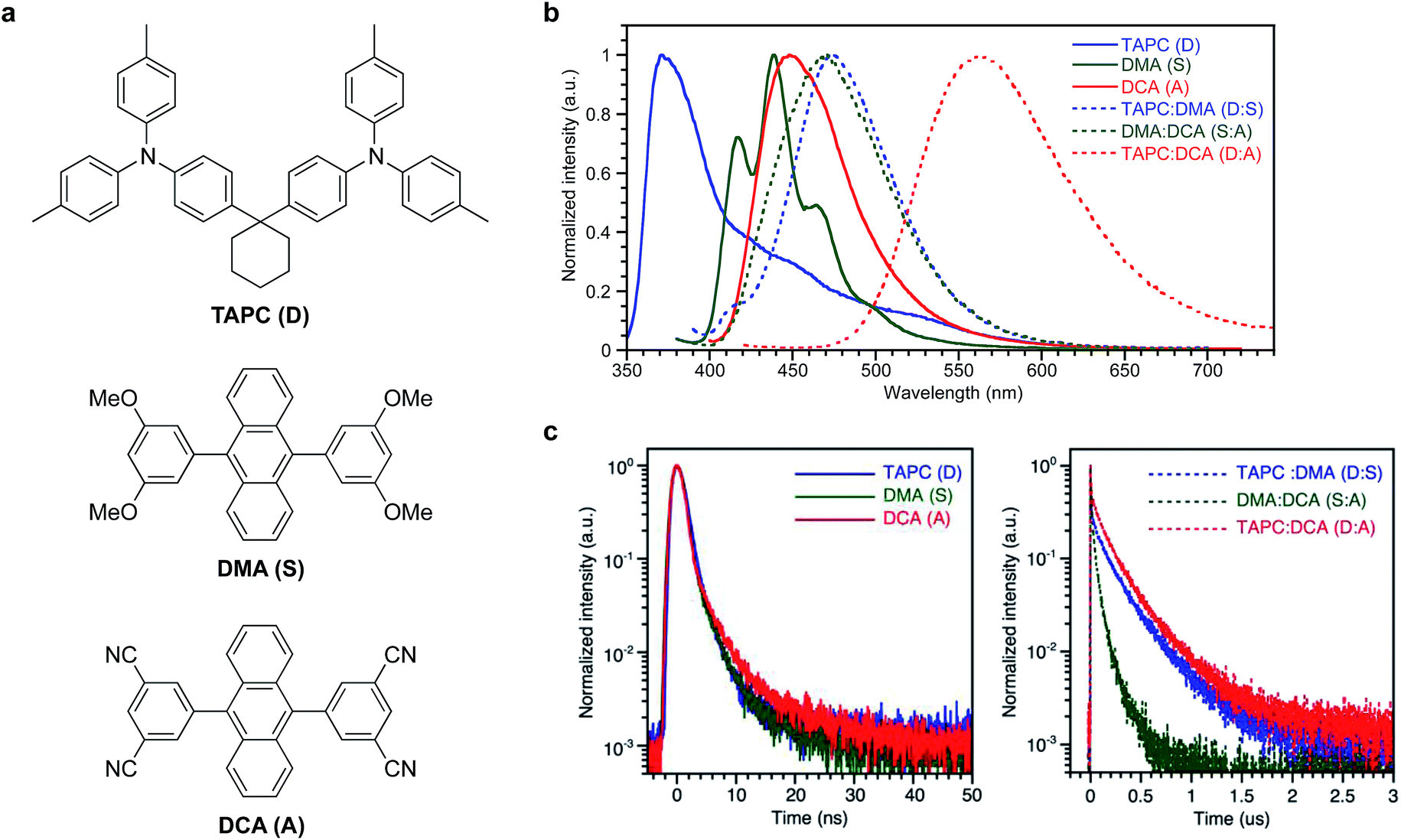 | ||
| Fig. 1 PL properties of the donor TAPC, the spacer DMA, and the acceptor DCA; (a) chemical structures of the compounds. (b) PL spectra of the neat films and the co-evaporated (1/1 wt/wt) films. TAPC (λex 330 nm), DMA (λex 360 nm), DCA (λex 380 nm), TAPC:DMA (λex 370 nm), DMA:DCA (λex 370 nm), and TAPC:DCA (λex 400 nm). (c) Transient PL decay of the neat films and the co-evaporated (1/1 wt/wt) films: TAPC (λex 280 nm, λem 380 nm), DMA (λex 370 nm, λem 440 nm), DCA (λex 370 nm, λem 470 nm), TAPC:DMA (λex 370 nm, λem 475 nm), DMA:DCA (λex 370 nm, λem 470 nm), and TAPC:DCA (λex 370 nm, λem 550 nm). | ||
OLEDs with a structure of ITO/poly(3,4-ethylenedioxythiophene):poly(4-styrenesulfonate) (PEDOT:PSS) (30 nm)/TAPC (20 nm)/DMA (0, 10, 20, 30, 40, 50, 60, 70 nm)/DCA (10 nm)/1,3-bis[3,5-di(pyridin-3-yl)phenyl]benzene20 (B3PyPB) (40 nm)/LiF (1 nm)/Al were fabricated for this study. B3PyPB was used as an ETL and its chemical structure is shown in Fig. S1.† The energy diagram, EL spectra, and transient EL decay of the device are shown in Fig. 2. EQEs of the devices are summarized in Table S4.† The device without the spacer DMA (i.e. 0 nm spacer) showed two EL emissions, in which the high-energy band emission with a λmax of 445 nm was identical to the PL spectrum of the acceptor DCA itself and the low-energy band emission with a λmax of 550 nm was similar to the PL spectrum of the exciplex between the donor TAPC and the acceptor DCA. The low-energy band emissions became smaller with increasing the spacer layer thickness, but surprisingly the emission did not totally disappear even with a 70 nm thick spacer. Transient EL decay was also measured. The decay lifetime was long and on the microsecond scale, demonstrating that residual excitons in the device originate from long lifetime species such as exciplexes. The microsecond-scale transient EL component became smaller with increasing the spacer layer thickness, but this delayed component did not totally disappear even with abnormally thick spacers (∼70 nm thickness) between the donor and the acceptor layers. To exclude any possibility that the acceptor DCA or the donor TAPC is not involved in the low-energy band emission, the device D/S without the acceptor DCA layer and the device S/A without the donor TAPC layer were fabricated, respectively, and their EL spectra are compared (Fig. S2†). The device D/S and the device S/A clearly did not show the low-energy band emission corresponding to those of the D/A and D/S/A devices. The transient EL of the D/S and S/A devices showed prompt decays to the half intensity, and these are clearly different from the delayed decay of the D/A and D/S/A devices.
