
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValency engineering of monomeric enzymes for self-assembling biocatalytic hydrogels†
Patrick
Bitterwolf
a,
Sabrina
Gallus
a,
Theo
Peschke
b,
Esther
Mittmann
a,
Claude
Oelschlaeger
c,
Norbert
Willenbacher
c,
Kersten S.
Rabe
 a and
Christof M.
Niemeyer
*a
a and
Christof M.
Niemeyer
*a
aInstitute for Biological Interfaces (IBG1), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany. E-mail: niemeyer@kit.edu
bNovartis AG, Kohlestrasse WSJ 103, CH-4002 Basel, Switzerland
cInstitute for Mechanical Process Engineering and Mechanics, Karlsruhe Institute of Technology (KIT), Gotthard-Franz-Straße 3, D-76131 Karlsruhe, Germany
First published on 6th September 2019
Abstract
All-enzyme hydrogels are efficient reagents for continuous flow biocatalysis. These materials can be obtained by self-assembly of two oligomeric enzymes, modified with the complementary SpyTag and SpyCatcher units. To facilitate access to the large proportion of biocatalytically relevant monomeric enzymes, we demonstrate that the tagging valency of the monomeric (S)-stereoselective ketoreductase Gre2p from Saccharomyces cerevisiae can be designed to assemble stable, active hydrogels with the cofactor-regenerating glucose 1-dehydrogenase GDH from Bacillus subtilis. Mounted in microfluidic reactors, these gels revealed high conversion rates and stereoselectivity in the reduction of prochiral methylketones under continuous flow for more than 8 days. The sequential use as well as parallelization by ‘numbering up’ of the flow reactor modules demonstrate that this approach is suitable for syntheses on the semipreparative scale.
Introduction
Biocatalysis is widely regarded as a key area of industrial (“white”) biotechnology to open the doors to sustainable ‘green’ production of value-added molecules.1 To implement enzymatic catalysis efficiently in technical production processes, the development of bioinspired, multistep cascade reactions is currently attracting much attention.2–7 In order to prevent the multiple reactions from spreading and unproductive crosstalk, compartmentalized microfluidic reactors are being developed, which spatially separate sequential transformations into individual reaction vessels that are fluidically coupled with each other.6,8–10 Moreover, the microfluidic reactors offer a high level of control over temperature profiles and diffusion-based mixing.11,12 This approach, dubbed as ‘flow biocatalysis’, is often based on isolated enzymes, which need to be immobilized on carrier structures inside the reactor space. Owing to the delicate nature of many enzymes, chemically mild immobilization chemistries are of increasing relevance for this purpose.12,13 Furthermore, in order to overcome the limitation of the amount of the immobilized enzyme by the effective surface area, carrier-free immobilization techniques are required that avoid using the valuable reactor space for unproductive particulate or polymer carrier structures. We have recently demonstrated the construction of self-assembling all-enzyme hydrogels by using two homotetrameric enzymes, a highly (R)-selective alcohol dehydrogenase LbADH from Lactobacillus brevis and the nicotinamide adenine dinucleotide phosphate (NADPH)-regenerating glucose 1-dehydrogenase GDH from Bacillus subtilis, each of which was genetically fused with either the SpyTag (ST) peptide or the SpyCatcher (SC) protein.14 Since the ST/SC system enables the rapid cross-linking of the two protein building blocks under physiological conditions through the formation of covalent isopeptide bonds,15 the ST/SC-tagged LbADH and GDH spontaneously polymerize into porous networks upon mixing. Mounted into microfluidic reactors, these biocatalytic hydrogels showed extraordinary high space-time-yields in the continuous production of chiral alcohols.14 Despite these promising results, the methodological approach is limited to multimeric enzymes that are required for polymerization. A survey of the PDB database shows that only about 10% of the enzymes are homotetramers, whereas 40% are monomers. In order to make monomeric enzymes accessible for polymerization, we report here on the engineering of tagging valency of the monomeric (S)-selective methylglyoxal reductase Gre2p (EC 1.1.1.283) from Saccharomyces cerevisiae YJM193 (Taxonomy ID: 1294304). We illustrate that genetic fusion of Gre2p with two or three ST peptides leads to oligovalent building blocks suitable for polymerization with homotetrameric GDH.GDH is used as a cofactor regeneration system to deliver NADPH by reducing NADP+ and oxidizing glucose to gluconolactone. The resulting hydrogel materials can be used for the continuous stereoselective reduction of prochiral ketones into S-configured alcohols inside microfluidic reactors.
