DOI:
10.1039/C9SC04151F
(Edge Article)
Chem. Sci., 2019,
10, 10802-10814
Bifunctional ligand design for modulating mutant p53 aggregation in cancer†
Received
20th August 2019
, Accepted 6th October 2019
First published on 7th October 2019
Abstract
Protein misfolding and aggregation contributes to the development of a wide range of diseases. In cancer, over 50% of diagnoses are attributed to p53 malfunction due to missense mutations, many of which result in protein misfolding and accelerated aggregation. p53 mutations also frequently result in alteration or loss of zinc at the DNA-binding site, which increases aggregation via nucleation with zinc-bound p53. Herein, we designed two novel bifunctional ligands, LI and LH, to modulate mutant p53 aggregation and restore zinc binding using a metallochaperone approach. Interestingly, only the incorporation of iodine function in LI resulted in modulation of mutant p53 aggregation, both in recombinant and cellular environments. Native mass spectrometry shows a protein–ligand interaction for LI, as opposed to LH, which is hypothesized to lead to the distinct difference in the p53 aggregation profile for the two ligands. Incorporation of a di-2-picolylamine binding unit into the ligand design provided efficient intracellular zinc uptake, resulting in metallochaperone capability for both LI and LH. The ability of LI to reduce mutant p53 aggregation results in increased restoration of p53 transcriptional function and mediates both caspase-dependent and -independent cell death pathways. We further demonstrate that LI exhibits minimal toxicity in non-cancerous organoids, and that it is well tolerated in mice. These results demonstrate that iodination of our ligand framework restores p53 function by interacting with and inhibiting mutant p53 aggregation and highlights LI as a suitable candidate for comprehensive in vivo anticancer preclinical evaluations.
Introduction
Amyloidogenic proteins are prone to endogenous misfolding and prion-like conversion from a soluble, folded protein into alternative oligomeric and fibrillar structures.1 Proteins susceptible to this process include amyloid-β, tau, TDP-43, SOD1, and α-synuclein and contribute to a wide range of diseases including Alzheimer's disease and ALS.2 These proteins exhibit toxic gain-of-function (GoF) effects by self-propagating and acting as seeds to initiate aggregation.3 Similar to neurodegenerative diseases, recent studies have demonstrated that protein misfolding and aggregation play a role in cancer development through misfolding of the tumour suppressor protein p53.4 Several reports have highlighted that p53 aggregation not only leads to loss of function, but that it can co-aggregate with homologous proteins p63 and p73 to form amyloid oligomers and fibrils.4a,5
p53 plays a critical role in controlling the cell cycle by initiating apoptosis, DNA repair, and cell cycle arrest of damaged cells.6 The core DNA-binding domain of p53 (p53C) contains a single Zn2+ ion that is essential for proper protein folding and function.7 However, p53 is mutated in over 50% of cancer diagnoses, the most common mutations affecting the protein's tertiary structure and frequently resulting in a loss or alteration of Zn-binding at the core site.8 This can lead to protein unfolding and enhanced aggregation due to exposure of amyloidogenic regions of the protein (residues 251–257).8a,9 Kinetic studies indicate that this occurs via a two-step process wherein the first involves relatively slow unfolding of p53C to expose the aggregation nucleus followed by a second, rapid aggregation step.10 Interestingly, apo p53C (zinc-free) increases the aggregation process via nucleation with zinc-bound p53C and contributes to loss of protein function.11 The common hotspot mutant p53-Y220C destabilizes the protein's tertiary structure due to an exposed cavity at the surface of the protein. This can result in loss of Zn2+ and causes accelerated protein aggregation.7,8,10a,12 While research regarding restoration of p53 function has largely focused on stabilization of mutant p53C,13 repopulating the metal-depleted site via metallochaperones14 has been shown to restore function to common p53 mutants.13a,14a–c,15
More broadly, targeted metal ion chelation and redistribution has shown utility both as an anticancer strategy16 and in modulating amyloidogenic protein aggregation.17 In addition, a number of small molecule/peptide inhibitors of p53 aggregation have been developed18 and a cell-penetrating peptide (ReACp53) developed by Eisenberg and co-workers rescued p53 function in high-grade serous ovarian carcinomas and led to decreased tumour proliferation in xenograft models.9 Given the increased propensity for aggregation and possible zinc loss in the common mutant p53-Y220C, we used this as a model for testing compounds targeted to modulate mutant p53 aggregation.
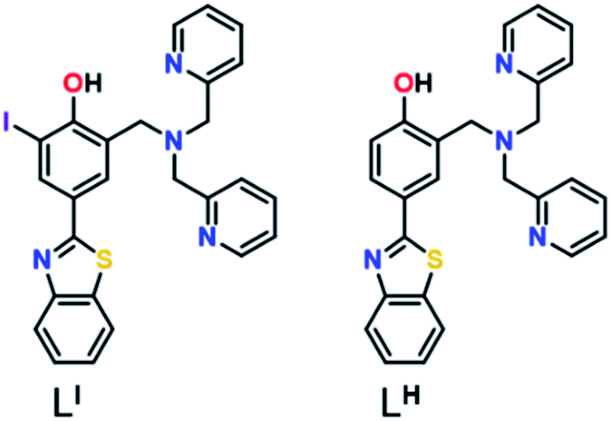
Herein, we describe two novel bifunctional ligands, LI and LH (Fig. 1), designed to reactivate p53 by inhibiting mutant p53 aggregation and restore zinc-binding using a metallochaperone approach. With reports showing that Zn-free p53 exhibits accelerated protein aggregation,5b,8a,11 the incorporation of a zinc metallochaperone unit to remetallate apo-p53 in combination with an aggregation-targeting moiety could provide advantages over reported single-target compounds. Furthermore, multifunctional agents are advantageous due to their ability to act on multiple targets, resulting in additive or synergistic effects, thereby increasing their therapeutic potential.19 The importance of the iodine in LI for inhibiting mutant p53 aggregation, activating specific cell death pathways, and exhibiting cytotoxic selectivity in cancer cells over non-cancerous organoids is highlighted in this work.
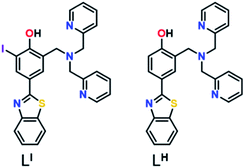 |
| | Fig. 1 Chemical structure of ligands LI and LH. | |
Results and discussion
Ligand design and synthesis
Amyloid aggregates of the p53 protein have been identified in tumour cell lines as well as patient biopsies and are correlated with tumour growth due to loss of protein function.4a,5b,20 Biophysical studies have characterized p53 aggregates as largely β-sheet fibrils and demonstrate their ability to bind to the fluorescent amyloid marker thioflavin T (ThT).4e As such, we designed amyloid interacting molecules based on a structure similar to thioflavin-T. Di-2-picolylamine groups were appended at the 2-position as the zinc chelator based on the frequent use of this moiety in zinc chemosensors.21 A similar ligand scaffold was reported by Mirica and co-workers to interact with and modulate the aggregation of the amyloid-β peptide involved in Alzheimer's disease.17cLI contains an iodine substituent at the ortho position of the phenol function to probe the role of halogen bonding interactions and/or differing steric and electronic effects on influencing p53 aggregation (Fig. 1). LI and LH were prepared via a Mannich reaction with 2-(4-hydroxy-3-iodo)benzathiazole (LI) or 2-(4-hydroxy)benzathiazole (LH) and di-2-picolylamine. LI required a prior iodination step of 4-hydroxybenzaldehyde with iodine monochloride.
Effects of LI and LH on p53 aggregation
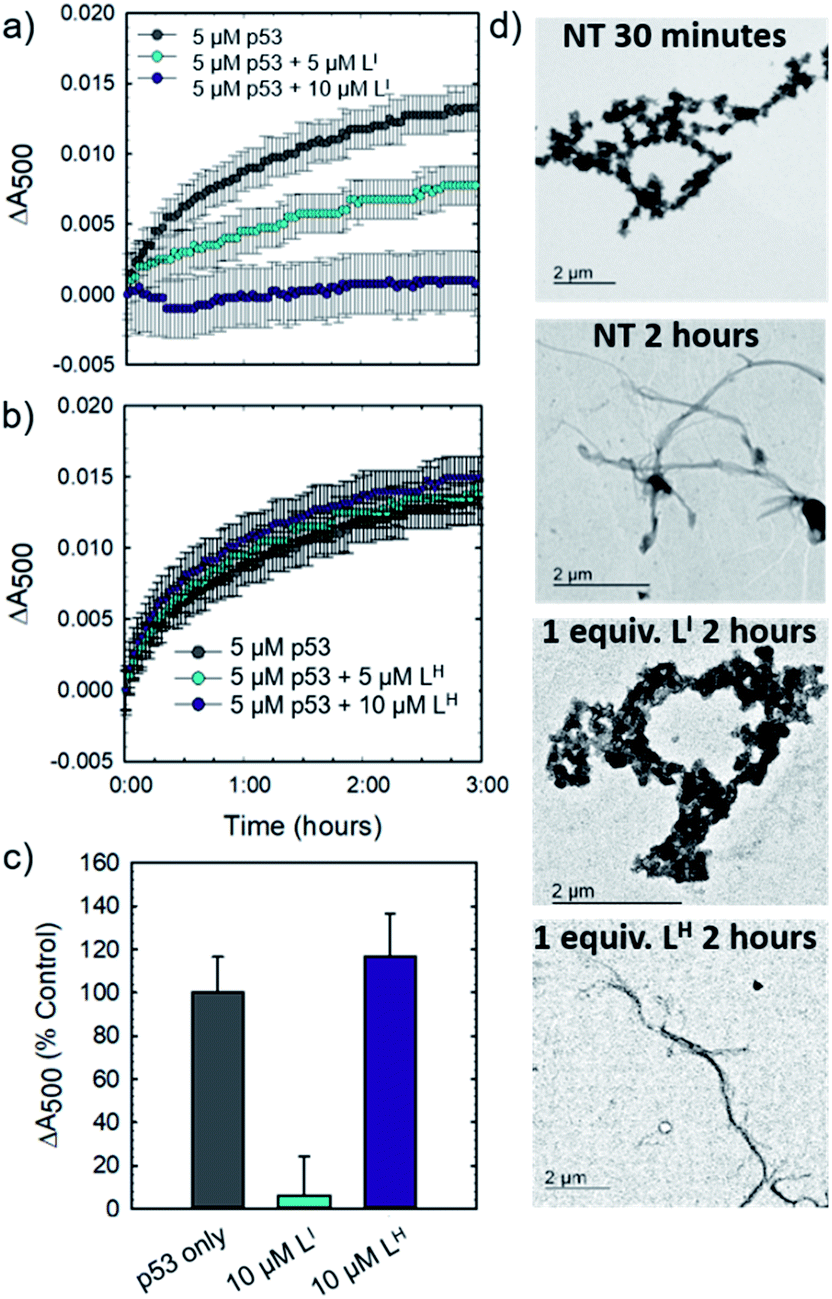
Initial studies to determine the effect of LI and LH on mutant p53 aggregation were carried out in vitro. The core DNA-binding domain of mutant p53-Y220C (p53C-Y220C) was incubated at 37 °C and light scattering at 500 nm was monitored over time. Protein alone (5 μM) demonstrated a rapid growth phase which continued to increase until two hours, where leveling off begins to occur, indicating a depletion of the substrate (Fig. 2a and b). Aggregation does not significantly increase past three hours up to six hours of monitoring (data not shown). Conversely, addition of one equivalent (5 μM) of LI significantly inhibits mutant p53 aggregation, resulting in nearly 50% reduction of aggregation after only 3 hours (Fig. 2a). This effect is concentration dependent as the addition of two equivalents of LI (10 μM) results in nearly complete inhibition of mutant p53 aggregation. Interestingly, neither the addition of 5 or 10 μM of LH cause inhibition of p53 aggregation (Fig. 2b). This result is unexpected given their structural similarity and hints at the importance of the iodine moiety in the LI structure. Increasing the LI or LH concentrations up to 25 μM afforded similar changes in aggregation compared to results at 10 μM (data not shown). To investigate whether this effect was exclusive to p53-Y220C, we also tested the effect of LI and LH on the aggregation of wild-type p53 under aggregating conditions. LI can also modulate the aggregation of wild-type p53, however, to a lesser extent in comparison to p53-Y220C. LH had no effect on the aggregation of wild-type p53, similar to the results for p53-Y220C (Fig. S5†). Overall, this suggests that LI could be used to modulate p53 aggregation beyond the specific p53-Y220C mutant. Similar aggregation profiles are obtained when monitoring aggregate size distribution via gel electrophoresis, wherein incubation of mutant p53 with two equivalents of LI for two hours limits aggregation and an intense band representative of monomeric species is observed (Fig. S6†). Conversely, protein alone and incubation with two equivalents of LH results in limited soluble species observable on the gel. These results were further corroborated upon investigation of aggregate morphology via transmission electron microscopy (TEM). After 30 minutes of incubation at 37 °C under constant agitation, p53 alone exhibits only amorphous aggregate structures by TEM, however, after 2 hours, mostly fibrillar structures are observed (Fig. 2d). Upon incubation with two equivalents (10 μM) of LI, fibrillar structures are inhibited and mostly amorphous aggregates were observed. Conversely, addition of 10 μM of LH resulted in the observation of largely fibrillar structures.
