
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwists or turns: stabilising alpha vs. beta turns in tetrapeptides†
Huy N.
Hoang
 a,
Timothy A.
Hill
a,
Gloria
Ruiz-Gómez
ab,
Frederik
Diness
ac,
Jody M.
Mason
ad,
Chongyang
Wu
a,
Giovanni
Abbenante
a,
Nicholas E.
Shepherd
a and
David P.
Fairlie
*a
a,
Timothy A.
Hill
a,
Gloria
Ruiz-Gómez
ab,
Frederik
Diness
ac,
Jody M.
Mason
ad,
Chongyang
Wu
a,
Giovanni
Abbenante
a,
Nicholas E.
Shepherd
a and
David P.
Fairlie
*a
aAustralian Research Council Centre of Excellence in Advanced Molecular Imaging, Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: d.fairlie@imb.uq.edu.au
bStructural Bioinformatics, BIOTEC, Technische Universität Dresden, Tatzberg 47-51, 01307 Dresden, Germany
cCenter for Evolutionary Chemical Biology, Department of Chemistry, University of Copenhagen, 2100 Copenhagen, Denmark
dDepartment of Biology & Biochemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 3rd October 2019
Abstract
Protein–protein interactions involve hotspots as small as 4 sequential amino acids. Corresponding tetrapeptides have no structure in water. Here we report linking side chains of amino acids X and Z to form 24 cyclic tetrapeptides, cyclo-[XAAZ]-NH2, and stabilise 14–18 membered rings that mimic different kinds of non-regular secondary structures found in protein hotspots. 2D NMR spectra allowed determination of 3D structures for 14 cyclic tetrapeptides in water. Five formed two (i, i + 3) hydrogen bonds and a beta/gamma (6, 7) or beta (9, 19, 20) turn; eight formed one (i, i + 4) hydrogen bond and twisted into a non-helical (13, 18, 21, 22, 24) or helical (5, 17, 23) alpha turn; one was less structured (15). A beta or gamma turn was favoured for Z = Dab, Orn or Glu due to a χ1 gauche (+) rotamer, while an alpha turn was favoured for Z = Dap (but not X = Dap) due to a gauche (−) rotamer. Surprisingly, an unstructured peptide ARLARLARL could be twisted into a helix when either a helical or non-helical alpha turn (5, 13, 17, 18, 21–24) with Z = Dap was attached to the N-terminus. These structural models provide insights into stability for different turns and twists corresponding to non-regular folds in protein hotspots.
Introduction
Protein structure is directed by inherent preferences of amino acids for different folds,1 and by packing and solvation effects. Protein–protein recognition is based on interactions between folded secondary structures like α-helices, β-strands, turns and loops.2 Thousands of protein–protein interfaces are now known to have bioactive ‘hotspots’ localised to just 4–8 amino acid segments (1–2 turns),3 but 50% of those sequences have nonregular secondary structures. They include different subtle variations of α-, β- and γ-turns (Fig. 1), including non-helical α-turns,4,5 underscoring the importance of gaining a better understanding of how such turn variations dictate local structure and, ultimately, determine function. New insights to peptide folding can also enable design of small molecules capable of mimicking different kinds of turn motifs in proteins. Here we investigate tetrapeptides for some new clues to how stereochemical and structural constraints can control folding of the peptide backbone into different turns and helical twists.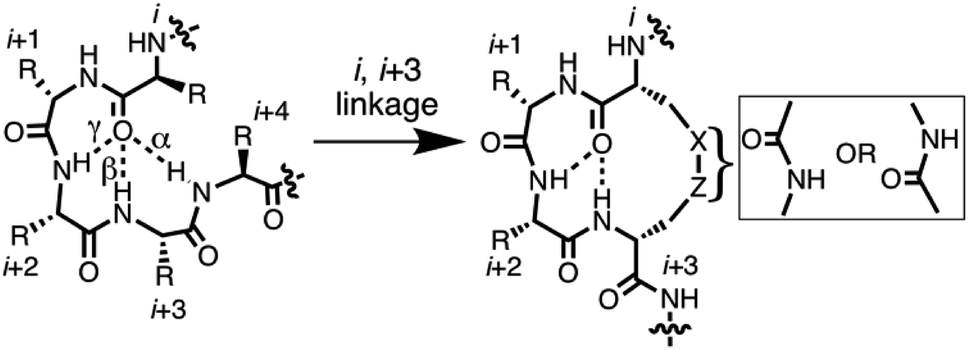 | ||
| Fig. 1 7-, 10-, 13-membered hydrogen bonded rings define γ-, β- and α-turns respectively (left) that potentially might be stabilised by cyclisation via side chains (right). | ||
Short peptides corresponding to bioactive sequences of proteins rarely show stable structures in water, but when artificially stabilised they can potently modulate protein–protein interactions. A number of strategies have been described for conformationally restricting peptides into beta-turns. One approach is cyclisation, which can promote intramolecular hydrogen bonds that stabilise an α-, β- or γ-turn (Fig. 1).6 Each of these turn structures is respectively sub-classified by phi and psi angles (Tables S1 and S2, ESI†) and by Cα(i)–Cα(i + 2), Cα(i)–Cα(i + 3), or Cα(i)–Cα(i + 4) distances between residues.
The use of crosslinks between side chains of amino acids in a peptide sequence is a robust and well-established approach to stabilising peptide structure. Cyclisation through linking amine and carboxylate side chains of L- or D-amino acids has been used to enhance peptide bioactivity for many years,7 but usually without any clear rationale or understanding of the effect of the length or positioning of the crosslink on three-dimensional structure. The effect of cyclisation on conformation is often inferred through improvements in activity, changes in circular dichroism (CD) spectra, or computer simulations that predict low energy structural ensembles. Incorporating a macrocycle within 10–20 residue peptides often results in local structural changes that are difficult to identify from CD spectra, which only sample the mix of all structures present. Consequently, key requirements for stabilising different turn types in short synthetic peptides are difficult to ascertain from the bulk of literature reports on short peptides. A better understanding about how specific macrocycles can influence structure can enable improved design of bioactive peptides for future mimicry of different folded components of polypeptides.