Results and discussion
Construction and characterization of the enzyme hydrogels
In analogy to the (R)-selective hydrogel comprised of SC-tagged LbADH and ST-tagged GDH,14 our first attempts to an (S)-selective hydrogel concerned the use of a Gre2p variant that is double tagged with two SC domains fused to the N- and C-terminus, respectively. Indeed, several doubly labeled enzymes have already been described in the literature, including monomers and SC/ST-modified variants that retain their activity after labeling.16–18 In our case, however, the resulting Gre2p-SC2 enzyme revealed only poor polymerization with GDH-ST (Fig. S1†). Therefore, the tagging approach was switched to fusing Gre2p with the small ST peptide, whereas the larger GDH homotetramer was modified with the bulkier SC domain.As shown in Fig. 1, we engineered Gre2p variants tagged with either two ST peptides at the N- and C-terminus (Gre2p-ST2) or with three ST peptides by inserting an additional internal ST in a loop structure amid the amino acid positions F86-D87 (Gre2p-ST3). Indeed, this strategy follows the “loop engineering” approach, which has been established as the standard method with which even sensitive proteins can be labeled at internal sites.19–21 The internal site was selected from four rationally designed candidates that had been tested for catalytic activity and expression yields (Table 1, Fig. S2†). The proteins were overexpressed in E. coli and purified to homogeneity by Ni-NTA affinity chromatography (Fig. S3†). Initial profiling of the specific biocatalytic activity of the enzyme variants indicated that GDH-SC and Gre2p-ST2 have a similar specific activity as the native enzymes, while the activity of the triple tagged variant was reduced by about 75% (Table 1).
 | ||
| Fig. 1 (a) Oligovalent Gre2p-ST variants, displaying ST peptides at the N- (red) and C-terminus (purple) or an internal loop (green). (b) Self-assembly of di- or trivalent Gre2p-ST variants with homotetrameric SC-tagged GDH (blue) leads to formation of hydrogels, as illustrated schematically and by photographic images of the materials. These polymers were mounted in fluidic microreactors to facilitate stereoselective reduction of prochiral ketones. | ||
| Specific activitya | Yield | MW | Subunits | ||
|---|---|---|---|---|---|
| [μmolsubstrate min−1 mgprotein−1] | [μmolsubstrate min−1 μmolsubunit−1] | [mg L−1] | [gprotein molsubunit−1] | ||
| a Specific activities of the enzymes using NDK 1 and glucose as substrates. Data represent the mean of at least triplicate analyses ± 1 SD (standard deviation). | |||||
| Gre2p | 5.9 ± 0.17 | 254 ± 7.2 | 54 | 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 333 333 |
1 |
| Gre2p-ST | 6.9 ± 1.4 | 281 ± 59 | 37 | 40804 |
|
| Gre2p-ST2 | 5.5 ± 0.07 | 236 ± 3 | 35 | 42574 |
|
| Gre2p-ST3 (F86-ST-D87) | 1.5 ± 0.07 | 67 ± 0.8 | 48 | 44660 |
|
| Gre2p-ST3 (K139-ST-S140) | 0.4 ± 0.05 | 17 ± 2.1 | <0.1 | 44660 |
|
| Gre2p-ST3 (E228-ST-D229) | 1.7 ± 0.03 | 75 ± 1.5 | 2 | 44660 |
|
| Gre2p-ST3 (S296-ST-T302) | 1 ± 0.06 | 48 ± 2.5 | 3 | 44179 |
|
| Gre2p-SC2 | 3.8 ± 0.03 | 240 ± 1.9 | 5 | 63917 |
|
| BsGDH | 3.6 ± 0.4 | 104 ± 12 | 79 | 29014 |
4 |
| BsGDH-ST | 4.3 ± 0.4 | 133 ± 14 | 86 | 31100 |
|
| BsGDH-SC | 3.5 ± 0.2 | 148 ± 7 | 74 | 41771 |
|
Initial assessment of the SC/ST-based coupling capabilities of Gre2p-ST2 and Gre2p-ST3 by gel electrophoretic analyses revealed that both variants crosslink with GDH-SC monomers (Fig. S4†). A more detailed analysis of the gelation process was conducted by dynamic light scattering (DLS) analysis at variable stoichiometric ratios of GDH-SC:Grep-ST2 and GDH-SC:Gre2p-ST3 (Fig. 2a and b).