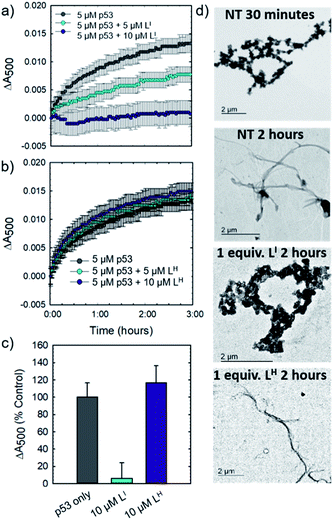 |
| | Fig. 2
LI inhibits mutant p53 aggregation in vitro. (a) 5 μM p53C-Y220C in 30 mM Tris–HCl, 150 mM NaCl, pH 7.4 was incubated at 37 °C. Light scattering at 500 nm was monitored over time from 0 to 6 hours. Absorbance readings were recorded every 3 minutes, with 30 seconds of agitation before each reading. Addition of 5 and 10 μM of LI shows concentration dependent inhibition of mutant p53 aggregation. (b) Addition of 5 and 10 μM of LH results in no significant changes in mutant p53 aggregation. (c) Percent changes in light scattering of mutant p53 upon addition of 10 μM LI/LH after 2 hours of incubation at 37 °C. (d) TEM images of mutant p53C-Y220C (8 μM) under specified conditions. | |
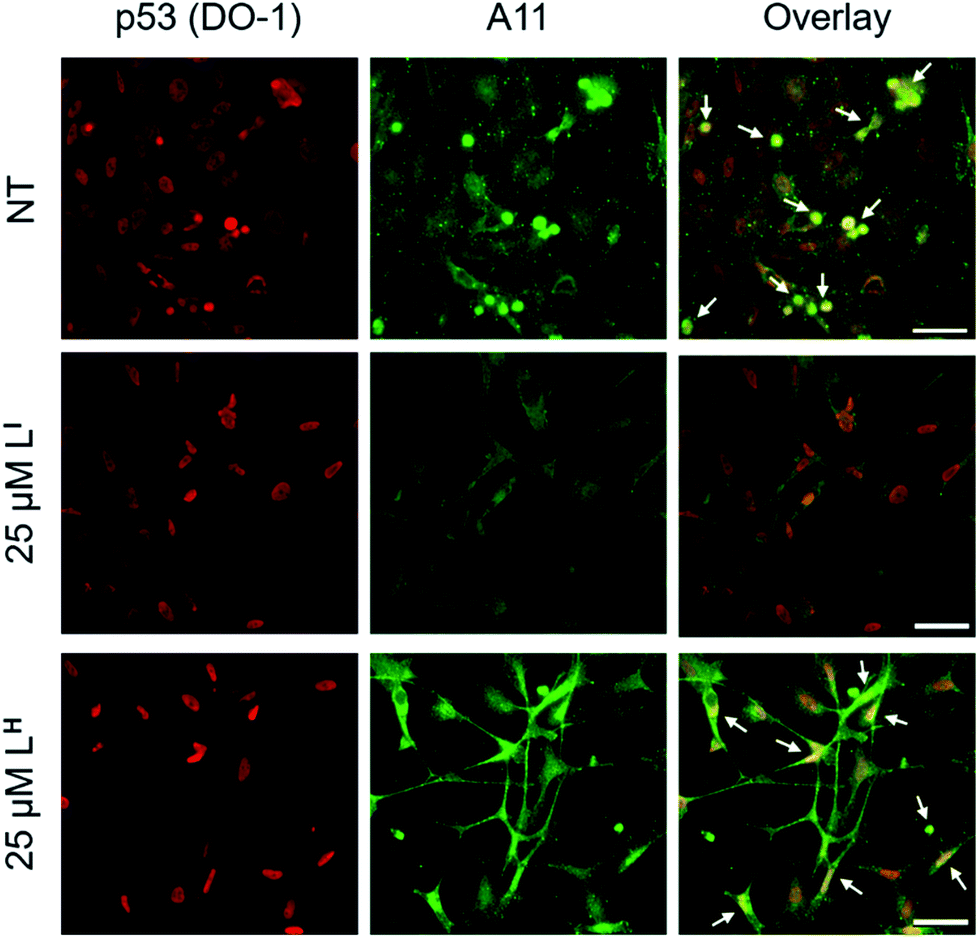
Using recombinant protein, we demonstrated the ability of LI to significantly inhibit mutant p53 aggregation in contrast to LH, which has no observable effect. We sought to determine whether this difference in aggregation inhibition is observed in cancer cell lines. It is important to study protein aggregation in the context of its complex cellular environment due to factors such as molecular chaperones and proteases, which are known to play a key role in protein folding.22 To this end, we used immunofluorescence to study the behavior of p53 aggregation upon treatment with LI/LH in the gastric cancer cell line NUGC3 expressing the p53-Y220C mutant. Further cell studies were carried out at 25 μM (vide infra), as this concentration corresponds to the IC50 value for LH (Table S5†). Using the p53 antibody DO-1, we detected high levels of p53 expression in NUGC3 (Fig. 3). Overexpression of p53 is commonly reported in cancer cell lines containing mutant p53 – the impaired function can often lead to changes in protein conformation and alter stability when targeted for degradation, leading to p53 accumulation.20a,23 We also observed high expression levels of aggregates in the NUGC3 cell line using the antibody A11, which has been used previously to label p53 aggregates.20,24 Under non-treated conditions, NUGC3 showed widespread protein aggregation via A11 that colocalized with p53. Interestingly, 24 hours of treatment with 25 μM LI significantly decreased the amount of A11-detected aggregates and resulted in decreased colocalization observed between DO-1 (p53) and A11. This indicates that LI is effective at reducing p53 aggregation in the mutant p53 cell line. In agreement with results obtained in the p53 aggregation experiment above, the addition of 25 μM LH did not decrease the levels of A11-detected aggregates in comparison to the control. In addition, significant co-localization between DO-1 (p53) and A11 is observed. We also investigated changes in p53 aggregation via co-immunofluorescence with DO-1 and ThT, a fluorescent dye that labels misfolded amyloid aggregates.25 Treatment with 25 μM LI led to an observable decrease in colocalization between p53 and ThT, whereas treatment with 25 μM LH exhibits high levels of colocalization similar to non-treated controls (Fig. S7†). Taken together, these results highlight the key importance of the iodine moiety of LI for modulation of mutant p53 aggregation even in cellular systems.
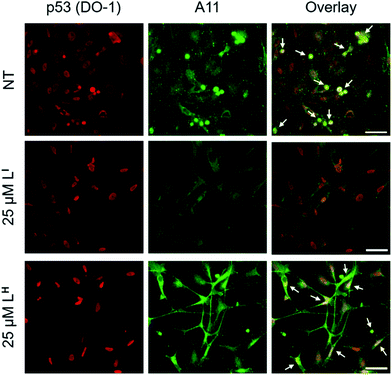 |
| | Fig. 3 Amyloid oligomer (A11) staining is reduced in mutant p53 cell line NUGC3 after treatment with LI. NUGC3 cells were treated with 25 μM LI/LH or 0.1% DMSO (NT) for 24 hours followed by labelling with anti-p53 (DO-1) and anti-oligomer (A11) antibodies at concentrations of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 and 1:100 respectively. Images were obtained using a fluorescence microscope. Columns from left to right include: DO-1 (anti-p53), A11 (anti-oligomer), and co-immunofluorescence of DO-1 and A11. White arrows representative of overlap between DO-1 and A11. The scale bar represents 50 μm. :1000 and 1:100 respectively. Images were obtained using a fluorescence microscope. Columns from left to right include: DO-1 (anti-p53), A11 (anti-oligomer), and co-immunofluorescence of DO-1 and A11. White arrows representative of overlap between DO-1 and A11. The scale bar represents 50 μm. | |
Interaction between LI/LH and mutant p53
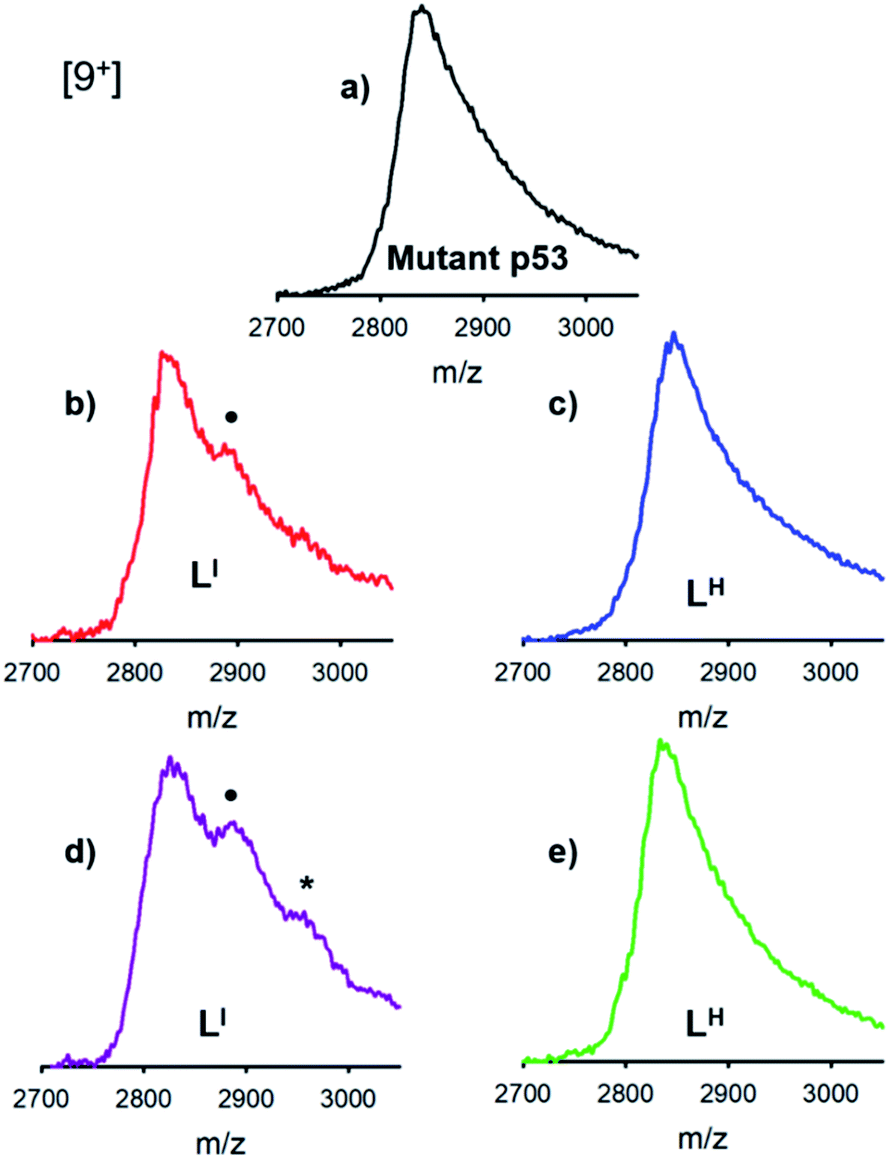
The distinct differences between LI and LH in modulating mutant p53 aggregation prompted an investigation into the potential differential binding of our bifunctional ligands with mutant p53. Using native mass spectrometry, we observed that the addition of increasing concentrations of LI affords an additional species in the mutant p53 spectrum. This corresponds to a mass increase of 564 Da, which is indicative of one equivalent of LI bound to p53 (Fig. 4). At higher equivalents of LI, a peak corresponding to a 1128 Da mass increase is evident, indicating the interaction of two LI ligands with mutant p53. In contrast, no evidence of LH interacting with mutant p53 was observed, even at higher concentrations. This suggests that the ability of LI to reduce mutant p53 aggregation is a result of direct interactions with the protein and could explain why mutant p53 aggregation is not perturbed in the presence of LH. While this result is both interesting and surprising due to their structural similarity, it is possible that the iodine is contributing to halogen bonding, which would result in favorable interactions with the exposed aggregation-prone, hydrophobic protein segment.26 Recent pharmaceutical advances have highlighted the advantages of halogen bonding and demonstrate significantly improved binding of many small molecules to their protein targets due to halogen bonding to carbonyl groups.13b,27 Furthermore, Eisenberg and co-workers have previously demonstrated that by binding the exposed hydrophobic segment, protein aggregation was prevented and therefore contributed to restored protein function.9