Results and discussion
Twenty-four cyclic tetrapeptides (cyclo-1,4)-[XAAZ]-NH2 (1–24) were synthesised, varying in ring size from 14–18 atoms (Table S3, ESI†). In each case X and Z have a side chain to side chain linkage between basic (L-Lys, L-Orn, L-Dab, L-Dap) and either an acidic (L-Asp, D-Asp, L-isoAsp, D-isoAsp, L-Glu, D-Glu) amino acid or a diacid (succinic, glutaric), with two intervening Ala residues. They were then systematically examined to discover how side chain-to-side chain cyclisation of residues at the ends of tetrapeptides can affect folding into different turn motifs in water, using circular dichroism (CD) spectra in conjunction with 2D 1H-NMR spectroscopy and molecular dynamics (MD) simulations to characterise peptide structure.Structural differences were first monitored by CD spectra in water (Fig. 2). Peptides 1–3, 5, 6, 9 and 13 produced two negative molar ellipticity minima between λ ∼ 200–220 nm, these wavelengths being expected for helical peptides.8 Peptides 1–3, 6 and 9 also had a positive maximum at ∼185 nm. Larger macrocycles (>16 atoms) 4, 8, 11, 12, 15, 16 gave less prominent CD line shapes, while 17–20 containing X = D-amino acid gave unusual CD spectra. For X = a carboxylic acid, where the first amide group (Ac-X) has been moved (21 and 22) or removed (23 and 24), molar ellipticities were more intense and spectral patterns were more similar to that for the structurally well-characterised α-helix of cyclic pentapeptide Ac-(cyclo-1,5)-[KAAAD]-NH2 (25),6d,e perhaps suggesting some type of helix.5 While 21–25 appear to be helical structures, the other compounds are of less certain structural identity but they do serve as useful fingerprints diagnostic of the structure or mix of structures present in water. Overall, there was however a trend where α-turn structures (i.e.5, 17, 18, 21, 22, 23 and 24) gave a more intense molar ellipticity at λ ∼ 215 nm than β-turn structures (i.e.6, 7, 9, 15, 19 and 20) (Fig. S1, ESI†).
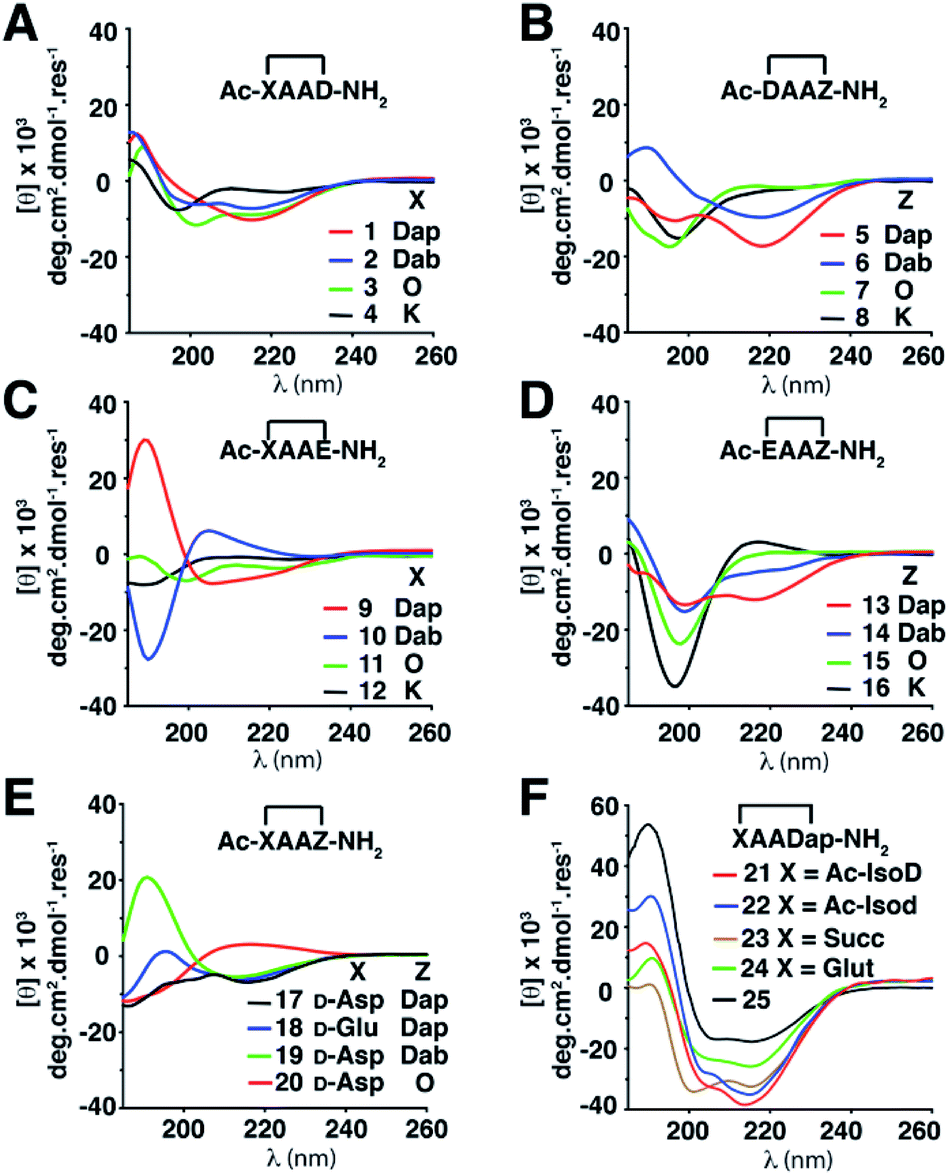 | ||
| Fig. 2 CD spectra of cyclic peptides (150 μM) in aqueous phosphate buffer (10 mM, pH 7.4, 25 °C). (A) 1–4. (B) 5–8. (C) 9–12. (D) 13–16. (E) 17–20. (F) 21–25. Standard 1-letter codes for L-amino acids unless otherwise indicated. Dap = L-2,3-diaminopropionic acid; Dab = L-2,4-diaminobutyric acid; O = L-ornithine; Succ = succinic acid; Glut = glutaric acid; IsoD and Isod are L- and D-aspartic acid respectively, where a side chain carbonyl instead forms the amide bond with the backbone nitrogen atom of the next residue. | ||
CD spectra are based on absorption of left versus right circularly polarized light by amide bonds formed between chiral amino acids, but tetrapeptides consist only of a few amides. While CD spectral traces are commonly used to interpret three-dimensional structure in peptides, they more often lead to incorrect interpretations of peptide structure,9 whereas 2D NMR spectra usually give rise to much more reliable structural characterisations.