 | ||
| Fig. 2 Stoichiometry-dependent increase of the hydrodynamic particle diameter observed in the initial 30 min after mixing of GDH-SC with (a) Gre2p-ST2 or (b) Gre2p-ST3 as determined by DLS. Subsequently the samples were denatured and subjected to SDS-PAGE analysis. The gels in (c and d) show the results of the ST/SC conjugation in the GDH-SC/Gre2p-ST2 and GDH-SC/Gre2p-ST3 mixtures, respectively. Note that the indication of molar subunit ratio reflects the number of monomeric enzyme subunits to illustrate the relative binding site ratio in the respective samples. | ||
DLS is well suited to determine the increase in hydrodynamic diameter of small particles formed in the early phase of the polymerization, which have typical sizes in the range of 16 to 31 nm, and we used this methodology to investigate the dependency of polymerization on the molar ratio of binding sites of GDH and the Gre2p variants.
As expected, the largest increase in hydrodynamic particle diameter was observed at a molar subunit ratio of 2:1 for the GDH-SC:Gre2p-ST2 mixture due to the presence of equal amounts of the complementary SC and ST binding sites. Surprisingly, the equimolar ratio of binding sites in the case of the GDH-SC:Gre2p-ST3 (subunit ratio of 3:1) did not lead to the largest increase in particle diameter. Instead, optimal gelation was observed for a binding site ratio of 2:3 (corresponding to a subunit ratio of 2:1). These results, which were confirmed by gel electrophoresis (Fig. 2c and d), suggest that the internal tag is not as good accessible as the terminal tags, presumably due to steric hindrance. Owing to the optimal gelation properties, a subunit ratio of 2:1 was used in all further experiments.
In the course of detailed investigations of an analogous hydrogel described previously14 we found that the use of scanning electron microscopy (SEM) was only of limited value, whereas the use of DLS and microrheology has proven to deliver quantitative insights on the material properties, such as the formation rate of the hydrogels and an estimation of the mesh size and determination of viscosity and entanglement values G0, respectively. We therefore applied these methodologies for characterization of the Gre2p-ST2 and Gre2p-ST3 hydrogels. In fact, DLS analyses indicated the kinetics of the gelation process of the enzyme hydrogels were comparable to those of the previously reported GDH/LbADH materials.14
Microrheology analysis of the viscoelastic properties of the hydrogels was achieved by Multiple Particle Tracking (MPT) using non-polymerized enzymes as controls (Fig. 3). To this end, mean square displacements (MSDs) of individual polystyrene microspheres of 500 nm diameter dispersed in a solution containing the soluble enzymes (Fig. 3a) and of 200 nm diameter tracer beads for the gelated GDH-SC/Gre2p-ST2 (Fig. 3b) and GDH-SC/Gre2p-ST3 (Fig. 3c) materials were recorded. The red curves show the ensemble-average MSD. In the control sample containing unpolymerized enzymes, MSD traces adopted a power-law behaviour as a function of time with a slope close to 1 throughout the probed time scales. This value indicated that the motion of the beads is purely diffusive and that the microenvironment surrounding the tracer particles responds like a viscous liquid. The apparent viscosity ηapp determined from the average MSD trace was 1.65 ± 0.65 mPa s and this value is in good agreement with the viscosity of pure water.22
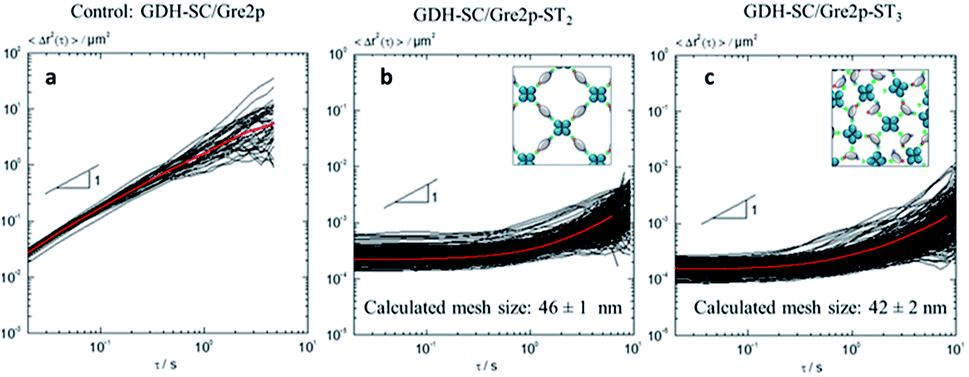 | ||