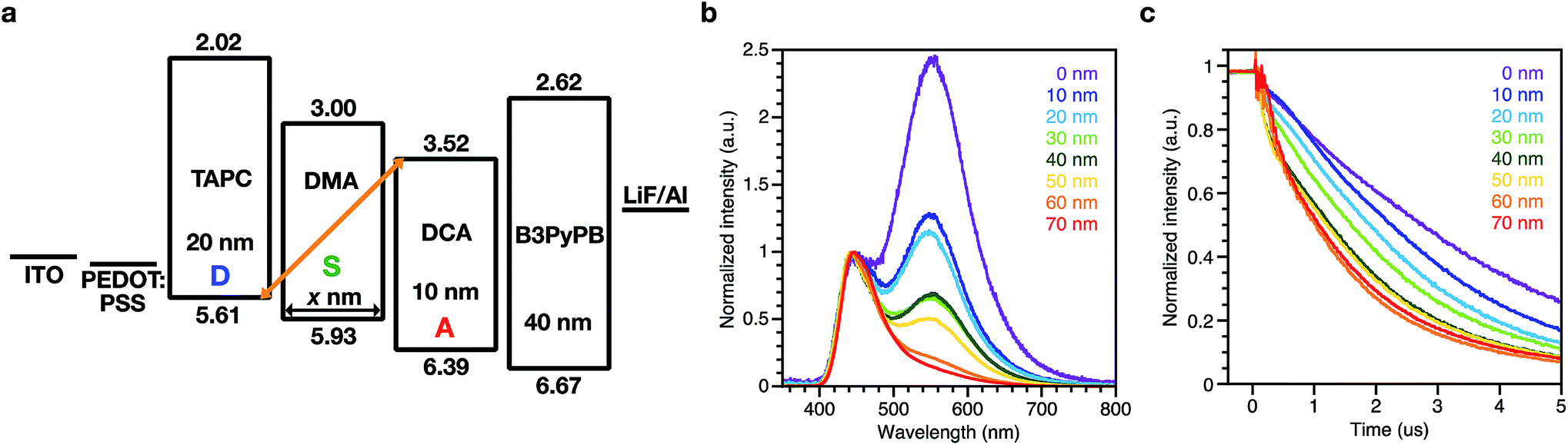 | ||
| Fig. 2 Spacer-thickness dependent EL in the devices with the DMA spacer; (a) device structure. (b) EL spectra at 25 mA cm−2 with different spacer thicknesses from 0 nm to 70 nm. (c) Transient EL decay at 50 mA cm−2 with different spacer thicknesses from 0 nm to 70 nm. The applied voltages are 5.50 V for 0 nm, 5.66 V for 10 nm, 6.41 V for 20 nm, 6.90 V for 30 nm, 7.60 V for 40 nm, 8.05 V for 50 nm, 8.58 V for 60 nm, and 9.14 V for 70 nm. | ||
The donor might be in close proximity to the acceptor if the spacer layer was not deposited uniformly. Therefore, we carefully checked the surface morphology of the donor layer, the spacer on the donor layer, and the acceptor layer on the spacer/donor stacked layer (Fig. S3†). They were sufficiently smooth with an average roughness (Ra) of 0.37 nm for the donor layer and 0.33 nm for the spacer layer. The acceptor layer on the spacer/donor stacked layer showed a relatively rough surface morphology with an Ra of 0.84 nm and the maximum depth of the depressions was 6 nm (Fig. S3c†). These are much shallower than the DCA thickness of 10 nm and DMA thickness of 30 nm. These results suggest that the donor TAPC and acceptor DCA are separated by the uniformly deposited spacer DMA and their distances are fairly constant in the stacked layers.
It should be noted that injected holes in the devices reach the acceptor layer, because one of the two EL emissions at the short wavelength is clearly ascribed to the emission of DCA. The onset of the DMA emission is located at an explicitly shorter wavelength than that of DCA, and the shape of the DMA emission shows three vibrational modes that differ from the unimodal shape of the DCA emission (Fig. 1b). This is also supported by the result that the hole mobility of DMA is larger than its electron mobility, as estimated by unipolar device experiments (Fig. S4†), and the charge recombination zone is located primarily in the acceptor layer. Therefore, partially accumulated holes at the interface between the donor layer and the spacer layer (D/S) interact with the electrons at the interface between the spacer layer and the acceptor layer (S/A) through the spacer layer.
It is known that TAPC sometimes shows distinctive electromer emissions in OLEDs at similar wavelength ranges to that of the exciplex emission of TAPC/DCA.21,22 However, we can rule out the possibility that the TAPC electromer is the origin of the above mentioned abnormal emissions for a number of reasons. These were confirmed by the device without both the spacer DMA and the acceptor DCA, fabricated with a structure of ITO/PEDOT:PSS (30 nm)/TAPC (15 nm)/B3PyPB (60 nm)/LiF (1 nm)/Al. First, the charge recombination zone is mainly located at the acceptor layer as mentioned above. Second, the maximum emission wavelength of the TAPC electromer is 581 nm, which clearly differs from that of the exciplex emissions (Fig. S5†). Third, the transient EL decay of the TAPC electromer is very rapid unlike the exciplex emissions. The slightly delayed component originates from the exciplex emission between TAPC and B3PyPB at 432 nm.