To investigate three dimensional structure, we next analysed 1H NMR spectra for 1–24 in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:D2O, cataloguing their amide coupling constants (3JNH–CHα) that inform on residue phi angles (Table S5, ESI†), and their amide proton temperature coefficients (Δδ/ΔT) suggestive of hydrogen bonds (Table S6, ESI†). Fifteen peptides had 1–3 β-strand-like (>8 Hz) or α-helix-like (<6 Hz) coupling constants (3JNH–CHα). Peptides 5–7, 9, 13 and 15 had a C-terminal amide proton with Δδ/ΔT < 4 ppb K−1, while 6, 7, 9 and 15 had an i + 3 amide proton with Δδ/ΔT < 4 ppb K−1, suggesting possible hydrogen bonds. 2D ROESY spectra for peptides 5–7, 9, 13, 15 in water showed 34–46 intramolecular ROEs, that is consistent with well-defined structure (Tables S9–S22, ESI†). NMR structure calculations indicated that these peptides were turn-like over the three residues AAZ, with peptides 5, 7, 13 giving prominent α-N(i, i + 2), α-N(i, i + 3), β-N(i, i + 4) ROEs. However, each N-terminal acetyl moiety projected outwards and was unable to form an intramolecular hydrogen bond for X to be in a helix.
:1 H2O:D2O, cataloguing their amide coupling constants (3JNH–CHα) that inform on residue phi angles (Table S5, ESI†), and their amide proton temperature coefficients (Δδ/ΔT) suggestive of hydrogen bonds (Table S6, ESI†). Fifteen peptides had 1–3 β-strand-like (>8 Hz) or α-helix-like (<6 Hz) coupling constants (3JNH–CHα). Peptides 5–7, 9, 13 and 15 had a C-terminal amide proton with Δδ/ΔT < 4 ppb K−1, while 6, 7, 9 and 15 had an i + 3 amide proton with Δδ/ΔT < 4 ppb K−1, suggesting possible hydrogen bonds. 2D ROESY spectra for peptides 5–7, 9, 13, 15 in water showed 34–46 intramolecular ROEs, that is consistent with well-defined structure (Tables S9–S22, ESI†). NMR structure calculations indicated that these peptides were turn-like over the three residues AAZ, with peptides 5, 7, 13 giving prominent α-N(i, i + 2), α-N(i, i + 3), β-N(i, i + 4) ROEs. However, each N-terminal acetyl moiety projected outwards and was unable to form an intramolecular hydrogen bond for X to be in a helix.
Inverting the chirality from L-Asp1 (5–7) or L-Glu1 (13) to D-Asp1 (17, 19–20) or D-Glu1 (18) caused the Ac-NH to reposition closer to the peptide backbone, so that the CH3CO– group might form another hydrogen bond. Peptides 17–20 with a D-residue all had 3JNH–CHα amide coupling constants < 6 Hz for Ala2 and Ala3, and a C-terminal amide proton with Δδ/ΔT < 4 ppb K−1. Peptides 19 and 20 also had Δδ/ΔT < 4 ppb K−1 for the amide proton of residue 4. 2D ROESY spectra of 17–20 showed 43–71 ROEs and calculated NMR structures (Fig. 3) revealed almost identical structures to their corresponding L-residue analogues (5–7 and 13) (Fig. 3). The Ac-NH moiety in 17–20 was now positioned closer to the helix/turn core, but the acetyl carbonyl oxygen atom was pointing outward. NMR structures suggested no i → i + 3 or i → i + 4 hydrogen bonds. To relocate or remove the Ac-NH moiety, we further synthesised 21–24. Peptides 21–23 had only one 3JNH–CHα amide coupling constant < 6 Hz (for Ala2), while 24 had amide coupling constants < 6 Hz for both Ala2 and Ala3. All these peptides (21–24) had two Δδ/ΔT < 4 ppb K−1 for the amide protons of residue 4 and C-terminal amide. Thus, NMR data indicated an α-turn structure4,5 for 21–24 in water.