| Fig. 3 Multiple Particle Tracking (MPT) microrheology analysis of local viscoelastic properties of unpolymerized enzyme solutions (a) and the enzyme hydrogels (b and c). The curves show individual (black) and average (red) mean square displacements (MSDs) of polystyrene tracer microspheres. Note that the decrease in MSD slope is indicative for an increase in viscosity of the medium (see text for details). | ||
In the hydrogels GDH-SC/Gre2p-ST2 (Fig. 3b) and GDH-SC/Gre2p-ST3 (Fig. 3c), the tracer particle motion was significantly different. All MSDs exhibited almost no time dependence indicating that tracer particles are highly constrained by the surrounding hydrogel. Moreover, the time-independent average MSD value was decreased thus indicating a higher degree of elasticity. Finally, we could directly determine the mesh size ξ of the network according to the classical theory of rubber elasticity.23 For example, MPT analysis revealed G0 values of 42 ± 2.5 Pa or 56 ± 8 Pa and average mesh sizes of ξ = 46 ± 1 nm or 42 ± 2 nm for the GDH-SC/Gre2p-ST2 and GDH-SC/Gre2p-ST3 gels, respectively, as compared to G0 of 20 ± 7 Pa and a mesh size ξ = 60 nm ± 7 for the GDH-ST/LbADH-SC gel. The slightly larger entanglement and smaller mesh size values clearly reflects the greater connectivity in the GDH-SC/Gre2p-ST3 gels originating from the trivalent building block Gre2p-ST3. All gels have pore sizes of <200 nm, which is in the range of typical microfiltration membranes.24
Microfluidic experiments
To benchmark the biocatalytic activity of the GDH-SC/Gre2p-ST2 and GDH-SC/Gre2p-ST3 gels, we chose the prochiral CS-symmetrical 5-nitrononane-2,8-dione (NDK) 1 (Fig. 4a) as the substrate because of its high relevance for stereochemistry and natural product synthesis.25,26 Either one or both carbonyl groups of 1 can be biocatalytically reduced to form diastereomeric hydroxyketones 2 or diols 3, respectively (Fig. 4a and S5†), and all products can be readily quantified by chiral HPLC analysis (Fig. S6†).25 | ||
| Fig. 4 Stereoselective reduction of NDK 1 in continuous flow micro reactors loaded with GDH-SC/Gre2p-ST2 or GDH-SC/Gre2p-ST3 hydrogels. (a) Reaction scheme of the (S)-selective biocatalytic reduction of NDK 1. (b and c) Time dependent NDK 1 conversion and distribution of 2 (green) and 3 (blue) products, determined in the reactor outflow by chiral HPLC. All error bars indicate the standard deviation, obtained from at least two independent analyses. | ||
To investigate the applicability of the novel (S)-selective hydrogels for flow biocatalysis, we used a microfluidic setup (Fig. S7†) consisting of syringe pumps to transfuse a substrate mix containing NADP+, glucose and NDK through a hydrogel-loaded linear PDMS micro reactor channel (V = 150 μL) with a flow rate of 5 μL min−1 (Fig. 4b and c). The outflow of the micro reactor was analyzed by HPLC. After mounting of the hydrogels in the microfluidic channel, swelling of the hydrogel inside the microreactor was typically observed in the initial phase. However, the gel always remained clear and showed no turbidity that could indicate precipitation of the proteins in the gel matrix. As indicated in Fig. 4, the hydrogel effectively retained the immobilized enzymes, resulting in stable conversion rates after an initial equilibration phase over more than 8 days. According to the specific activity of the enzymes (Table 1) the Gre2p-ST2 gel was about 3.6-fold more active than the Gre2p-ST3 gel.