Considering the exciplex emission between the donor and the acceptor through the spacer layer, the type of donor molecule must influence the emission wavelength of the exciplex due to the change of the energy difference between the donor and acceptor. Four different donor molecules were used for comparison with TAPC: tris(4-carbazoyl-9-ylphenyl)amine (TCTA), N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPD), N,N′-bis[4-[bis(3-methylphenyl)amino]phenyl]-N,N′-diphenyl-[1,1′-biphenyl]-4,4′-diamine (DNTPD), and 4,4′,4′′-tris[(3-methylphenyl)phenylamino]triphenylamine (MTDATA). Chemical structures, energy diagrams, PL spectra of the co-evaporated films with the acceptor DCA, and EL spectra of the devices with the same structure as the TAPC donor device are shown in Fig. 3. The PL spectra of the co-evaporated films were bathochromically shifted with decreased energy gaps between the HOMO of the donor and the LUMO of the acceptor DCA. TCTA and NPD showed emissions corresponding to their exciplex with the acceptor DCA, in addition to the emission from the acceptor DCA itself. In the devices with DNTPD and MTDATA, the emission corresponding to their exciplex with the acceptor DCA also appeared, but a blue-shifted emission corresponding to their exciplex with the spacer DMA appeared more strongly because of their shallower HOMO energy level compared to that of NPD, TCTA, and TAPC, resulting in a larger hole injection barrier to the spacer layer, efficiently forming the exciplex with the spacer DMA. These results showing that the lowest-energy band emissions are clearly dependent on the energy difference between the donor and the acceptor species demonstrate that these emissions are related to the exciplex between the donor and the acceptor species.
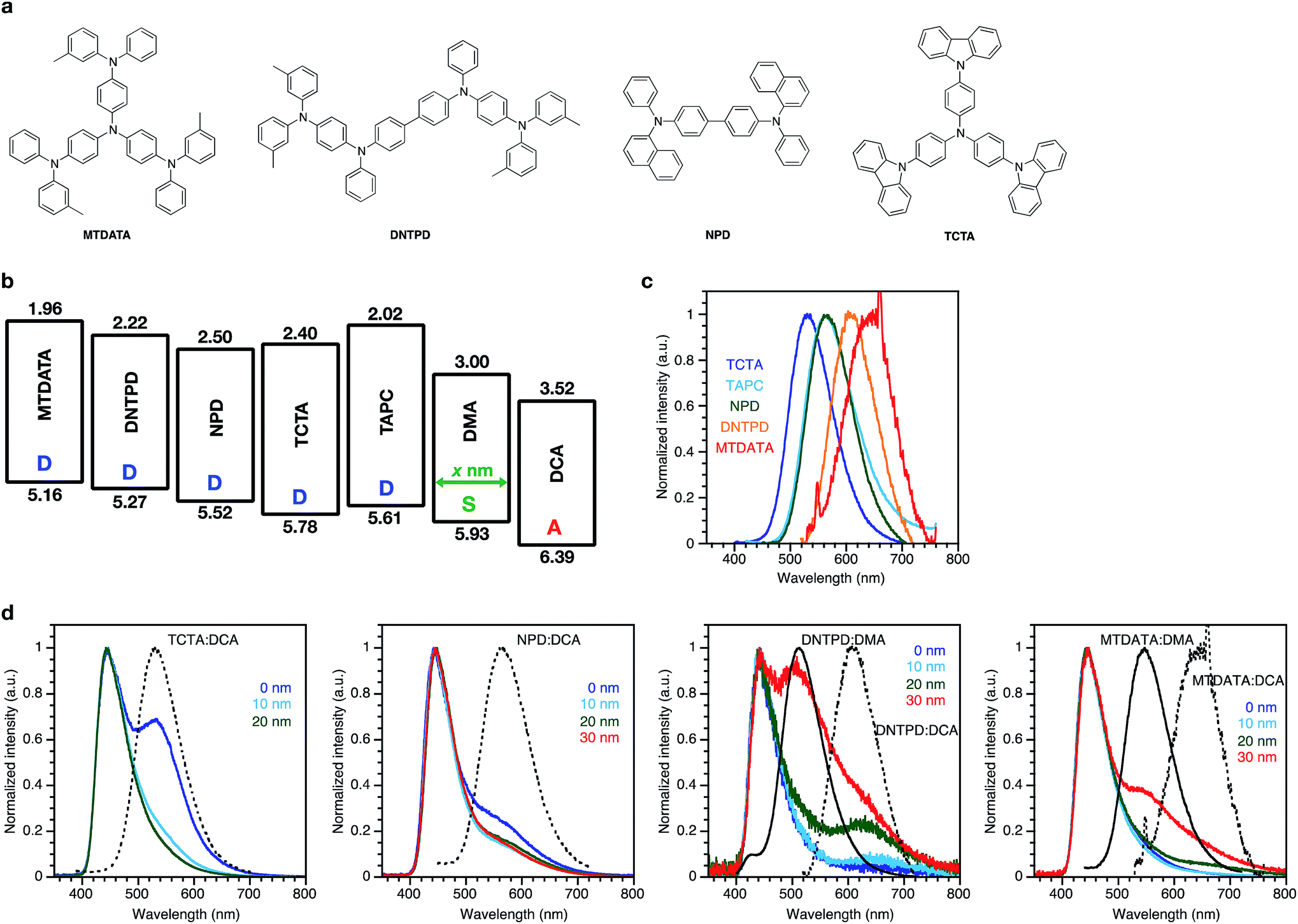 | ||
| Fig. 3 EL properties of the devices with different types of donor compounds; (a) chemical structures of the compounds. (b) Energy diagrams of the compounds. (c) PL spectra of the co-evaporated (1/1 wt/wt) films of DCA and the different donors: TCTA (λex 370 nm), NPD (λex 380 nm), DNTPD (λex 380 nm), and MTDATA (λex 400 nm). (d) EL spectra at 25 mA cm−2 with different spacer thicknesses. The black solid and dashed lines represent the PL spectra of the corresponding exciplexes. | ||