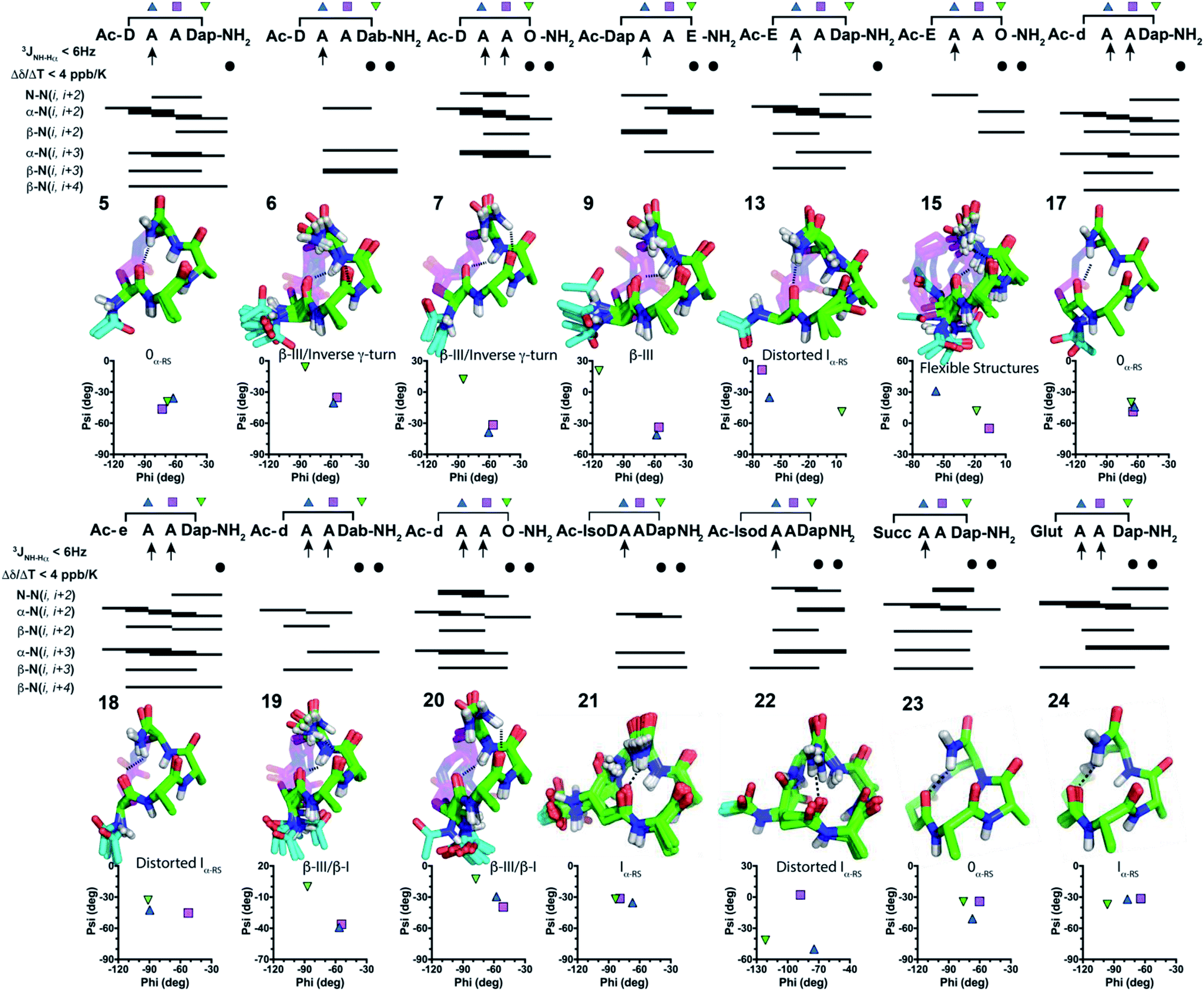 | ||
| Fig. 3 NMR summaries, 20 lowest energy structures, Ramachandran plots and turn types in water for fourteen cyclic tetrapeptides: 5–7, 9, 13, 15, 17–24 in H2O/D2O (9:1) at 298 K. Compounds 5, 13, 17, 18, 21–24 form one hydrogen bond; while 6, 7, 9, 15, 19, 20 form two hydrogen bonds. | ||
To further characterise structure ensembles for 5–7, 9, 13, 15 and 17–24, average phi and psi angles were compared with those found in classical or idealised α- and 310-helices, β- and specialised α-turns4 (where phi, psi = −58°, −47° or −60°, −30° define idealised classical α- or 310-helical turns, respectively) (Tables S1, S2 and S8, ESI†). NMR-derived structures were superimposed upon these idealised turns and the average backbone RMSDs were calculated to support their structural classifications. In all cases, the first residue X1 did not correlate with any structure. This fits well with our ability to stereo-invert or relocate or remove the first residue, whilst maintaining similar overall structures in residues Ala2–Ala3–Z4. Phi and psi angles across these three residues correlated best with an 0α-RS turn for 5, 17 and 23, but with a β(III) turn across residues Ala2–Ala3 for 6, 7, 9, 19, 20. These angles were variable in residue Z4, in some cases indicating an inverse γ-turn (6, 7) or β(I)-turn (19, 20). Peptides 13, 18, 21, 22, 24 correlated with an Iα-RS turn across Ala2–Ala3. Peptide 15 only showed α-helix or β(III)-turn character in residue Ala2, all other residues were variable.
Since the cyclisation constraint affects structure stabilisation, we also examined the χ1 angle for residue Z4 (Fig. 4). The gauche (−) rotamer was adopted for Dap4-containing 5 (+58°) and 17 (+59°). This χ1 angle optimally positions the constraint for i → i + 4 hydrogen bond formation. Peptides 13 (χ1 = +50°) and 18 (χ1 = +41°) were next closest. For the remaining peptides with a larger side chain in residue Z4, the χ1 angle gradually shifted towards the gauche (+) rotamer: e.g. Dab-containing 6 (χ1 = −28°) and 19 (χ1 = −23°). For β(III)-like Orn-containing 7 (χ1 = −58°) and 20 (χ1 = −59°), this rotamer enabled two i → i + 3 hydrogen bonds to form. MD simulations for 5–7, 9, 13, 15 and 17–20 supported this finding (Fig. 4A).