We had previously demonstrated that both unbound, free25 and particle-immobilized27 Gre2p converts NDK 1 with very high stereoselectivity into the (S)-anti-hydroxyketone 2 (e.r. > 99:1), whereas the (S,S)-configured pseudo C2 diol 3 is only formed with substantially slower rates at high enzyme concentrations. Given that Gre2p generates the hydroxyketone 2 with a much higher efficiency than the diol 3,25 it is important to note that, due to the high enzyme concentration in the hydrogels, substantial amounts of diol 3 were produced. For example, running the reactor at low flow rates of 5 μL min−1 led to formation of about 65% of diol 3, (Fig. 4 and S8†), whereas less than 10% of 3 were produced with particle-supported enzyme in a packed-bed reactor format.27 Importantly, the present study revealed a high dependency of the product distribution on the set flow rate (Fig. S8†). In particular, we found that the relatively high amounts of 3 formed at low flowrates (65%@5 μL min−1) were significantly decreased at high flowrates (<25%@100 μL min−1, see Fig. S8†).
The above results clearly show that the enzymatic activity can be influenced by the formulation of the biocatalyst and the process conditions. In order to further demonstrate that the reactor performance can also be influenced by the material properties of the hydrogels, two reactors containing either the GDH-SC/Gre2p-ST2 or the GDH-SC/Gre2p-ST3 hydrogels were run at a flow rate of 5 μL min−1, resulting in a residence time of 30 minutes for the substrate 1 (Fig. 4b and c, respectively).
Because of the lower activity of Gre2p-ST3 the respective gel produced lesser amounts of 3 than the gel obtained from Gre2p-ST2. As expected, analysis of the product distribution revealed that the GDH-SC/Gre2p-ST2 hydrogel initially produced more than 50% of diol 3, thus confirming that the high enzyme concentration inside the hydrogel favors the second reduction step (Fig. 4b). Due to its lower specific activity, the diol production of the GDH-SC/Gre2p-ST3 gel was reduced (Fig. 4c and S9†) resulting in higher initial conversion rates of 1 to the hydroxyketone 2. Both hydrogel microreactors showed a first phase of activity loss, which passed into a second phase of stable conversion rates. Of note, the comparison with previously published packed-bed microreactors bearing particle-immobilized Gre2p,27 showed that an about 40-fold higher amount of enzyme could be accommodated in the present hydrogel microreactor. Using the same substrate concentration, this enabled a more than 2.5-fold higher conversion of 1. Furthermore, at high flow rates (100 μL min−1), a substantially improved space-time-yield (STY) of about 320 g L−1 d−1 can be obtained as compared to the packed-bed reactor (STY = 36 g L−1 d−1).
Subsequently, the GDH-SC/Gre2p-ST2 hydrogel-based system was used for the continuous stereoselective reduction of prochiral ketones to their corresponding (S)-configured alcohols (2, 3, 5 and 7). To this end, the microreactor was sequentially perfused with three different substrate solutions containing either 1, acetophenone 4, or 4′-chloroacetophenone 6. Each substrate administration was conducted for 2 h and reaction products in the outflow were analyzed by chiral HPLC (Fig. S5†). All substrates were reduced to the corresponding (S)-configured alcohols with near quantitative conversion and stereoselectivities of >99% (Fig. 5a).