We performed steady-state PL measurements, to further probe the emission from the long-distance coupled exciplex, on a stacked film of TAPC (2 nm)/DMA (10 nm)/DCA (2 nm), a stacked film of TAPC (2 nm)/DCA (2 nm), and a mixed film of TAPC:DCA (20 nm). Excitation light (380–405 nm) was incident on the side of the DCA layer. Without any electric field, the stacked TAPC/DMA/DCA film exhibited an intense low-energy band PL with a λmax of 534 nm (Fig. S6†), ruling out the possibility of an electromer (or electroplex) emission. For comparison, the stacked TAPC/DCA film and the mixed TAPC:DCA film exhibited low-energy band PL with a λmax of 548 nm and 561 nm, respectively. This observed blue shift in the PL spectra is consistent with the increased spatial separation of the coupled hole and electron in the following order: the mixed TAPC:DCA film < the stacked TAPC/DCA film < the stacked TAPC/DMA/DCA film, because the spatial separation leads to a decrease in the coulombic attraction energy in the hole–electron pair and thus increases the exciplex emission energy.14,23
Furthermore, the net intensity of the low-energy band emission was decreased in the presence of atmospheric oxygen (Fig. S7†), indicating that the triplet excited state is involved in the emission mechanism, as in the case of the previously reported exciplex systems exhibiting TADF.5,18 The excited state can be quenched by energy transfer and/or electron transfer to oxygen. However, the PL intensity was recovered by removing oxygen in a vacuum, although photodegradation was still observed even under vacuum. This result suggests that the quenching process is unlikely based on an irreversible chemical reaction with oxygen and likely governed by the triplet energy transfer to oxygen. The peak intensity of the high-energy band emission at 437 nm corresponding to the PL of DMA itself (Fig. S7c†) did not show reversible oxygen quenching, also supporting the exciplex nature of the low-energy band emission.