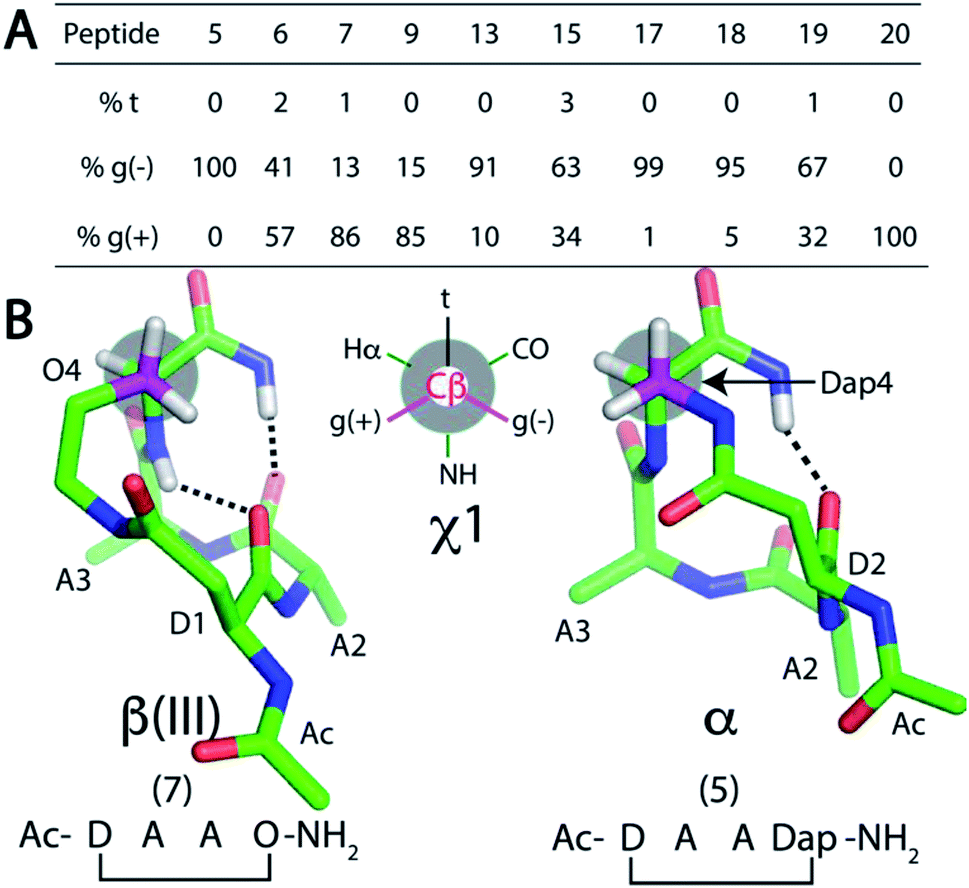 | ||
| Fig. 4 (A) Rotamer populations for residue Z4 from 50 ns MD simulations of cyclic peptides 5–20. NMR structures of: (B) 7 and 5 show how the χ1 angle of residue Z4 controls β(III)-turn vs. α-turn structures. | ||
To summarise Fig. 4, cyclic tetrapeptides with a small side chain at the fourth residue (Z = Dap) favoured χ1 in a gauche (−) configuration, which stabilised a psi dihedral angle of −40° ± 10° and a C-terminal amide NH was involved in an i → i + 4 hydrogen bond. By contrast, a larger side chain for Z = Dab, Orn, Glu favoured a lower energy conformation, χ1 for gauche (+). NMR structures of peptides where Z = Dap (5, 13, 17, 18, 21–24) had residues Ala2–Ala3–Dap4 and the C-terminal amide folded into α-turn structures. Amide carbonyls were aligned to potentially accept hydrogen bonds from amide protons in a peptide appended to the C-terminus of Dap. A distinctive difference between these structures was the intramolecular hydrogen bonding network. One i → i + 4 hydrogen bond was found in peptides 5, 13, 17, 18 and 21–24, whereas two i → i + 3 hydrogen bonds were present in 6, 7, 9, 15, 19 and 20 (Fig. 3). We therefore considered that 5, 13, 17, 18 and 21–24 could be potential N-terminal helix nucleators as their structures support an i → i + 4 hydrogen bond that is crucial in an α-helical structure.
Next, we searched the PDB for examples of α-helix, α-turn, β-III and β-I turns in hotspots of proteins. We superimposed (PyMOL) our cyclic peptide structures from Fig. 3, calculated by NMR and validated by MD simulations (Fig. 4A), upon three consecutive residues of the protein secondary structures (Fig. 5). Examples spanned classical α-helix (0α-RS), α-turn (Iα-RS), β-turn type III (310-helix), β-turn type I and γ-turn in 12 different protein crystal structures. These different protein turns play important structural roles within human, mammalian and bacterial proteins of very diverse functions. The successful structural mimicry here of these important protein structural motifs by small cyclic peptides provides new capacity to study these structures independent of packing influences in proteins.