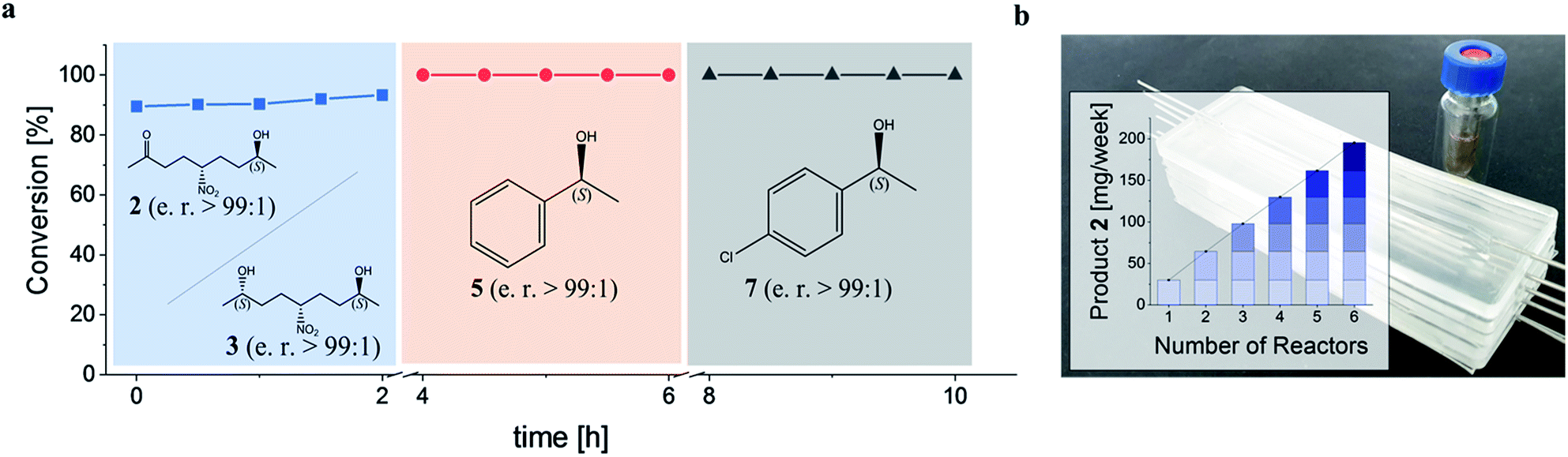 | ||
| Fig. 5 Continuous flow, biocatalytic production of chiral alcohols using GDH-SC/Gre2p-ST2 hydrogel-loaded microreactors. (a) Sequential continuous conversion of (i) NDK 1, (ii) acetophenone 4 and (iii) 4′-chloroacetophenone 6 to their corresponding (S)-configured alcohols 2, 3, 5, and 7 using a single hydrogel-loaded microreactor. (b) Reactor stack used for semipreparative biocatalytic reduction of 1. The vial contains 141 mg purified 2. The inset shows that all 6 modules have a comparable performance. | ||
To illustrate the scalability of the hydrogel microreactors for semipreparative applications, we demonstrate the ‘numbering up’ of reactor modules. In fact, this is the widely accepted approach in microreaction technology, as it allows a direct upscaling of reactions without having to re-evaluate and optimize the flow system and process. Numbering up is more efficient than the use of larger reactors, which would lead to altered temperature distributions and diffusion processes, so that the process would have to be re-evaluated and optimized in terms of temperature, reactant concentration, pressure and flow conditions. By parallel operation of 6 chips containing the GDH-SC/Gre2p-ST2 gel (Fig. 5b), isolated product yields of 141 mg (46%) and 83 mg (27%) of 2 and 3, respectively, were obtained.