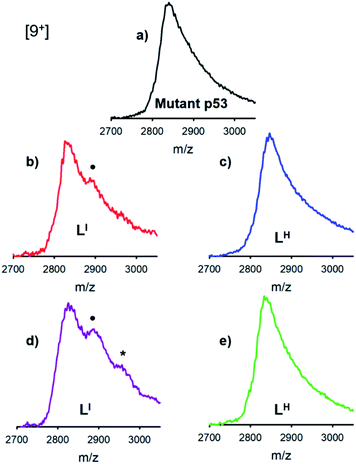 |
| | Fig. 4 Interactions between LI and mutant p53 detected using native mass spectrometry, whereas no interaction is observed for LH even at high concentrations. Recombinant p53-Y220C (3 μM) was incubated with (a) 0.2% DMSO (b) 30 μM LI (c) 30 μM LH (d) 75 μM LI and (e) 75 μM LH for 2 hours at room temperature. • indicates a 564 Da mass increase representing an interaction between mutant p53 and LI, * indicates an 1128 Da mass increase representing an interaction between mutant p53 and 2LI. | |
Zinc binding affinities for LI and LH
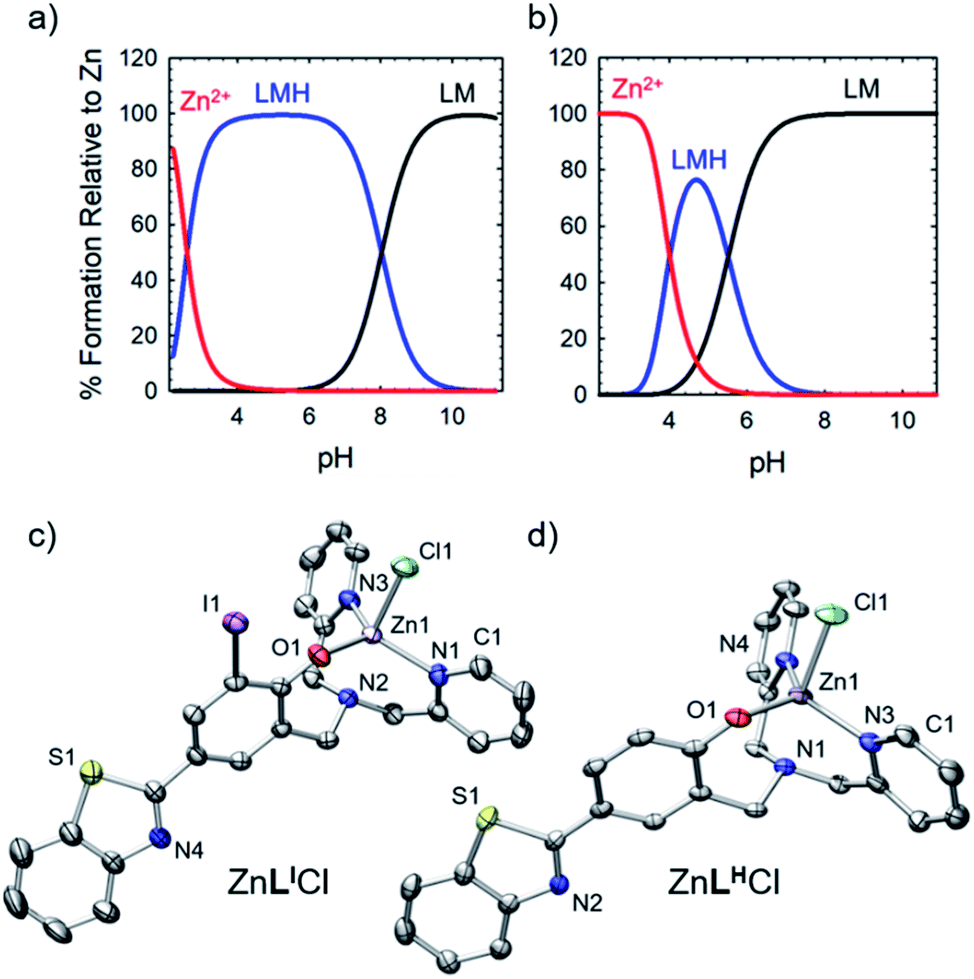
To further explore the bifunctional nature of LI and LH, we investigated their ability to serve as zinc metallochaperones. Previously reported models for Zn-binding in p53 describe two possible ligation sites, the native binding site (Kd1) estimated to bind on the order of 10−12 M, and non-native (Kd2) sites with an estimated affinity on the order of 10−6 M.11,14b,c,15a,28 Metallochaperones designed to rescue zinc-binding in p53 mutants should therefore have Zn2+ affinities that are in between that of Kd1 and Kd2. While the exact value of Kd1 for p53-Y220C is unknown, we have previously shown that Zn metallochaperones on the order of 10−12 M have appropriate affinities to increase intracellular levels of zinc in cells expressing this mutant.13a Spectrophotometric (UV-visible) pH titrations carried out to characterize ligand speciation (see Table S1† for pKa values) and Zn-affinity for LI and LH demonstrate that at biological pH (7.4), the speciation diagrams describe a model with only 1:1 ligand to metal species present (Fig. 5a and b). The concentration of free Zn2+ present in solution at a given pH, referred to as pM (pZn = −log[Znunchelated]), is a direct estimate of the metal–ligand affinity when all species in solution are considered.29 Calculated pM values for LI and LH (Table S2†) are very similar (8.4 and 8.2) and are consistent with previously published reports containing similar ligands.13a,17c These values afford approximate dissociation constants (Kd) in the nanomolar range, an affinity appropriate for functioning as Zn metallochaperones for p53-Y220C. Complete models and simulations are detailed in the ESI (Fig. S8–S15†). Zn complexes of LI and LH were also isolated and characterized using NMR and X-ray crystallography (Fig. 5c and d) and are in accord with the 1:1 binding of LI and LH to Zn2+ modelled above for the variable pH titrations. Complete crystallographic information is provided in Table S3.†
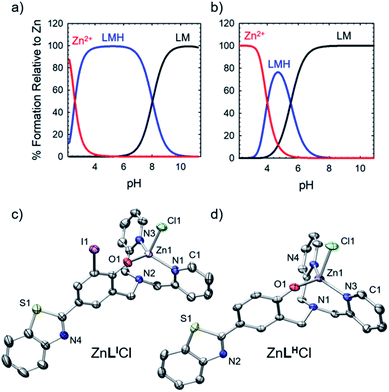 |
| | Fig. 5 (a) Simulated species distribution plot of Zn2+ + LI. (b) Simulated species distribution plot of Zn2+ + LH. Speciation diagrams were made using HySS2009. (c) ORTEP of ZnLICl (50% probability) using POV-Ray, excluding hydrogen atoms and solvent. Selected interatomic distances [Å]: Zn(1)–N(1): 2.271(3), Zn(1)–N(3): 2.090(2), Zn(1)–N(4): 2.088(3), Zn(1)–O(1): 1.954(2), Zn(1)–Cl(1): 2.2978(8). (d) ORTEP of ZnLHCl (50% probability) using POV-Ray, excluding hydrogen atoms and solvent. Selected interatomic distances [Å]: Zn(1)–N(1): 2.251(5), Zn(1)–N(3): 2.067(4), Zn(1)–N(4): 2.090(5), Zn(1)–O(1): 1.964(4), Zn(1)–Cl(1): 2.341(2). | |
Metallochaperone ability of LI and LH
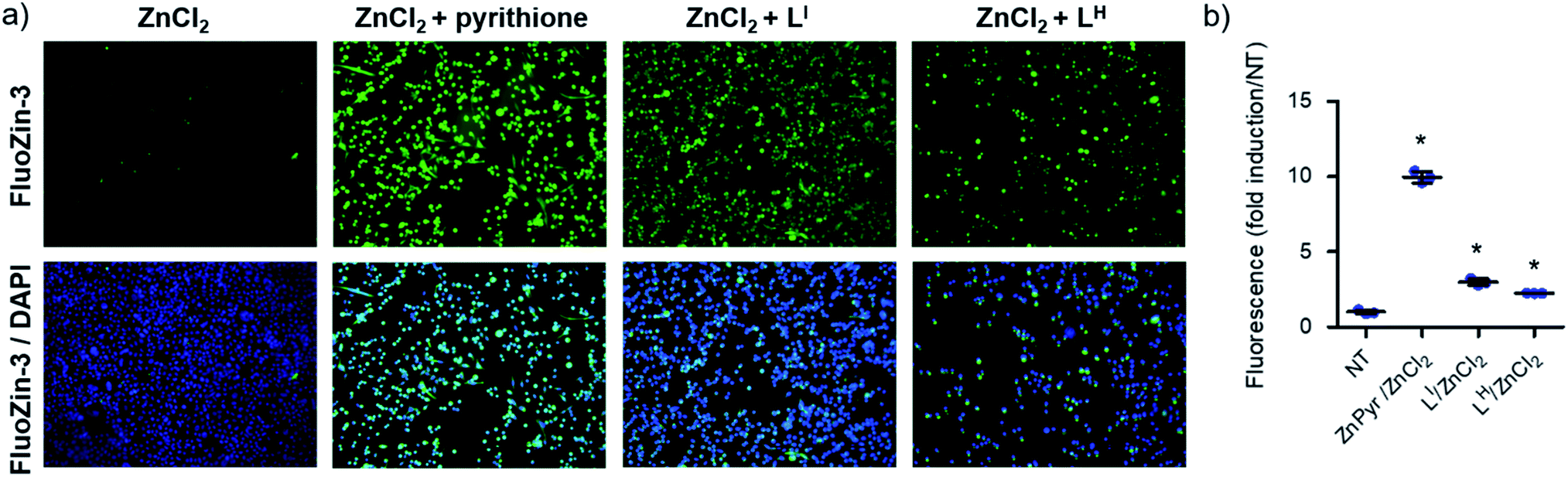
To determine whether LI and LH could serve as Zn metallochaperones, we analyzed the changes in levels of intracellular Zn2+ in NUGC3 cells. The cells were incubated with the fluorescent Zn2+ sensitive probe FluoZin-3 (1 μM),30 50 μM of ZnCl2, and either DMSO for non-treated (NT) controls or 50 μM of LI and LH. Pyrithione was used as a positive control for intracellular uptake of Zn2+.31 Subsequent fluorescence imaging revealed that treatment with ZnCl2 alone or LI/LH alone does not result in significant increase in intracellular Zn2+ (Fig. S16†). However, addition of ZnCl2 in combination with LI/LH significantly increased intracellular Zn2+ levels as indicated by increased fluorescence (Fig. 6), thus demonstrating that both ligands and extracellular Zn2+ are required. However, due to the similar Zn Kd values of FluoZin-3 and LI/LH (Zn2+Kd = ∼15 nM for FluoZin-3,30 4 nM for LI and 6.3 nM for LH) Zn-binding to the fluorophore in this experiment is likely restricted, and thus total Zn uptake is underestimated. Quantification of fluorescent signals indicates that LI and LH increase intracellular Zn2+ by more than 3 and 2-fold respectively compared to the non-treated control. These results demonstrate the potential of LI and LH to serve as Zn-metallochaperones for mutant p53 by increasing intracellular Zn2+ concentrations.