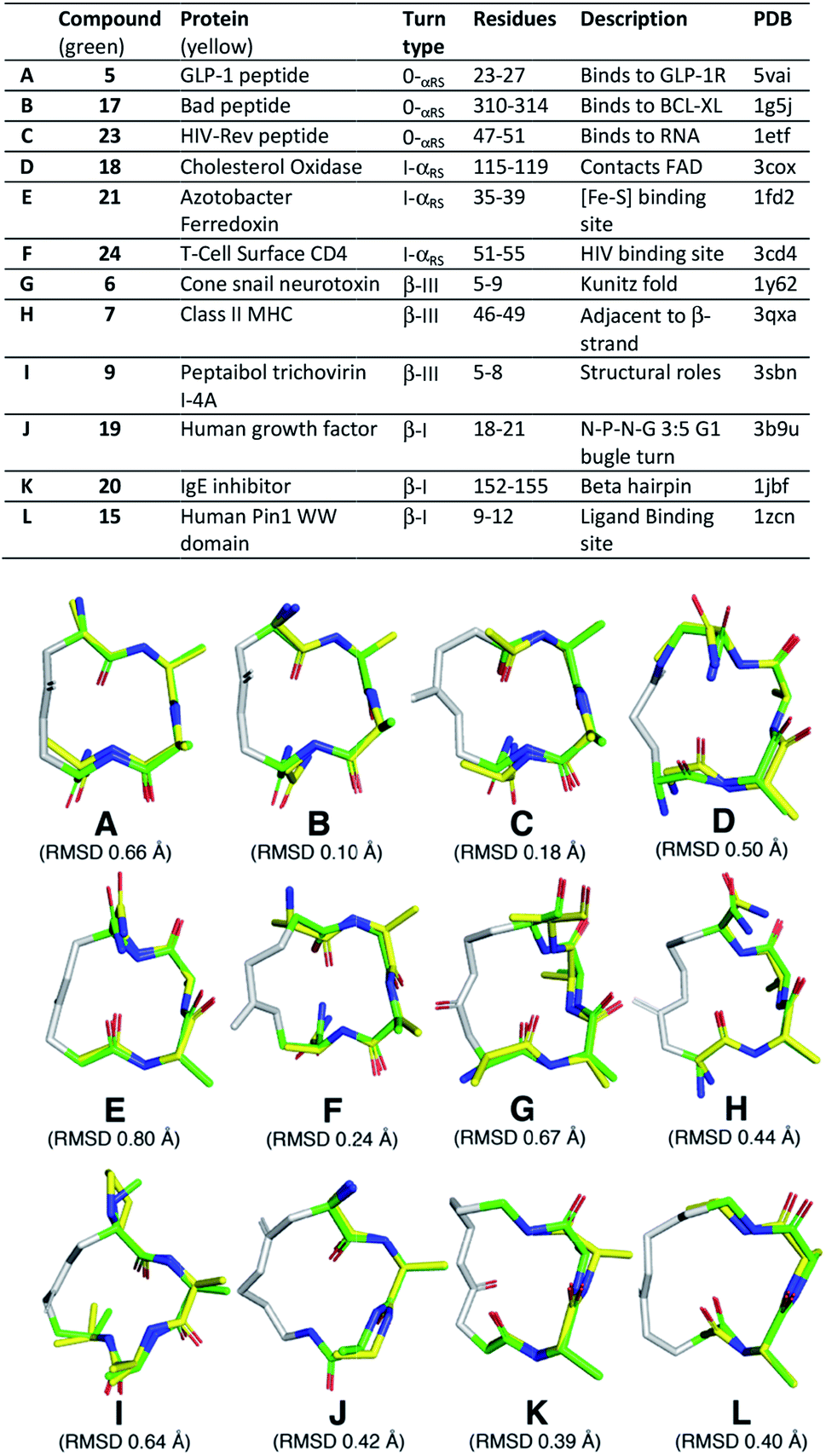 | ||
| Fig. 5 Average backbone solution structures of 5–7, 9, 15, 17–21 and 23–24 (green; residues: A, A, X/Z) superimposed on the three central, consecutive, residues of an α-helix, α-turn, β-III turn and β-I turn from protein crystal structures (yellow). The linkers in these compounds are coloured grey. | ||
Finally, we appended these cyclic tetrapeptides to the N-terminus of nonapeptide ARLARLARL-NH2 (26) to give 27–39. For comparison, we also appended cyclic pentapeptide Ac-[KAAAD]- (25) to 26 to give 40. The relative percent α-helicity was measured from CD spectra using fractional helicities (fH) calculated in 10 mM phosphate buffer (Table 1). Each cyclic tetrapeptide had a different α-helix nucleating capacity (fH 0.29–0.66) when attached to the N-terminus of the non-helical peptide 26 (fH = 0.12). The N-capped 9-residue peptides with the best helix induction (Table 1) were 36–38, 40 (fH ≥ 0.59) > 33, 39 (fH ∼0.53) > 27, 31, 34 (fH ∼0.43) where an α-turn was appended. These data show how subtle differences in cyclic tetrapeptide structure permit or prevent helix nucleation. None of the cyclic tetrapeptides alone has its key carbonyl groups perfectly aligned for helix induction within the cycle itself, yet some are sufficiently aligned to nucleate helicity in attached peptides.