Transient PL decay measurements on the stacked TAPC/DMA/DCA film and the mixed TAPC:DCA film revealed multi-exponential decays with significant contribution of the submicrosecond delayed emission, along with nanosecond prompt emission (Fig. S8 and Table S5–S7†). The lifetimes of these delayed emissions decreased with increasing the temperature, suggesting thermal activation of the triplet excited state back to the singlet excited state through reverse intersystem crossing (RISC). The rate constants of RISC follow Arrhenius-type temperature dependence with an activation energy of 10 meV for the mixed TAPC:DCA film and 4 meV for the stacked TAPC/DMA/DCA film (Fig. S9†). The decreased activation energy of RISC is attributed to the increased spatial separation of the coupled hole and electron in the stacked film, having a very small electron exchange energy (i.e., energy difference between the singlet and triplet excited states).24,25 We note that the exciplex emission intensity showed a linear dependence on the excitation power (Fig. S10†), confirming that the emission is caused through a unimolecular process, rather than a bimolecular process, such as triplet–triplet annihilation.
The Onsager radius for typical organic materials is known to be 16–19 nm with a dielectric constant of 3.0–3.5 at room temperature.26 It is unlikely that electrons and holes can be coupled over a distance of 70 nm without any spacer molecules to form the exciplex state. Therefore, the role of the spacer molecules would be a key factor in understanding the mechanism of the abnormally long-distance exciplex formation; we could propose a conceivable mechanism to explain this behaviour. As an initial step, two exciplex states at the D/S and S/A interfaces, [Dδ+·Sδ−] and [Sδ+·Aδ−], are formed, respectively. Then, the negative and positive charges on the spacer species, Sδ− and Sδ+, diffuse in the spacer layer, as expressed in (i) and (ii), towards the cathode and anode, respectively. This could be supported by the recent report that bound electron–hole pairs move to a distance of 10 nm at the p–n interface.19 The diffusion in this study is in the opposite direction of the applied bias. These long-distance excited CT states, [Dδ+·Sn·Sδ−] and [Sδ+·Sm·Aδ−], encounter each other, and a further long-distance excited CT state [Dδ+·Sn+m·Aδ−] is formed as expressed in (iii).
| [Dδ+·Sδ−] → [Dδ+·S·S·Sδ−] → [Dδ+·Sn·Sδ−] | (i) |
| [Sδ+·Aδ−] → [Sδ+·S·S·Aδ−] → [Sδ+·Sm·Aδ−] | (ii) |
| [Dδ+·Sn·Sδ−] + [Sδ+·Sm·Aδ−] → [Dδ+·Sn+m·Aδ−] | (iii) |
In the proposed emission mechanism, the exciplex formation at both the D/S and S/A interfaces is important to form long-distance electron–hole coupling. We used 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) as a spacer instead of the DMA spacer. The chemical structure, PL spectra, EL spectra and transient decay of CBP are shown in Fig. S11.† The co-evaporated film of TAPC:CBP (D:S) showed bathochromically shifted PL spectra compared to each neat film of TAPC and CBP, while the co-evaporated film of CBP:DCA (S:A) showed the same PL spectra as that of DCA itself. However, the PL lifetime of the CBP:DCA (S:A) film at 550 nm was relatively long compared to the TAPC:CBP (D:S) film, although the PL lifetime at 450 nm, which is the wavelength at the peak top in the PL spectrum of the CBP:DCA (S:A) film, showed much faster decay. The decays of the neat film of CBP and DCA themselves are also fast. Therefore, the delayed lifetime of the CBP:DCA mixed film at 550 nm does not result from the excimer of CBP or DCA, but is considered to be ascribed to the less generated exciplex species between CBP and DCA, although the PL of the CBP:DCA film was the same as that of the DCA neat film without any other low-energy band emission. The D/S/A-type OLEDs with the CBP spacer also showed two EL emissions. One with a λmax of 445 nm is ascribed to emissions from the acceptor DCA itself, while the other at λmax = 550 nm is ascribed to the exciplex emission between the donor TAPC and the acceptor DCA. The device with spacer distances greater than 15 nm did not show the exciplex EL. This spacer thickness is still exceptionally large, but is smaller than that of the device with the DMA spacer. This result is attributed to the less exciplex formation at the S/A interface.