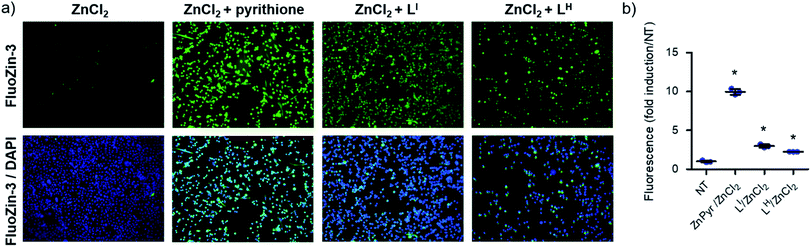 |
| | Fig. 6 Treatment of NUGC3 (p53-Y220C) with LI and LH increases intracellular Zn2+. (a) Imaging of intracellular Zn2+ levels in complete serum-free media. NUGC3 cells were incubated with 1 μM FluoZin-3 for 20 minutes at 37 °C, followed by incubation with indicated treatment (ZnCl2 = 50 μM, LI = LH = 50 μM, 50 μM PYR) for 2 hours. Cells were imaged using a fluorescence microscope and fluorescence-quantified using ImageJ. (b) Fluorescence intensity of FluoZin-3 at 488 nm demonstrating relative Zn2+ levels. Black line indicates mean values, while black error bars demonstrate the 95% confidence interval. Statistical differences were analyzed using 1-way ANOVA with multiple comparisons (Dunnett test). * indicates statistical differences from control with p < 0.001. | |
Restoration of p53 function
To determine if the ligands could restore wild-type function to mutant p53, we first investigated whether they reduced mutant p53 levels via immunoprecipitation with the mutant-specific PAb240 anti-p53 antibody. Using native NUGC3 lysate in the absence or presence of increasing concentrations of LI and LH, we immunoprecipitated mutant p53. As shown in representative experiments, overnight treatment with LI and LH were able to reduce levels of mutant p53 by 54% and 47% respectively, indicating that upon treatment with our bifunctional ligands, p53 conformation is altered wherein the antigen recognized by PAb240 is now buried within the protein's core (Fig. S17†).
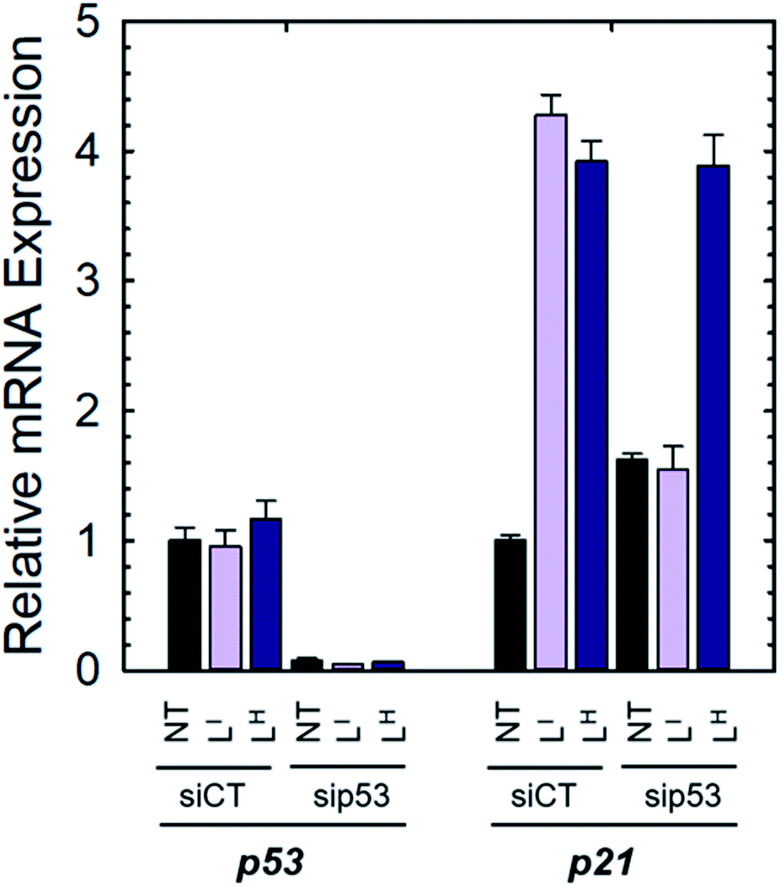
We next investigated whether treatment with LI or LH could increase the expression levels of several p53 target genes that are involved in either cell cycle arrest (P21) or apoptosis (NOXA, PUMA).6a,32 Expression levels were also monitored in p53-silenced NUGC3 cells to probe the extent of p53 dependence. NUGC3 cells were treated for 6 hours with 25 μM of LI or LH and the expression level of p53 and three of its representative target genes (P21, NOXA, PUMA) were measured by RT-PCR. Strikingly, treatment of NUGC3 with LI and LH in the presence of non-targeting siRNA (SiControl) resulted in a 4.3- and 3.9-fold increase in P21, a common indicator of p53 function (Fig. 7).33 A 2.0- and 1.5-fold increase in NOXA was also observed upon treatment with LI and LH respectively (Fig. S18†). These results are obtained without alteration of p53 levels, suggesting restored p53 function in mutant p53 wherein cell cycle arrest and apoptotic pathways are activated. Silencing of p53 with siRNA resulted in a significant decrease in P21 levels in the presence of LI (Fig. 7). P21 expression was not completely abolished, which may be a result of a baseline level of the p53 protein, or the ability of mutant p53 to bind and inactivate p63 and p73, both of which regulate target genes P21 and NOXA.34 Thus, knock-out of mutant p53 could lead to increased activity of p63 and p73, causing an upregulation in P21/NOXA expression levels. Interestingly, the addition of sip53 to LH treatment resulted in no significant difference in P21 and NOXA expression levels from SiCtl conditions. These results can be rationalized when considering the aggregation experiments above, as LI interacts directly with mutant p53 and reduces aggregation whereas LH has no observable effect, suggesting that in contrast to LI, LH may act largely through a p53-independent mechanism.
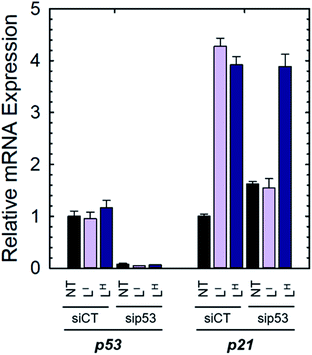 |
| | Fig. 7 NUGC3 cells were transfected for 6 hours with control siRNA (siCT) or siRNA directed against p53 (sip53) and then treated for 6 hours with 25 μM of indicated compounds. Total RNAs were extracted and RT-qPCR performed to measure the expression of TP53, P21, PUMA and NOXA. | |
In vitro cytotoxicity
The cytostatic and cytotoxic activity of LI and LH was evaluated against a panel of 60 cancer cell lines (NCI-60 screening program) containing a wide range of cancer types. Both ligands showed high activity across all 60 cell lines as demonstrated by their average growth inhibitory (GI50) concentrations (Table S4†). LI exhibited a 3-fold increase in average GI50 compared to both LH and cisplatin and a 6-fold increase in average GI50 compared to oxaliplatin. The corresponding zinc complexes ZnLICl and ZnLHCl were also tested but exhibited lower biological activity at the initial test concentration of 10 μM and were not subjected to further studies. A heat map summarizing the patterns of in vitro cytostatic activity from low activity (blue) to high activity (red) is shown in Fig. S19.† The in vitro cytotoxicity of ligands LI and LH was also tested in the gastric cancer cell line NUGC3. Gastric cancer is the second leading cause of cancer-related deaths worldwide and contains a wide range of p53 mutations that are present in up to 77% of gastric carcinomas. Standard treatment protocols for gastric cancer include oxaliplatin.35 Using standard MTT protocols,36 we determined that LI exhibited a significant 11-fold increase in cytotoxicity compared to oxaliplatin and a 5-fold increase in cytotoxicity over LH (Table S5†).
Apoptotic effects of LI and LH in human gastric cancer cell lines
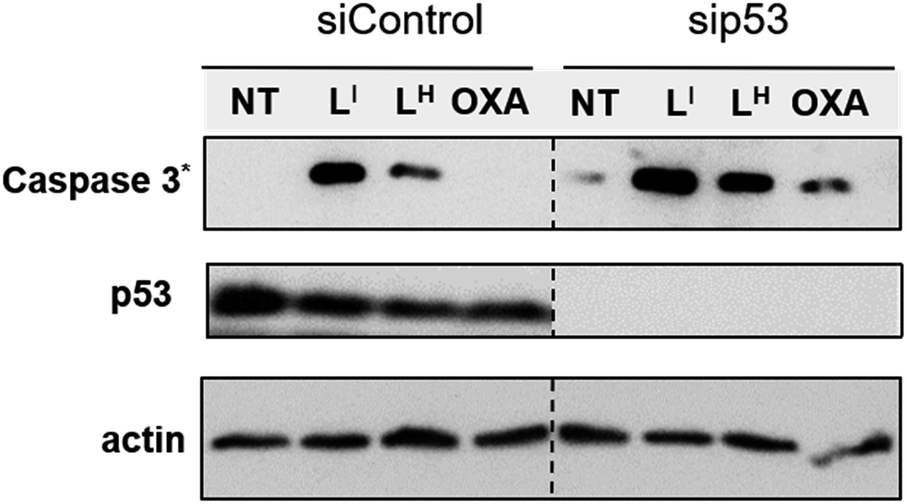
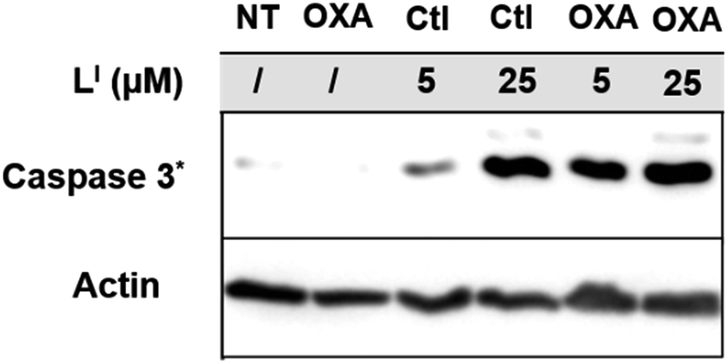
To investigate the molecular basis for the observed cytotoxicity, we examined whether LI and LH could induce apoptosis in gastric cancer cell lines containing wild-type (AGS) and mutant p53 (NUGC3). AGS cells were treated with IC50 and IC75 concentrations of ligands LI and LH and oxaliplatin for 48 hours and then cleavage of caspase-3 and p53 protein levels were assessed via Western blot. Oxaliplatin exhibits a significant increase in p53 levels, however, very little caspase-3 is observed at selected concentrations (Fig. S20†). In the wild-type p53 cell line, neither LI or LH induced any changes in either p53 expression levels or cleavage of caspase 3, even at the IC75 concentration. In the NUGC3 cell line, cells were treated with 25 μM of oxaliplatin, LI, or LH. This concentration of LI resulted in reduction of mutant p53 aggregation in NUGC3 cells, and therefore we investigated whether this was coupled with an induction of apoptosis. Upon treatment of NUGC3 cells with 25 μM of indicated compound for 48 hours, western blot analysis revealed that LI resulted in a strong presence of cleaved-caspase-3 as indicated by the intense band in Fig. 8. Treatment with LH also resulted in cleavage of caspase-3, however to a significantly lesser extent than treatment with LI (see Fig. S21† for quantification). Oxaliplatin did not induce caspase-dependent apoptosis in NUGC3 cells at 25 μM. Interestingly, this induction of apoptosis by LI and LH in NUGC3 cells was not abolished by treatment with sip53, but rather an increase in cleaved caspase-3 is observed. While this could be an indication of off-target effects, the result is suggestive of a removal of toxic gain-of-function effects from mutant p53 aggregates.37 Furthermore, although faint, the presence of cleaved caspase-3 under sip53 NT conditions suggests removal of toxic gain-of-function effects rather than off-target mechanisms. Gain of function effects for mutant p53 have been well characterized, and structurally destabilized mutants have been documented to co-aggregate with the homologous protein p73 thereby inactivating its function.34a,38 Thus, removal of mutant p53 in this case could result in p73-mediated apoptosis. Although this hypothesis is attractive and highly plausible, it is likely that additional mechanisms are also involved.