| # | Sequence | Cycle | f H (buffer) | [θ222]/[θ208] |
|---|---|---|---|---|
| a 10 mM phosphate buffer (pH 7.4, 25 °C), [peptide] = 125 μM. | ||||
| 27 | Ac-cyclo-[DAADap]-(ARL)3-NH2 | 5 | 0.42 | 1.15 |
| 28 | Ac-cyclo-[DAADab]-(ARL)3-NH2 | 6 | 0.31 | 0.84 |
| 29 | Ac-cyclo-[DAAO]-(ARL)3-NH2 | 7 | 0.29 | 0.74 |
| 30 | Ac-cyclo-[DapAAE]-(ARL)3-NH2 | 9 | 0.29 | 0.73 |
| 31 | Ac-cyclo-[EAADap]-(ARL)3-NH2 | 13 | 0.42 | 0.85 |
| 32 | Ac-cyclo-[EAAO]-(ARL)3-NH2 | 15 | 0.36 | 0.75 |
| 33 | Ac-cyclo-[dAADap]-(ARL)3-NH2 | 17 | 0.54 | 0.98 |
| 34 | Ac-cyclo-[eAADap]-(ARL)3-NH2 | 18 | 0.45 | 0.96 |
| 35 | Ac-cyclo-[dAAO]-(ARL)3-NH2 | 20 | 0.36 | 0.77 |
| 36 | Ac-cyclo-[isoDAADap]-(ARL)3-NH2 | 21 | 0.66 | 1.08 |
| 37 | Ac-cyclo-[isodAADap]-(ARL)3-NH2 | 22 | 0.61 | 0.98 |
| 38 | Cyclo-[SuccAADap]-(ARL)3-NH2 | 23 | 0.65 | 1.03 |
| 39 | Cyclo-[GlutAADap]-(ARL)3-NH2 | 24 | 0.52 | 0.91 |
| 40 | Ac-cyclo-[KAAAD]-(ARL)3-NH2 | 25 | 0.59 | 1.02 |
Conclusions
We synthesised and characterised 24 cyclic tetrapeptides, Ac-(cyclo-1,4)-[XAAZ]-NH2, using different i → i + 3 side chain amide crosslinks for cyclization. Such crosslinks have been widely used to improve the bioactivity of peptides, however the cyclic tetrapeptide structures were unknown or inferred on the basis of CD spectral line-shapes, which usually lead to incorrect structure assignments. Here we combined 2D ROESY spectra, amide coupling constants, temperature coefficients and MD simulations to determine their three-dimensional structures in water. Fourteen crosslinked macrocycles were studied with a ring size of 14–16 atoms (5–7, 9, 13, 15, 17–24) that gave stable structures in water (Fig. 3). The smaller 14-membered rings tended to be α-turns; 5, 17 and 23 were 0α-RS turns,5 while 21 and 22 were Iα-RS turns4 and 1 was unstructured. The 15-membered rings adopted β-turns (6, 9, 19) or Iα-RS turns4 (13, 18, 24), with 2 being unstructured. The 16-membered rings were either β-III turns (7, 20) or did not show stable structures (3, 10, 14). Larger macrocycles with 17- (4, 8, 11, 15) or 18- (12, 16) membered rings gave unstable or less stable structures in water (Table S3, ESI†). All α-turn structures (5, 13, 17, 18, 21–24) had a residue with a small side chain Dap at position Z, with its χ1 angle defining a gauche (−) configuration and favoured one i → i + 4 hydrogen bond. On the other hand, β-turn structures were observed for peptides (6, 7, 9, 19, 20) with residues having larger side chains (Dab, Orn, Glu) at this position and favoured two i → i + 3 hydrogen bonds. These results contrast with a previous report that suggested some of these bridges form β-II and γ-turns, but those peptides also contained turn-promoting residues (proline, glycine).7c The Ac-NH moiety in 5–7, 9, 13, 15 did not stabilise the structures. Inverting chirality from L- to D- (17–20), or repositioning (21, 22) or removing (23, 24) the N-terminal Ac-NH moiety, preserved turn structure in the last three residues 2–4 (AAZ).Each of these model cyclic tetrapeptides were found to successfully mimic corresponding α-helix, α-turn, β-III turn and β-I turn secondary structural components found in biologically important locations in proteins, suggesting the potential for future mimicry of functional properties of such important irregular structures at hotspots in proteins.
We also investigated helix nucleation by these cyclic tetrapeptides at the N-terminus of a non-helical 9-residue peptide sequence ARLARLARL-NH2 (Table 1). Tetrapeptides forming β(III)-turns (e.g.6, 7, 9, 20), favouring a gauche (+) conformation and two i → i + 3 hydrogen bonds, induced less helicity than α-turn forming 5 and 13, which showed a gauche (−) conformation and one i → i + 4 hydrogen bond. Cyclic tetrapeptides with D- rather than L-residues at the (i) position X were better helix nucleators (17vs.5, 18vs.13). Cyclic tetrapeptides Ac-[(isoD or d)AADap]-NH2 (21 or 22)10 and cyclic pentapeptide 256e have been reported previously. We find that they were better nucleators than 17 and 18. Achiral diacid(i) → Dap(i + 3) crosslinks were also efficient helix nucleators. Succinate at the i position (23) was as good, or better, a helix nucleator than isoaspartate 22 or 25. Interestingly, the glutarate linker (24) was also an effective nucleator (equal to 17), and such dicarboxylate(i) → Dap(i + 3) linked tetrapeptides are important new N-terminal helix-nucleating templates easily synthesised, requiring fewer protecting group strategies and cheaper to make than most macrocycles herein.
These new insights into stereochemical and structural influences on peptide folding have led to better definition of peptide turns and helical twists, which mimic the nonregular secondary structures found in bioactive hotspots of proteins, and can inspire design of small molecules to structurally and functionally mimic some of the many subtly different turns and helices in proteins.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank NHMRC (Senior Principal Research Fellowship 1117017) and ARC (DP130100629, DP180103244, CE14010001-1) for grants to D. F., Engineering and Physical Sciences Research Council for an Overseas Travel Grant to J. M. (EP/M001873/2), and Carlsberg Foundation (Denmark) for supporting F. D.Notes and references
- (a) E. G. Hutchinson and J. M. Thornton, Protein Sci., 1994, 3, 2207–2216 CrossRef CAS PubMed; (b) C. M. Wilmot and J. M. Thornton, Protein Eng., 1990, 3, 479–493 CrossRef CAS PubMed.
- (a) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed; (b) L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC; (c) J. D. Tyndall, B. Pfeiffer, G. Abbenante and D. P. Fairlie, Chem. Rev., 2005, 105, 793–826 CrossRef CAS PubMed.
- (a) R. S. Harrison, PhD dissertation, University of Queensland, 2011; (b) C. M. Bergey, A. M. Watkins and P. S. Arora, Bioinformatics, 2013, 29, 2806–2807 CrossRef CAS PubMed; (c) J. Gavenonis, B. A. Sheneman, T. R. Siegert, M. R. Eshelman and J. A. Kritzer, Nat. Chem. Biol., 2014, 10, 716–722 CrossRef CAS PubMed.