Conclusions
In summary, we demonstrate here that engineering of tagging valency of a monomeric (S)-selective ketoreductase enzyme can be used to expand the scope of self-assembling all-enzyme hydrogels.In order to ensure rapid access to application-relevant enzymes, the genetically encoded SC/ST system was used here. Despite the larger (13 amino acid) SpyTag, this system is distinguished by its simpler implementation and handling compared to bioorthogonal coupling methods based on small-molecule tags.28 The high productivity and stereoselectivity of the gels can be used advantageously for continuous flow biocatalysis. Furthermore, our demonstration of the sequential and parallel use of flow reactor modules suggests that this approach is also suitable for semipreparative syntheses on the gram scale. We therefore believe that such controlled, mild and selective immobilization of other enzyme classes like cofactor-dependent transaminases, imine reductases and monooxygenases should lead to new, powerful production systems for industrial biotechnology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Helmholtz programme “BioInterfaces in Technology and Medicine” and DFG project Ni399/15-1. PB and SG are grateful for a Kekulé fellowship by Fonds der Chemischen Industrie (FCI). We thank Inés Roque González and Christin Lederer for experimental help.References
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2017, 118, 801–838 CrossRef.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef CAS.
- A. Kuchler, M. Yoshimoto, S. Luginbuhl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409–420 CrossRef CAS.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- K. S. Rabe, J. Muller, M. Skoupi and C. M. Niemeyer, Angew. Chem., Int. Ed. Engl., 2017, 56, 13574–13589 CrossRef CAS.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2018, 23, 9–18 CrossRef.
- C. Schmid-Dannert and F. Lopez-Gallego, Curr. Opin. Chem. Biol., 2018, 49, 97–104 CrossRef.
- S. Gandomkar, A. Żądło-Dobrowolska and W. Kroutil, ChemCatChem, 2019, 11, 225–243 CrossRef CAS.
- B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS.
- R. Wohlgemuth, I. Plazl, P. Znidarsic-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS.
- D. Kim and A. E. Herr, Biomicrofluidics, 2013, 7, 041501 CrossRef.
- T. Peschke, P. Bitterwolf, S. Gallus, Y. Hu, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2018, 57, 17028–17032 CrossRef CAS.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS.
- T. H. Wang, Y. Y. Chen, H. H. Pan, F. P. Wang, C. H. Cheng and W. C. Lee, BMC Biotechnol., 2009, 9, 63 CrossRef.
- C. Schoene, J. O. Fierer, S. P. Bennett and M. Howarth, Angew. Chem., Int. Ed., 2014, 53, 6101–6104 CrossRef CAS.
- M. Si, Q. Xu, L. Jiang and H. Huang, PLoS One, 2016, 11, e0162318 CrossRef.
- S. R. Sauter, S. Diekmann and V. Braun, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 786, 33–37 CrossRef CAS.
- C. Richez, J. Boetzel, N. Floquet, K. Koteshwar, J. Stevens, B. Badet and M. A. Badet-Denisot, Protein Expression Purif., 2007, 54, 45–53 CrossRef CAS.
- D. M. Paul, F. Beuron, R. B. Sessions, A. Brancaccio and M. G. Bigotti, Sci. Rep., 2016, 6, 20696 CrossRef CAS.
- A. Kowalczyk, C. Oelschlaeger and N. Willenbacher, Polymer, 2015, 58, 170–179 CrossRef CAS.
- J. C. Crocker and D. G. Grier, J. Colloid Interface Sci., 1996, 179, 298–310 CrossRef CAS.
- Y. Pouliot, Int. Dairy J., 2008, 18, 735–740 CrossRef CAS.
- M. Skoupi, C. Vaxelaire, C. Strohmann, M. Christmann and C. M. Niemeyer, Chem.–Eur. J., 2015, 21, 8701–8705 CrossRef CAS.
- T. C. Nugent, F. T. Najafian, H. A. Hussein and I. Hussain, Chem.–Eur. J., 2016, 22, 14342–14348 CrossRef CAS.
- T. Peschke, M. Skoupi, T. Burgahn, S. Gallus, I. Ahmed, K. S. Rabe and C. M. Niemeyer, ACS Catal., 2017, 7, 7866–7872 CrossRef CAS.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Contains details about plasmid construction, protein expression, DLS measurements, hydrogel preparation, MPT measurements, microreactor preparation, synthesis of reduced methylketones, chiral HPLC analysis, determination of enzyme activities, microfluidic setups and product purification conducted during this study. See DOI: 10.1039/c9sc04074a |
| This journal is © The Royal Society of Chemistry 2019 |