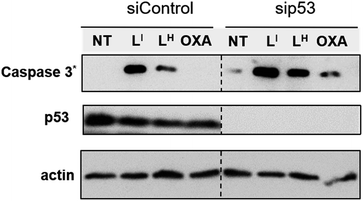 |
| | Fig. 8
LI results in increased activation of apoptosis over LH in mutant p53 cells. NUGC3 cells were treated with 25 μM of indicated compound for 24 hours. Proteins were extracted, and 40 μg was separated on SDS PAGE. Cleaved caspase-3 (Caspase 3*), p53, and actin were detected by western blot. | |
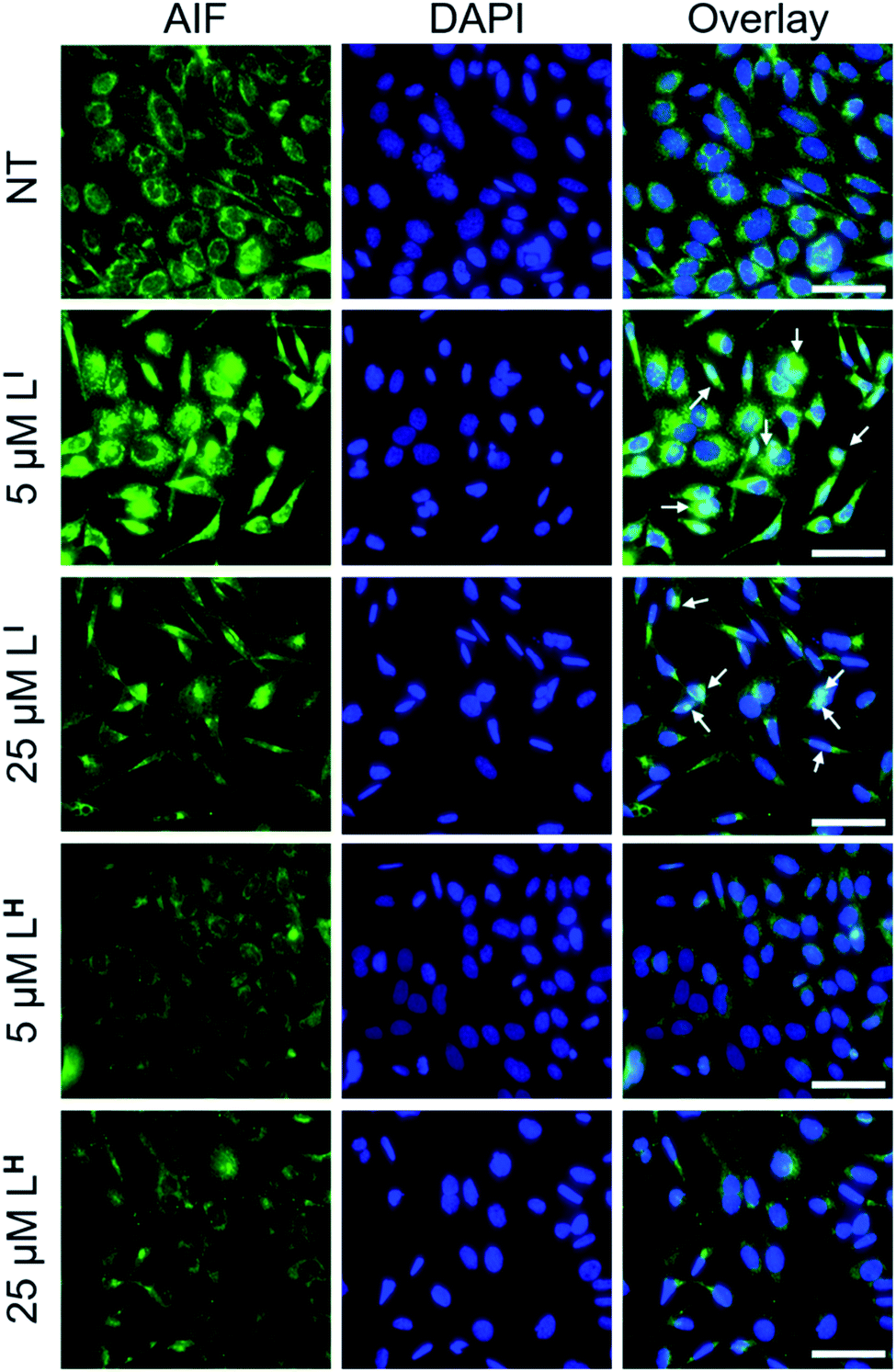
To explore possible caspase-independent cell death pathways activated by LI and LH, we investigated whether they participated in translocation of the apoptosis inducing factor (AIF). During apoptosis, AIF moves through the outer mitochondrial membrane into the cytosol and participates in chromatin condensation and DNA-fragmentation upon translocation into the nucleus.39 Using immunofluorescence, we monitored AIF localization upon treatment of LI and LH in NUGC3 cells. In NT conditions, AIF punctate staining is observed that spares the nucleus, exhibiting mitochondrial localization (Fig. 9). Upon treatment with 5 and 25 μM LI for 24 hours, AIF expression is largely upregulated, and diffuse nuclear localization is observed, indicative of apoptotic cells. In contrast, very little change in AIF expression levels or localization is observed upon treatment with LH (see Fig. S22† for quantification of nuclear localization), again highlighting the increased pro-cell death activity of LI in mutant p53 cell lines. Importantly, studies have shown that in mutant p53 cell lines, caspase-independent cell death is compromised, and that functional p53 could regulate AIF expression and result in activation of cell death.40 This provides further indication that by reducing mutant p53 aggregation, LI can restore protein function and activate otherwise compromised cell death pathways.
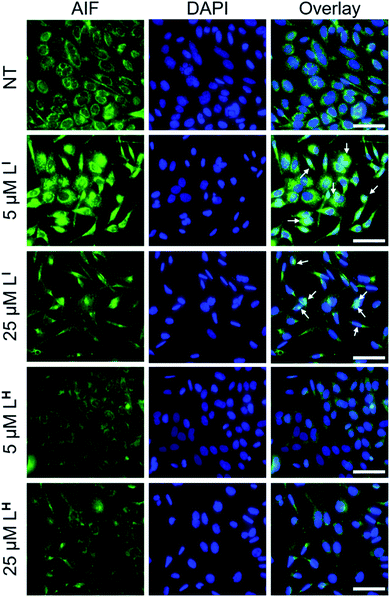 |
| | Fig. 9 Redistribution of AIF upon treatment with LI indicates activation of caspase-independent cell death. NUGC3 cells were treated with 5 and 25 μM LI/LH or 0.1% DMSO (NT) for 24 hours followed by labelling with anti-AIF antibody (1:1000). Nuclei were stained with DAPI, followed by imaging using a Nikon ApoTome microscope. Columns from left to right include: AIF, DAPI, and coimmunofluorescence of AIF and DAPI. White arrows representative of overlap between AIF and DAPI. The scale bar represents 50 μm. | |
Co-treatment with platinum chemotherapeutics
Platinum based chemotherapy is among the first line standard of care for gastric cancer patients, however, cancers harboring p53 mutations often exhibit decreased sensitivity and increased resistance to platinum agents.41 This is due to the fact that the mechanism of these agents includes induction of DNA damage, which leads to downstream activation of the p53 pathway, providing that functional p53 is present.9,42 With up to 77% of gastric cancers exhibiting p53 mutations,35 co-administration of agents that first restore p53 function and thereby increase the activity of platinum agents are highly desirable. To this end, we tested whether pre-incubation of NUGC3 cells with LI could result in an activation of apoptosis with oxaliplatin. Incubation of NUGC3 cells for 48 hours with 25 μM oxaliplatin or 5 μM LI separately results in no to very little cleavage of caspase 3 (Fig. 10). However, pre-incubation with 5 μM LI for two hours followed by subsequent incubation with oxaliplatin for 48 hours results in a significant increase in cleaved caspase 3 (see Fig. S23† for quantification). Similarly, pre-incubation with LI followed by additional incubation with oxaliplatin for 24 hours further induced PUMA expression and the ratio of BAX (proapoptotic) and BCl2 (antiapoptotic) expression compared to individual treatments (Fig. S24†). The ratio of BAX/BCl2 has been described as an indicator of apoptosis sensitivity as an increased ratio results in a higher response to apoptotic signals.43 The results for BAX/BCl2 highlights a possible synergistic mechanism between LI and oxaliplatin and provides a potential application for LI in co-administration with platinum agents for tumours harboring malfunctioning p53.
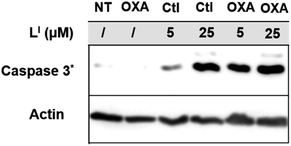 |
| | Fig. 10 Cotreatment with LI and oxaliplatin results in increased activation of apoptosis over treatment with LI or oxaliplatin alone. NUGC3 cells were treated with 5 or 25 μM of indicated compound for 48 hours. In the case of cotreatment, cells were incubated with LI for 2 hours followed by a total 48 hour incubation with 25 μM oxaliplatin. Proteins were extracted, and 40 μg were separated on SDS PAGE. Cleaved caspase-3 (Caspase 3*) and actin were detected by western blot. | |
Cytotoxicity in non-cancerous organoids
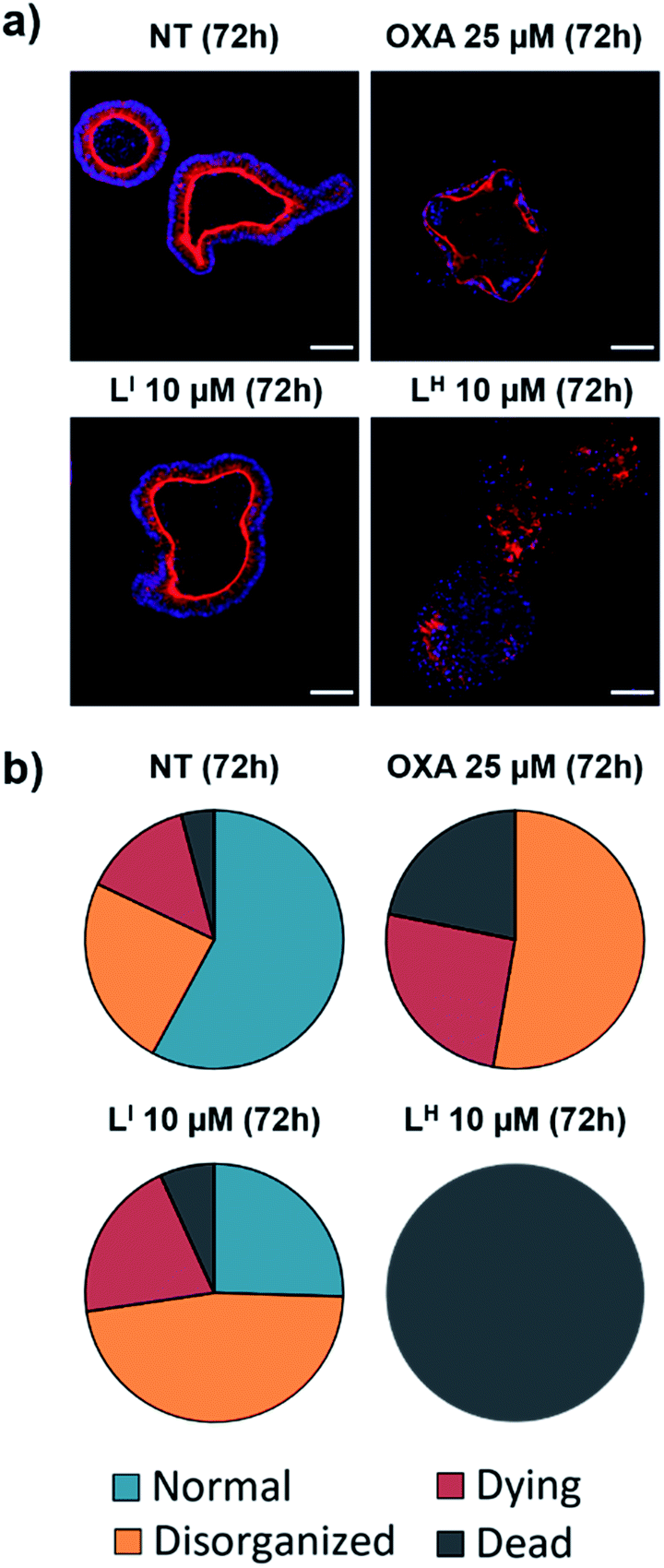
Despite new anti-cancer drugs representing one of the largest areas in pharmaceutical development, the onset of adverse side effects from chemotherapeutics still presents a major clinical hurdle.44 Promisingly, pharmacological restoration of p53 function has been associated with increased protection of normal cells from cytotoxicity due to the selective nature of targeting mutant p53-bearing tumours.45 To this end, we tested the cytotoxicity of LI and LH in non-cancerous cells using mouse small intestine organoids. Intestinal organoids are three-dimensional multicellular structures that comprise of crypts and villi to reproduce an intestinal organization. They are an important aspect in drug discovery due to their increased similarities to physiological models over their two-dimensional counterparts – the three-dimensional organization with budding is an indication of a healthy and viable organoid.46 To analyse the cytotoxicity of our compounds on normal organoids, we performed a long-term survival assay by monitoring organoid viability over a 23 day period. To elucidate the impact of treatment on the organoid organization, we performed an immunofluorescence assay and monitored organoid shape upon treatment with LI and LH. Organoids were classified as normal, disorganized, dying, or dead based on the images obtained (see Fig. S25 and Table S5† for description and quantification of each classification system). Interestingly, treatment with LI retained mostly viable organoids after a 72 hour period resembling normal and disorganized structures similar to NT control (Fig. 11). In contrast, treatment with oxaliplatin resulted in about 50% of organoids either dying or dead, and strikingly, after 72 hours all organoids treated with LH were dead. To further probe their differing cytotoxicity in our non-cancerous organoid model and investigate the mechanism by which LH imparts cytotoxicity, we incubated cells treated with oxaliplatin, LI, and LH with an antibody recognizing cleaved caspase 3. In agreement with the results obtained above, treatment with LI resembled that of non-treated conditions with no significant cleaved caspase 3 detected (Fig. S26†). In contrast, significant cleavage of caspase 3 was detected upon treatment with oxaliplatin and LH, indicating that both treatments induce apoptosis in non-cancerous organoids.