We investigated the effect of spacer thickness on the exciplex species generated by photoexcitation (Fig. S12 and Table S8–S9†). Stacked films were prepared as quartz/TAPC (2 nm)/DMA or CBP (x nm)/DCA (2 nm). Excitation light was irradiated on the acceptor DCA side at 380–405 nm. When DMA was used as a spacer, incident photons were absorbed in the DCA layer and unabsorbed photons passing through the DCA layer were also absorbed in the DMA layer. This is supported by the PL spectra showing DMA emissions at shorter wavelengths than those of DCA. Therefore, the exciplex states at both the D/S and S/A interfaces can be generated initially, and then the D/A exciplex forms as well as the electrical excitation. Up to a 70 nm DMA spacer layer, an emission corresponding to the D/A exciplex at 530–550 nm appeared. This thickness is larger than the 5–20 nm CBP spacer in which the D/A exciplex emission was observed. As shown in Fig. S11b,† the S/A exciplex state is less generated. Therefore, for the formation of the D/A exciplex, Sδ− needs to move to the acceptor DCA layer, which is why the CBP spacer thickness was smaller than that of the DMA spacer in the formation of the long-distance D/S/A exciplexes.
Finally, the current density and voltage dependence of the D/A exciplex formation as shown in Fig. S13† also supports this proposed mechanism. Under a positive bias, the negative and positive spacer species Sδ− and Sδ+ become less diffusive toward the cathode and anode, respectively, as the electric field increases. Therefore, it becomes difficult to form long-distance coupled [Dδ+·Sn+m·Aδ−], in which n and m become small, and consequently the emission corresponding to the D/A exciplex with the spacer decreases.
Conclusion
Exciplex emissions were observed with exceptionally long-distance donor–acceptor coupling through spacer molecules in OLEDs, with a maximum spacer distance of 70 nm. The observed low-energy band EL component was congruent with the exciplex PL in the co-evaporated films of the donor and acceptor molecules, even when the donor and acceptor were separated by a long-distance spacer up to around 70 nm. Four different types of donor molecules having different HOMO levels also showed different low-energy band EL components with the acceptor DCA, corresponding to the difference of their HOMO levels and LUMO levels of the acceptor DCA. The delayed transient decay and large oxygen dependency of the low-energy band emission are considered to originate from the feature of the exciplex species. The effect of the different spacer molecules suggests that the exciplex formation both at the donor/spacer and the spacer/acceptor interface results in long-distance coupling between the donor and the acceptor through such a thick spacer layer. This finding of the exceptionally long-distance donor–acceptor coupling in OLEDs is far beyond previous perception and helps to eliminate preconceptions of photophysics in organic semiconductors, and would have the possibility to lead to totally different highly sensitive sensors or photodetectors to external stimulus, with various applications other than OLED applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.-J. Pu gratefully acknowledges PRESTO (13417316) from JST, and a Grant-in-Aid for Young Scientists [A] (24685029) and a Grant-in-Aid for Scientific Research [B] (17H03103) from the JSPS.References
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS PubMed.
- S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283–2297 RSC.
- N. Matsumoto and C. Adachi, J. Phys. Chem. C, 2010, 114, 4652–4658 CrossRef CAS.