- (a) V. Pavone, G. Gaeta, A. Lombardi, F. Nastri, O. Maglio, C. Isernia and M. Saviano, Biopolymers, 1996, 38, 705–721 CrossRef CAS PubMed; (b) D. V. Nataraj, N. Srinivasan, R. Sowdhamini and C. Ramakrishnan, Curr. Sci., 1995, 69, 434–447 CAS.
- H. N. Hoang, R. W. Driver, R. L. Beyer, A. K. Malde, G. T. Le, G. Abbenante, A. E. Mark and D. P. Fairlie, Angew. Chem., Int. Ed., 2011, 50, 11107–11111 CrossRef CAS PubMed.
- (a) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 ( Angew. Chem. , 2014 , 126 , 13234–13257 ) CrossRef CAS PubMed; (b) O. Jacobsen, H. Maekawa, N. H. Ge, C. H. Gorbitz, P. Rongved, O. P. Ottersen, M. Amiry-Moghaddam and J. Klaveness, J. Org. Chem., 2011, 76, 1228–1238 CrossRef CAS PubMed; (c) Y. W. Kim, P. S. Kutchukian and G. L. Verdine, Org. Lett., 2010, 12, 3046–3049 CrossRef CAS PubMed; (d) A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 ( Angew. Chem. , 2014 , 126 , 7085–7089 ) CrossRef CAS PubMed; (e) N. E. Shepherd, H. N. Hoang, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 2974–2983 CrossRef CAS PubMed; (f) J. W. Taylor, Biopolymers, 2002, 66, 49–75 CrossRef CAS PubMed; (g) H. Lin, Y. Jiang, Q. Zhang, K. Hu and Z. G. Li, Chem. Commun., 2016, 52, 10389–10391 RSC.
- (a) P. W. Schiller, T. M. D. Nguyen and C. Lemieux, Tetrahedron, 1988, 44, 733–743 CrossRef CAS; (b) A. Miranda, S. C. Koerber, J. Gulyas, S. L. Lahrichi, A. G. Craig, A. Corrigan, A. Hagler, C. Rivier, W. Vale and J. Rivier, J. Med. Chem., 1994, 37, 1450–1459 CrossRef CAS PubMed; (c) H. R. Marepalli, O. Antohi, J. M. Becker and F. Naider, J. Am. Chem. Soc., 1996, 118, 6531–6539 CrossRef CAS; (d) S. Arttamangkul, J. E. Ishmael, T. F. Murray, D. K. Grandy, G. E. DeLander, B. L. Kieffer and J. V. Aldrich, J. Med. Chem., 1997, 40, 1211–1218 CrossRef CAS PubMed; (e) K. A. Carpenter, R. Schmidt, S. Y. Yue, L. Hodzic, C. Pou, K. Payza, C. Godbout, W. Brown and E. Roberts, Biochemistry, 1999, 38, 15295–15304 CrossRef CAS PubMed; (f) A. Giolitti, M. Altamura, F. Bellucci, D. Giannotti, S. Meini, R. Patacchini, L. Rotondaro, S. Zappitelli and C. A. Maggi, J. Med. Chem., 2002, 45, 3418–3429 CrossRef CAS PubMed; (g) S. De Luca, M. Saviano, R. Della Moglie, G. Digilio, C. Bracco, L. Aloj, L. Tarallo, C. Pedone and G. Morelli, ChemMedChem, 2006, 1, 997–1006 CrossRef CAS PubMed; (h) M. Der Torossian Torres, A. F. Silva, F. L. Alves, M. L. Capurro, A. Miranda and V. X. O. Junior, Int. J. Pept. Res. Ther., 2014, 20, 277–287 CrossRef CAS; (i) R. Perlikowska, J. Piekielna, L. Gentilucci, R. De Marco, M. C. Cerlesi, G. Calo, R. Artali, C. Tomboly, A. Kluczyk and A. Janecka, Eur. J. Med. Chem., 2016, 109, 276–286 CrossRef CAS PubMed; (j) R. S. Harrison, N. E. Shepherd, H. N. Hoang, G. Ruiz-Gómez, T. A. Hill, R. W. Driver, V. S. Desai, P. R. Young, G. Abbenante and D. P. Fairlie, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11686–116891 CrossRef CAS PubMed; (k) F. Plisson, T. A. Hill, J. M. Mitchell, H. N. Hoang, A. D. de Araujo, W. Xu, A. Cotterell, D. J. Edmonds, R. V. Stanton, D. R. Derksen, P. M. Loria, D. A. Griffith, D. A. Price, S. Liras and D. P. Fairlie, J. Med. Chem., 2017, 127, 703–714 CrossRef CAS PubMed.
- C. Toniolo, F. Formaggio and R. W. Woody, in Comprehensive Chiroptical Spectroscopy, Wiley & Sons, Inc., 2012, pp. 499–544 Search PubMed.
- R. W. Driver, H. N. Hoang, G. Abbenante and D. P. Fairlie, Org. Lett., 2009, 11, 3092–3095 CrossRef CAS.
- H. Zhao, Q. S. Liu, H. Geng, Y. Tian, M. Cheng, Y. H. Jiang, M. S. Xie, X. G. Niu, F. Jiang, Y. O. Zhang, Y. Z. Lao, Y. D. Wu, N. H. Xu and Z. G. Li, Angew. Chem., Int. Ed., 2016, 55, 12088–12093 ( Angew. Chem. , 2016 , 128 , 12267–12272 ) CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, materials, chemical synthesis (peptide assembly, cyclisation, cleavage), purification (HPLC), characterisation (CD, NMR), structure calculations, molecular dynamics simulations, and extensive NMR parameters and spectra are available (Tables S1–S22, Fig. S1–S40; 41 pages). See DOI: 10.1039/c9sc04153b |
| This journal is © The Royal Society of Chemistry 2019 |