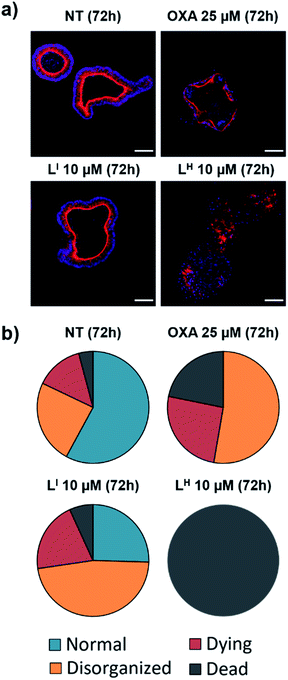 |
| | Fig. 11 Treatment of non-cancerous intestinal organoids results in increased survival upon treatment with LI compared to LH. (a) Intestinal organoids were treated with indicated compound and monitored for 72 hours. F-actin (red) and nuclei (blue) were stained using phalloidin and DAPI respectively at recommended concentrations and imaged using a Zeiss ApoTome microscope. (b) Characterization and quantification of organoids as either normal, disorganized, dying or dead upon indicated treatment based on images obtained in part (a). ca. 150 organoids were counted and analyzed for each condition. The scale bar represents 50 μm. | |
Importantly, even over a period of 23 days, organoid treatment with LI (2.5 and 5 μM) has a similar survival profile to NT organoids, resulting in substantial viability after 23 days (Fig. S27†). In contrast, treatment with LH resulted in complete cell death after 5 days (5 μM). Treatment with LI also results in organoids with more budding than those treated with LH. Finally, given that chemotherapeutic regiments involve treating patients at the maximum tolerated dose (MTD), we sought to determine the maximum dose of LI tolerated in normal C57BL/6 mice. Promisingly, no decrease in weight is observed up to ca. 25 μmol kg−1 (13 mg kg−1) LI treatment (Fig. S28†), a concentration which corresponds to that wherein p53 aggregation is inhibited and cell death pathways are activated in our cancer cell experiments.
Overall, these results highlight that by iodination of our bifunctional scaffold, we have increased cytotoxicity in cancerous cells lines yet have significantly decreased cytotoxicity in a non-cancerous organoid model. Taken together, these results highlight LI as a suitable candidate for in vivo testing in xenograft models.
Summary
Restoration of p53 function holds significant promise in the search for effective chemotherapeutics as over 50% of cancer diagnoses are attributed to mutant p53. Given that a large proportion of these mutations result in accelerated protein aggregation and contribute to loss of function, molecules aimed to inhibit protein aggregation are of particular interest. Importantly, the loss of zinc within the protein's DNA-binding core further increases the aggregation process via nucleation with zinc-bound p53C. In this work, we have designed bifunctional ligands (LI and LH) aimed to restore p53 function by modulating mutant p53 aggregation and incorporating zinc-binding fragments for metallochaperone activity. We demonstrate that only the iodinated framework LI was effective at inhibiting p53 aggregation, as demonstrated in recombinant systems via light scattering, TEM, and gel electrophoresis, and in cellular systems using immunofluorescence. These results prompted an investigation into the binding capabilities of these ligands with mutant p53. Using native MS, we showed that only LI interacted with mutant p53. This result can explain the differing abilities of LI and LH to modulate mutant p53 aggregation and leads us to hypothesize that the iodine in LI is contributing to favorable interactions with the hydrophobic aggregation-prone segment. After extensive characterization of the zinc-binding capability of LI and LH, we show that our bifunctional ligands significantly increase intracellular levels of zinc in cells, thus demonstrating their potential for metallochaperone function. We further demonstrate the increased cytotoxicity of LI in cancer cells compared to LH and demonstrate that by restoring protein function, LI contributes to activation of apoptotic pathways. Remarkably, LI is well tolerated in non-cancerous organoids, while LH was highly toxic.
Overall, these results demonstrate that by iodination of our bifunctional framework, we have substantially changed the biological properties of our molecules. LI can modulate mutant p53 aggregation, activate otherwise inaccessible apoptotic pathways, and is well tolerated in non-cancerous 3D models and normal mice. The utility of LI in restoring p53-dependent pathways presents an interesting opportunity for co-administration with clinically approved platinum agents. Cancers with mutant p53 status have been associated with increased resistance, and thus restoring p53 function to increase the effectiveness of platinum agents is an interesting opportunity that we plan to further investigate.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the National Cancer Institute for NCI-60 screening, and gratefully acknowledge the assistance of David Weber in visualization of the NCI-60 heat map. This work was supported by a Natural Sciences and Engineering Research Council (NSERC) Discovery Grant and a Michael Smith Career Investigator Award (T. S.). J. J. M. thanks NSERC and SFU for postgraduate fellowships. R. M. C. acknowledges NSERC for a postgraduate fellowship. Prof. Jeffrey Warren is thanked for helpful discussions. Work at the Université de Strasbourg is supported by the CNRS, Inserm, and ARC.
References
-
(a) P. Brundin, R. Melki and R. Kopito, Prion-like transmission of protein aggregates in neurodegenerative diseases, Nat. Rev. Mol. Cell Biol., 2010, 11(4), 301–307 CrossRef CAS;
(b) M. D. Geschwind, Prion Diseases, Continuum, 2015, 21(6), 1612–1638 Search PubMed;
(c) D. S. Eisenberg and M. R. Sawaya, Structural Studies of Amyloid Proteins at the Molecular Level, Annu. Rev. Biochem., 2017, 86(1), 69–95 CrossRef CAS;
(d) T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, The amyloid state and its association with protein misfolding diseases, Nat. Rev. Mol. Cell Biol., 2014, 15, 384 CrossRef CAS.
-
(a) F. Chiti and C. M. Dobson, Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade, Annu. Rev. Biochem., 2017, 86(1), 27–68 CrossRef CAS PubMed;
(b) A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Misfolded proteins in Alzheimer's disease and type II diabetes, Chem. Soc. Rev., 2012, 41(2), 608–621 RSC;
(c) D. Eisenberg and M. Jucker, The amyloid state of proteins in human diseases, Cell, 2012, 148(6), 1188–1203 CrossRef CAS PubMed;
(d) Y.-R. Chen, Recombinant Tdp-43 Forms Toxic and Stable Amyloid-like Oligomers, Alzheimer's Dementia, 2010, 6(4), e44 CrossRef.
-
(a) M. Jucker and L. C. Walker, Self-propagation of pathogenic protein aggregates in neurodegenerative diseases, Nature, 2013, 501(7465), 45–51 CrossRef CAS;
(b) R. F. Sowade and T. R. Jahn, Seed-induced acceleration of amyloid-β mediated neurotoxicity in vivo, Nat. Commun., 2017, 8(1), 512 CrossRef PubMed.
-
(a) J. Xu, J. Reumers, J. R. Couceiro, F. De Smet, R. Gallardo, S. Rudyak, A. Cornelis, J. Rozenski, A. Zwolinska, J.-C. Marine, D. Lambrechts, Y.-A. Suh, F. Rousseau and J. Schymkowitz, Gain of function of mutant p53 by coaggregation with multiple tumor suppressors, Nat. Chem. Biol., 2011, 7, 285 CrossRef CAS PubMed;
(b) G. Hao, Y. Xin, Z. Yudan, B. P. Robert, L. Xinran, L. Yang and H. Kun, Amyloidogenicity of p53: A Hidden Link Between Protein Misfolding and Cancer, Curr. Protein Pept. Sci., 2015, 16(2), 135–146 CrossRef;
(c) L. P. Rangel, D. C. F. Costa, T. C. R. G. Vieira and J. L. Silva, The aggregation of mutant p53 produces prion-like properties in cancer, Prion, 2014, 8(1), 75–84 CrossRef CAS PubMed;
(d) D. Ishimaru, L. R. Andrade, L. S. P. Teixeira, P. A. Quesado, L. M. Maiolino, P. M. Lopez, Y. Cordeiro, L. T. Costa, W. M. Heckl, G. Weissmüller, D. Foguel and J. L. Silva, Fibrillar Aggregates of the Tumor Suppressor p53 Core Domain, Biochemistry, 2003, 42(30), 9022–9027 CrossRef CAS PubMed;
(e) A. P. D. Ano Bom, L. P. Rangel, D. C. F. Costa, G. A. P. de Oliveira, D. Sanches, C. A. Braga, L. M. Gava, C. H. I. Ramos, A. O. T. Cepeda, A. C. Stumbo, C. V. De Moura Gallo, Y. Cordeiro and J. L. Silva, Mutant p53 Aggregates into Prion-like Amyloid Oligomers and Fibrils: Implications for Cancer, J. Biol. Chem., 2012, 287(33), 28152–28162 CrossRef CAS PubMed.
-
(a) C. A. Lasagna-Reeves, A. L. Clos, D. Castillo-Carranza, U. Sengupta, M. Guerrero-Muñoz, B. Kelly, R. Wagner and R. Kayed, Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity, Biochem. Biophys. Res. Commun., 2013, 430(3), 963–968 CrossRef CAS;
(b) J. L. Silva, C. V. D. M. Gallo, D. C. F. Costa and L. P. Rangel, Prion-like aggregation of mutant p53 in cancer, Trends Biochem. Sci., 2014, 39(6), 260–267 CrossRef CAS PubMed;
(c) S. Kehrloesser, C. Osterburg, M. Tuppi, B. Schäfer, K. H. Vousden and V. Dötsch, Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family, Cell Death Differ., 2016, 23, 1952 CrossRef CAS PubMed;
(d) D. C. F. Costa, G. A. P. de Oliveira, E. A. Cino, I. N. Soares, L. P. Rangel and J. L. Silva, Aggregation and Prion-Like Properties of Misfolded Tumor Suppressors: Is Cancer a Prion Disease?, Cold Spring Harbor Perspect. Biol., 2016, 8(10), a023614 CrossRef PubMed.
-
(a) A. C. Joerger and A. R. Fersht, The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches, Annu. Rev. Biochem., 2016, 85(1), 375–404 CrossRef CAS PubMed;
(b) P. A. Lazo, Reverting p53 activation after recovery of cellular stress to resume with cell cycle progression, Cell. Signalling, 2017, 33, 49–58 CrossRef CAS PubMed;
(c) D. P. Lane, p53, guardian of the genome, Nature, 1992, 358(6381), 15–16 CrossRef CAS PubMed;
(d) T. Riley, E. Sontag, P. Chen and A. Levine, Transcriptional control of human p53-regulated genes, Nat. Rev. Mol. Cell Biol., 2008, 9(5), 402–412 CrossRef CAS PubMed.
- S. N. Loh, The missing Zinc: p53 misfolding and cancer, Metallomics, 2010, 2, 442–449 RSC.
-
(a) G. Wang and A. R. Fersht, First-order rate-determining aggregation mechanism of p53 and its implications, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(34), 13590–13595 CrossRef CAS PubMed;
(b) B. Vogelstein, D. Lane and A. J. Levine, Surfing the p53 network, Nature, 2000, 408(6810), 307–310 CrossRef CAS PubMed;
(c) P. A. J. Muller and K. H. Vousden, P53 mutations in cancer, Nat. Cell Biol., 2013, 15, 2–8 CrossRef CAS PubMed;
(d) M. Hollstein, D. Sidransky, B. Vogelstein and C. Harris, p53 mutations in human cancers, Science, 1991, 253(5015), 49–53 CrossRef CAS PubMed.