- E. Skuodis, A. Tomkeviciene, R. Reghu, L. Peciulyte, K. Ivaniuk, D. Volyniuk, O. Bezvikonnyi, G. Bagdziunas, D. Gudeika and J. V. Grazulevicius, Dyes Pigm., 2017, 139, 795–807 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- Y. S. Park, S. Lee, K. H. Kim, S. Y. Kim, J. H. Lee and J. J. Kim, Adv. Funct. Mater., 2013, 23, 4914–4920 CrossRef CAS.
- J. W. Sun, J. H. Lee, C. K. Moon, K. H. Kim, H. Shin and J. J. Kim, Adv. Mater., 2014, 26, 5684–5688 CrossRef CAS PubMed.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- C. Deibel, T. Strobel and V. Dyakonov, Adv. Mater., 2010, 22, 4097–4111 CrossRef CAS PubMed.
- J. Baek, T. Umeyama, K. Stranius, H. Yamada, N. V. Tkachenko and H. Imahori, J. Phys. Chem. C, 2017, 121, 13952–13961 CrossRef CAS.
- T. Umeyama, T. Hanaoka, J. Baek, T. Higashino, F. Abou-Chahine, N. V. Tkachenko and H. Imahori, J. Phys. Chem. C, 2016, 120, 28337–28344 CrossRef CAS.
- J. R. Miller, L. T. Calcaterra and G. L. Closs, J. Am. Chem. Soc., 1984, 106, 3047–3049 CrossRef CAS.
- H. Nakanotani, T. Furukawa, K. Morimoto and C. Adachi, Sci. Adv., 2016, 2, e1501470 CrossRef.
- M. Chapran, E. Angioni, N. J. Findlay, B. Breig, V. Cherpak, P. Stakhira, T. Tuttle, D. Volyniuk, J. V. Grazulevicius, Y. A. Nastishin, O. D. Lavrentovich and P. J. Skabara, ACS Appl. Mater. Interfaces, 2017, 9, 4750–4757 CrossRef CAS.
- B. B. Li, L. Gan, X. Y. Cai, X. L. Li, Z. H. Wang, K. Gao, D. C. Chen, Y. Cao and S. J. Su, Adv. Mater. Interfaces, 2018, 5, 1800025 CrossRef.
- G. Sych, J. Simokaitiene, O. Bezvikonnyi, U. Tsiko, D. Volyniuk, D. Gudeika and J. V. Grazulevicius, J. Phys. Chem. C, 2018, 122, 14827–14837 CrossRef CAS.
- M. Colella, A. Danos and A. P. Monkman, J. Phys. Chem. Lett., 2019, 10, 793–798 CrossRef CAS PubMed.
- P. B. Deotare, W. Chang, E. Hontz, D. N. Congreve, L. Shi, P. D. Reusswig, B. Modtland, M. E. Bahlke, C. K. Lee, A. P. Willard, V. Bulovic, T. Van Voorhis and M. A. Baldo, Nat. Mater., 2015, 14, 1130–1134 CrossRef CAS PubMed.
- H. Sasabe, E. Gonmori, T. Chiba, Y. J. Li, D. Tanaka, S. J. Su, T. Takeda, Y. J. Pu, K. I. Nakayama and J. Kido, Chem. Mater., 2008, 20, 5951–5953 CrossRef CAS.
- S. Y. Yang and M. X. Jiang, Chem. Phys. Lett., 2009, 484, 54–58 CrossRef CAS.
- J. Kalinowski, G. Giro, M. Cocchi, V. Fattori and P. Di Marco, Appl. Phys. Lett., 2000, 76, 2352–2354 CrossRef CAS.
- M. Cocchi, D. Virgili, C. Sabatini and J. Kalinowski, Chem. Phys. Lett., 2006, 421, 351–355 CrossRef CAS.
- A. Kohler and D. Beljonne, Adv. Funct. Mater., 2004, 14, 11–18 CrossRef.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 83302 CrossRef.
- L. Onsager, Phys. Rev., 1938, 54, 554–557 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04262h |
| This journal is © The Royal Society of Chemistry 2019 |