- A. Soragni, D. M. Janzen, L. M. Johnson, A. G. Lindgren, A. Thai-Quynh Nguyen, E. Tiourin, A. B. Soriaga, J. Lu, L. Jiang, K. F. Faull, M. Pellegrini, S. Memarzadeh and D. S. Eisenberg, A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas, Cancer Cell, 2016, 29(1), 90–103 CrossRef CAS PubMed.
-
(a) R. Wilcken, G. Wang, F. M. Boeckler and A. R. Fersht, Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(34), 13584–13589 CrossRef CAS PubMed;
(b) E. A. Cino, I. N. Soares, M. M. Pedrote, G. A. P. de Oliveira and J. L. Silva, Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds, Sci. Rep., 2016, 6, 32535 CrossRef CAS PubMed.
- J. S. Butler and S. N. Loh, Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain, Biochemistry, 2003, 42(8), 2396–2403 CrossRef CAS PubMed.
- G. D'Orazi and D. Givol, p53 reactivation: The link to zinc, Cell Cycle, 2012, 11(14), 2581–2582 CrossRef PubMed.
-
(a) J. J. Miller, C. Orvain, S. Jozi, R. M. Clarke, J. R. Smith, A. Blanchet, C. Gaiddon, J. J. Warren and T. Storr, Multifunctional Compounds for Activation of the p53-Y220C Mutant in Cancer, Chem.–Eur. J., 2018, 24(67), 17734–17742 CrossRef CAS PubMed;
(b) R. Wilcken, X. Liu, M. O. Zimmermann, T. J. Rutherford, A. R. Fersht, A. C. Joerger and F. M. Boeckler, Halogen-Enriched Fragment Libraries as Leads for Drug Rescue of Mutant p53, J. Am. Chem. Soc., 2012, 134(15), 6810–6818 CrossRef CAS PubMed;
(c) M. F. Lavin and N. Gueven, The complexity of p53 stabilization and activation, Cell Death Differ., 2006, 13, 941 CrossRef CAS PubMed;
(d) S. Chuikov, J. K. Kurash, J. R. Wilson, B. Xiao, N. Justin, G. S. Ivanov, K. McKinney, P. Tempst, C. Prives, S. J. Gamblin, N. A. Barlev and D. Reinberg, Regulation of p53 activity through lysine methylation, Nature, 2004, 432(7015), 353–360 CrossRef CAS PubMed;
(e) A. C. Joerger, M. R. Bauer, R. Wilcken, M. G. J. Baud, H. Harbrecht, T. E. Exner, F. M. Boeckler, J. Spencer and A. R. Fersht, Exploiting Transient Protein States for the Design of Small-Molecule Stabilizers of Mutant p53, Structure, 2015, 23(12), 2246–2255 CrossRef CAS PubMed;
(f) R. G. Doveston, A. Kuusk, S. A. Andrei, S. Leysen, Q. Cao, M. P. Castaldi, A. Hendricks, L. Brunsveld, H. Chen, H. Boyd and C. Ottmann, Small-molecule stabilization of the p53 – 14-3-3 protein–protein interaction, FEBS Lett., 2017, 591(16), 2449–2457 CrossRef CAS PubMed;
(g) J. D. Amaral, D. Silva, C. M. P. Rodrigues, S. Solá and M. M. M. Santos, A Novel Small Molecule p53 Stabilizer for Brain Cell Differentiation, Front. Chem., 2019, 7(15), a00015 Search PubMed;
(h) S. Hietanen, S. Lain, E. Krausz, C. Blattner and D. P. Lane, Activation of p53 in cervical carcinoma cells by small molecules, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(15), 8501–8506 CrossRef CAS PubMed;
(i) A. C. Joerger, I. S. Chuckowree, J. Amin, J. Spencer, R. Wilcken, X. Liu and A. R. Fersht, Small molecule induced reactivation of mutant p53 in cancer cells, Nucleic Acids Res., 2013, 41(12), 6034–6044 CrossRef;
(j) M. G. J. Baud, M. R. Bauer, L. Verduci, F. A. Dingler, K. J. Patel, D. Horil Roy, A. C. Joerger and A. R. Fersht, Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines, Eur. J. Med. Chem., 2018, 152, 101–114 CrossRef CAS.
-
(a) A. R. Blanden, X. Yu, A. J. Wolfe, J. A. Gilleran, D. J. Augeri, R. S. O'Dell, E. C. Olson, S. D. Kimball, T. J. Emge, L. Movileanu, D. R. Carpizo and S. N. Loh, Synthetic Metallochaperone ZMC1 Rescues Mutant p53 Conformation by Transporting Zinc into Cells as an Ionophore, Mol. Pharmacol., 2015, 87(5), 825–831 CrossRef CAS;
(b) A. R. Blanden, X. Yu, S. N. Loh, A. J. Levine and D. R. Carpizo, Reactivating mutant p53 using small molecules as zinc metallochaperones: awakening a sleeping giant in cancer, Drug Discovery Today, 2015, 20(11), 1391–1397 CrossRef CAS PubMed;
(c) X. Yu, A. R. Blanden, S. Narayanan, L. Jayakumar, D. Lubin, D. Augeri, S. D. Kimball, S. N. Loh and D. R. Carpizo, Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism, OncoTargets Ther., 2014, 5, 8879–8892 Search PubMed;
(d) S. Zaman, X. Yu, A. F. Bencivenga, A. R. Blanden, Y. Liu, T. Withers, B. Na, A. J. Blayney, J. Gilleran, D. A. Boothman, S. N. Loh, S. D. Kimball and D. R. Carpizo, Combinatorial therapy of zinc metallochaperones with mutant p53 reactivation and diminished copper binding, Mol. Cancer Ther., 2019, 8(8), 1355–1365 CrossRef PubMed.
-
(a) X. Yu, A. Blanden, A. T. Tsang, S. Zaman, Y. Liu, J. Gilleran, A. F. Bencivenga, S. D. Kimball, S. N. Loh and D. R. Carpizo, Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53, Mol. Pharmacol., 2017, 91(6), 567–575 CrossRef CAS PubMed;
(b) M. Cirone, A. Garufi, L. Renzo, M. Granato, A. Faggioni and G. D'Orazi, Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells, OncoImmunology, 2013, 2, e26198 CrossRef PubMed;
(c) A. Garufi, V. D'Orazi, A. Crispini and G. D'Orazi, Zn(II)-curc targets p53 in thyroid cancer cells, Int. J. Oncol., 2015, 47(4), 1241–1248 CrossRef CAS PubMed;
(d) A. Garufi, D. Pucci, V. D'Orazi, M. Cirone, G. Bossi, M. L. Avantaggiati and G. D'Orazi, Degradation of mutant p53H175 protein by Zn(II) through autophagy, Cell Death Dis., 2014, 5, e1271 CrossRef CAS PubMed.
-
(a) C. M. Weekley and C. He, Developing drugs targeting transition metal homeostasis, Curr. Opin. Chem. Biol., 2017, 37(Suppl. C), 26–32 CrossRef CAS PubMed;
(b) A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, Zinc(II)–Thiosemicarbazone Complexes Are Localized to the Lysosomal Compartment Where They Transmetallate with Copper Ions to Induce Cytotoxicity, J. Med. Chem., 2016, 59(10), 4965–4984 CrossRef CAS PubMed;
(c) C. Mertens, E. A. Akam, C. Rehwald, B. Brüne, E. Tomat and M. Jung, Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression, PLoS One, 2016, 11(11), 1–19 CrossRef PubMed;
(d) K. C. Park, L. Fouani, P. J. Jansson, D. Wooi, S. Sahni, D. J. R. Lane, D. Palanimuthu, H. C. Lok, Z. Kovacevic, M. L. H. Huang, D. S. Kalinowski and D. R. Richardson, Copper and conquer: copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics, Metallomics, 2016, 8(9), 874–886 RSC;
(e) A. P. King, H. A. Gellineau, J.-E. Ahn, S. N. MacMillan and J. J. Wilson, Bis(thiosemicarbazone) Complexes of Cobalt(III). Synthesis, Characterization, and Anticancer Potential, Inorg. Chem., 2017, 56(11), 6609–6623 CrossRef CAS PubMed;
(f) E. A. Akam and E. Tomat, Targeting Iron in Colon Cancer via Glycoconjugation of Thiosemicarbazone Prochelators, Bioconjugate Chem., 2016, 27(8), 1807–1812 CrossRef CAS PubMed.
-
(a) M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. Wee Yong, S. Tsutsui, P. K. Stys and T. Storr, Multi-target-directed phenol–triazole ligands as therapeutic agents for Alzheimer's disease, Chem. Sci., 2017, 8(8), 5636–5643 RSC;
(b) A. Sharma, V. Pachauri and S. J. S. Flora, Advances in Multi-Functional Ligands and the Need for Metal-Related Pharmacology for the Management of Alzheimer Disease, Front. Pharmacol., 2018, 9, 1247 CrossRef CAS;
(c) A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42 Peptide, J. Am. Chem. Soc., 2012, 134(15), 6625–6636 CrossRef CAS PubMed;
(d) M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer's Disease, Parkinson's Disease,
and Amyotrophic Lateral Sclerosis in the Last Decade, Chem. Rev., 2019, 119(2), 1221–1322 CrossRef CAS.
-
(a) L. P. Rangel, G. D. S. Ferretti, C. L. Costa, S. M. M. V. Andrade, R. S. Carvalho, D. C. F. Costa and J. L. Silva, p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells, J. Biol. Chem., 2019, 3670–3682 CrossRef CAS;
(b) D. C. Ferraz da Costa, N. P. C. Campos, R. A. Santos, F. H. Guedes-da-Silva, M. M. D. C. Martins-Dinis, L. Zanphorlin, C. Ramos, L. P. Rangel and J. L. Silva, Resveratrol prevents p53 aggregation in vitro and in breast cancer cells, OncoTargets Ther., 2018, 9(49), 29112–29122 Search PubMed;
(c) M. Kanapathipillai, Treating p53 Mutant Aggregation-Associated Cancer, Cancers, 2018, 10(6), 154 CrossRef;
(d) C. Zhaolin and K. Mathumai, Inhibition of p53 Mutant Peptide Aggregation In Vitro by Cationic Osmolyte Acetylcholine Chloride, Protein Pept. Lett., 2017, 24(4), 353–357 CrossRef PubMed;
(e) Z. Chen, J. Chen, V. G. Keshamouni and M. Kanapathipillai, Polyarginine and its analogues inhibit p53 mutant aggregation and cancer cell proliferation in vitro, Biochem. Biophys. Res. Commun., 2017, 489(2), 130–134 CrossRef CAS PubMed;
(f) J. L. Silva, E. A. Cino, I. N. Soares, V. F. Ferreira and G. A. P. de Oliveira, Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer, Acc. Chem. Res., 2018, 51(1), 181–190 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, Designed Multiple Ligands. An Emerging Drug Discovery Paradigm, J. Med. Chem., 2005, 48(21), 6523–6543 CrossRef CAS.
-
(a) C. B. Levy, A. C. Stumbo, A. P. D. Ano Bom, E. A. Portari, Y. Carneiro, J. L. Silva and C. V. De Moura-Gallo, Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors, Int. J. Biochem. Cell Biol., 2011, 43(1), 60–64 CrossRef CAS;
(b) S. Ghosh, S. Salot, S. Sengupta, A. Navalkar, D. Ghosh, R. Jacob, S. Das, R. Kumar, N. N. Jha, S. Sahay, S. Mehra, G. M. Mohite, S. K. Ghosh, M. Kombrabail, G. Krishnamoorthy, P. Chaudhari and S. K. Maji, p53 amyloid formation leading to its loss of function: implications in cancer pathogenesis, Cell Death Differ., 2017, 24, 1784 CrossRef CAS.
-
(a) S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, Fluorescent Sensors for Zn2+ Based on a Fluorescein Platform: Synthesis, Properties and Intracellular Distribution, J. Am. Chem. Soc., 2001, 123(32), 7831–7841 CrossRef CAS;
(b) E. M. Nolan and S. J. Lippard, Small-molecule fluorescent sensors for investigating zinc metalloneurochemistry, Acc. Chem. Res., 2009, 42(1), 193–203 CrossRef CAS;
(c) C. J. Chang, E. M. Nolan, J. Jaworski, S. C. Burdette, M. Sheng and S. J. Lippard, Bright Fluorescent Chemosensor Platforms for Imaging Endogenous Pools of Neuronal Zinc, Chem. Biol., 2004, 11(2), 203–210 CrossRef CAS PubMed;
(d) Z. Xu, J. Yoon and D. R. Spring, Fluorescent chemosensors for Zn2+, Chem. Soc. Rev., 2010, 39(6), 1996–2006 RSC.
- D. Ami, A. Natalello, M. Lotti and S. M. Doglia, Why and how protein aggregation has to be studied in vivo, Microb. Cell Fact., 2013, 12, 17 CrossRef CAS PubMed.
-
(a) N. Lukashchuk and K. H. Vousden, Ubiquitination and degradation of mutant p53, Mol. Cell. Biol., 2007, 27(23), 8284–8295 CrossRef CAS PubMed;
(b) N. R. Rodrigues, A. Rowan, M. E. Smith, I. B. Kerr, W. F. Bodmer, J. V. Gannon and D. P. Lane, p53 mutations in colorectal cancer, Proc. Natl. Acad. Sci. U. S. A., 1990, 87(19), 7555–7559 CrossRef CAS PubMed;
(c) D. P. Lane and S. Benchimol, p53: oncogene or anti-oncogene?, Genes Dev., 1990, 4(1), 1–8 CrossRef CAS;
(d) M. Oren and V. Rotter, Mutant p53 gain-of-function in cancer, Cold Spring Harbor Perspect. Biol., 2010, 2(2), a001107 Search PubMed;
(e) V. Rotter, p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells, Proc. Natl. Acad. Sci. U. S. A., 1983, 80(9), 2613 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis, Science, 2003, 300(5618), 486–489 CrossRef CAS PubMed.
- G. Fine and S. M. Saeed, Thioflavin-T for Amyloid Detection, Am. J. Clin. Pathol., 1967, 47(5), 588–593 CrossRef.
- J. Xu, J. Reumers, J. R. Couceiro, F. De Smet, R. Gallardo, S. Rudyak, A. Cornelis, J. Rozenski, A. Zwolinska, J.-C. Marine, D. Lambrechts, Y.-A. Suh, F. Rousseau and J. Schymkowitz, Gain of function of mutant p53 by coaggregation with multiple tumor suppressors, Nat. Chem. Biol., 2011, 7(5), 285–295 CrossRef CAS PubMed.
-
(a) P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Halogen bonds in biological molecules, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(48), 16789 CrossRef CAS PubMed;
(b) M. B. Shah, J. Liu, Q. Zhang, C. D. Stout and J. R. Halpert, Halogen–π Interactions in the Cytochrome P450 Active Site: Structural Insights into Human CYP2B6 Substrate Selectivity, ACS Chem. Biol., 2017, 12(5), 1204–1210 CrossRef CAS PubMed;
(c) M. Erdélyi, Application of the Halogen Bond in Protein Systems, Biochemistry, 2017, 56(22), 2759–2761 CrossRef PubMed.
- A. N. Bullock, J. Henckel, B. S. DeDecker, C. M. Johnson, P. V. Nikolova, M. R. Proctor, D. P. Lane and A. R. Fersht, Thermodynamic stability of wild-type and mutant p53 core domain, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(26), 14338–14342 CrossRef CAS PubMed.
-
(a)
A. E. Martell and R. D. Hancock, Metal Complexes in Aqueous Solutions, Springer, New York, US, 1996 CrossRef;
(b) J.-S. Choi, J. J. Braymer, R. P. R. Nanga, A. Ramamoorthy and M. H. Lim, Design of small molecules that target metal-Aβ species and regulate metal-induced Aβ aggregation and neurotoxicity, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(51), 21990–21995 CrossRef CAS PubMed;
(c) W. R. Harris, C. J. Carrano, S. R. Cooper, S. R. Sofen, A. E. Avdeef, J. V. McArdle and K. N. Raymond, Coordination chemistry of microbial iron transport compounds. 19. Stability constants and electrochemical behavior of ferric enterobactin and model complexes, J. Am. Chem. Soc., 1979, 101(20), 6097–6104 CrossRef CAS.
- K. R. Gee, Z. L. Zhou, D. Ton-That, S. L. Sensi and J. H. Weiss, Measuring zinc in living cells.: A new generation of sensitive and selective fluorescent probes, Cell Calcium, 2002, 31(5), 245–251 CrossRef CAS PubMed.
- R. E. Carraway and P. R. Dobner, Zinc pyrithione induces ERK- and PKC-dependent necrosis distinct from TPEN-induced apoptosis in prostate cancer cells, Biochim. Biophys. Acta, 2012, 1823(2), 544–557 CrossRef CAS PubMed.
- K. K. Hoe, C. S. Verma and D. P. Lane, Drugging the p53 pathway: understanding the route to clinical efficacy, Nat. Rev. Drug Discovery, 2014, 13(3), 217–236 CrossRef.
-
(a) K. F. Macleod, N. Sherry, G. Hannon, D. Beach, T. Tokino, K. Kinzler, B. Vogelstein and T. Jacks, p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage, Genes Dev., 1995, 9(8), 935–944 CrossRef CAS PubMed;
(b) E. K. Benson, S. K. Mungamuri, O. Attie, M. Kracikova, R. Sachidanandam, J. J. Manfredi and S. A. Aaronson, p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes, Oncogene, 2014, 33(30), 3959–3969 CrossRef CAS.
-
(a) C. Gaiddon, M. Lokshin, J. Ahn, T. Zhang and C. Prives, A Subset of Tumor-Derived Mutant Forms of p53 Down-Regulate p63 and p73 through a Direct Interaction with the p53 Core Domain, Mol. Cell. Biol., 2001, 21(5), 1874–1887 CrossRef CAS;
(b) Y. Li and C. Prives, Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function?, Oncogene, 2007, 26(15), 2220–2225 CrossRef CAS PubMed;
(c) P. Muller and K. Vousden, Mutant p53 in Cancer: New Functions and Therapeutic Opportunities, Cancer Cell, 2014, 25(3), 304–317 CrossRef CAS PubMed;
(d) E. C. Pietsch, S. M. Sykes, S. B. McMahon and M. E. Murphy, The p53 family and programmed cell death, Oncogene, 2008, 27(50), 6507–6521 CrossRef CAS.
- C. M. Fenoglio-Preiser, J. Wang, G. N. Stemmermann and A. Noffsinger, TP53 and gastric carcinoma: A review, Hum. Mutat., 2003, 21(3), 258–270 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1), 55–63 CrossRef CAS PubMed.
- H.-B. Zhu, K. Yang, Y.-Q. Xie, Y.-W. Lin, Q.-Q. Mao and L.-P. Xie, Silencing of mutant p53 by siRNA induces cell cycle arrest and apoptosis in human bladder cancer cells, World J. Surg. Oncol., 2013, 11, 22 CrossRef PubMed.
-
(a) A. E. Sayan, B. S. Sayan, V. Gogvadze, D. Dinsdale, U. Nyman, T. M. Hansen, B. Zhivotovsky, G. M. Cohen, R. A. Knight and G. Melino, p73 and caspase-cleaved p73 fragments localize to mitochondria and augment TRAIL-induced apoptosis, Oncogene, 2008, 27, 4363 CrossRef CAS PubMed;
(b) J. Liu, H. Uematsu, N. Tsuchida and M.-A. Ikeda, Essential role of caspase-8 in p53/p73-dependent apoptosis induced by etoposide in head and neck carcinoma cells, Mol. Cancer, 2011, 10, 95 CrossRef CAS;
(c) A. K. Frank, E. C. Pietsch, P. Dumont, J. Tao and M. E. Murphy, Wild-type and mutant p53 proteins interact with mitochondrial caspase-3, Cancer Biol. Ther., 2011, 11(8), 740–745 CrossRef CAS PubMed;
(d) D. Bergamaschi, M. Gasco, L. Hiller, A. Sullivan, N. Syed, G. Trigiante, I. Yulug, M. Merlano, G. Numico, A. Comino, M. Attard, O. Reelfs, B. Gusterson, A. K. Bell, V. Heath, M. Tavassoli, P. J. Farrell, P. Smith, X. Lu and T. Crook, p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis, Cancer Cell, 2003, 3(4), 387–402 CrossRef CAS PubMed;
(e) M. C. Marin, C. A. Jost, L. A. Brooks, M. S. Irwin, J. O'Nions, J. A. Tidy, N. James, J. M. McGregor, C. A. Harwood, I. G. Yulug, K. H. Vousden, M. J. Allday, B. Gusterson, S. Ikawa, P. W. Hinds, T. Crook and W. G. Kaelin Jr, A common polymorphism acts as an intragenic modifier of mutant p53 behaviour, Nat. Genet., 2000, 25, 47 CrossRef CAS PubMed.
-
(a) S. Elmore, Apoptosis: a review of programmed cell death, Toxicol. Pathol., 2007, 35(4), 495–516 CrossRef CAS PubMed;
(b) C. Candé, N. Vahsen, C. Garrido and G. Kroemer, Apoptosis-inducing factor (AIF): caspase-independent after all, Cell Death Differ., 2004, 11, 591 CrossRef;
(c) I. F. Sevrioukova, Apoptosis-inducing factor: structure, function, and redox regulation, Antioxid. Redox Signaling, 2011, 14(12), 2545–2579 CrossRef CAS.
-
(a) P. Stambolsky, L. Weisz, I. Shats, Y. Klein, N. Goldfinger, M. Oren and V. Rotter, Regulation of AIF expression by p53, Cell Death Differ., 2006, 13, 2140 CrossRef CAS PubMed;
(b) L. Shen, X. Sun, Z. Fu, G. Yang, J. Li and L. Yao, The Fundamental Role of the p53 Pathway in Tumor Metabolism and Its Implication in Tumor Therapy, Clin. Cancer Res., 2012, 18(6), 1561–1567 CrossRef CAS PubMed.
-
(a) K. Hientz, A. Mohr, D. Bhakta-Guha and T. Efferth, The role of p53 in cancer drug resistance and targeted chemotherapy, OncoTargets Ther., 2016, 8(5), 8921–8946 Search PubMed;
(b) X. Zhou, Q. Hao and H. Lu, Mutant p53 in cancer therapy—the barrier or the path, J. Mol. Cell Biol., 2018, 11, 293–305 CrossRef;
(c) G. Blandino, A. J. Levine and M. Oren, Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy, Oncogene, 1999, 18, 477 CrossRef CAS.
- Z. H. Siddik, Cisplatin: mode of cytotoxic action and molecular basis of resistance, Oncogene, 2003, 22(47), 7265–7279 CrossRef CAS.
-
(a) H. Perlman, X. Zhang, M. W. Chen, K. Walsh and R. Buttyan, An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis, Cell Death Differ., 1999, 6(1), 48–54 CrossRef CAS PubMed;
(b) L. D. Walensky, Targeting BAX to drug death directly, Nat. Chem. Biol., 2019, 15(7), 657–665 CrossRef CAS PubMed.
-
(a) A. Pearce, M. Haas, R. Viney, S.-A. Pearson, P. Haywood, C. Brown and R. Ward, Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study, PLoS One, 2017, 12(10), e0184360 CrossRef;
(b) C. F. Cheok, Protecting normal cells from the cytotoxicity of chemotherapy, Cell Cycle, 2012, 11(12), 2227–2228 CrossRef CAS.
-
(a) I. M. M. van Leeuwen, B. Rao, M. C. C. Sachweh and S. Laín, An evaluation of small-molecule p53 activators as chemoprotectants ameliorating adverse effects of anticancer drugs in normal cells, Cell Cycle, 2012, 11(9), 1851–1861 CrossRef CAS;
(b) D. P. Lane, C. F. Cheok and S. Lain, p53-based cancer therapy, Cold Spring Harbor Perspect. Biol., 2010, 2(9), a001222 Search PubMed.
-
(a) T. E. Wallach and J. R. Bayrer, Intestinal Organoids: New Frontiers in the Study of Intestinal Disease and Physiology, J. Pediatr. Gastroenterol. Nutr., 2017, 64(2), 180–185 CrossRef PubMed;
(b) A. Fatehullah, S. H. Tan and N. Barker, Organoids as an in vitro model of human development and disease, Nat. Cell Biol., 2016, 18, 246 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic schemes, 1H and 13C NMR characterization, pKa values, speciation diagrams, Zn affinity determination and respective HypSpec data fitting, and NCI-60 in vitro cytotoxicity data for LI and LH (PDF). Crystallographic data for ZnLICl and ZnLHCl (CIF). CCDC 1947980 and 1947981. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04151f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Diego
Martelino
a,
Derek
Wilson
c,
Christian
Gaiddon
a,
Diego
Martelino
a,
Derek
Wilson
c,
Christian
Gaiddon