
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst experimental evidence for the elusive tetrahedral cations [EP3]+ (E = S, Se, Te) in the condensed phase†
Philippe
Weis
a,
David Christopher
Röhner
a,
Richard
Prediger
a,
Burkhard
Butschke
a,
Harald
Scherer
a,
Stefan
Weber
b and
Ingo
Krossing
 *a
*a
aInstitut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
bInstitut für Physikalische Chemie, Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany
First published on 7th October 2019
Abstract
Condensed phase access to the unprecedented tetrahedral cations [EP3]+ (E = S, Se, Te) was achieved through the reaction of ECl3[WCA] with white phosphorus ([WCA]− = [Al(ORF)4]− and [F(Al(ORF)3)2]−; –RF = –C(CF3)3). Previously, [EP3]+ was only known from gas phase MS investigations. By contrast, the reaction of ECl3[A] with the known P33− synthon Na[Nb(ODipp)3(P3)] (enabling AsP3 synthesis), led to formation of P4. The cations [EP3]+ were characterized by multinuclear NMR spectroscopy in combination with high-level quantum chemical calculations. Their bonding situation is described with several approaches including Atoms in Molecules and Natural Bond Orbital analysis. The first series of well-soluble salts ECl3[WCA] was synthesized and fully characterized as starting materials for the studies on this elusive class of [EP3]+ cations. Yet, with high [ECl3]+ fluoride ion affinity values between 775 (S), 803 (Se) and 844 (Te) kJ mol−1, well exceeding typical phosphenium ions, these well-soluble ECl3[WCA] salts could be relevant in view of the renewed interest in strong (also cationic) Lewis acids.
Introduction
The chemistry of simple tetrahedral main group clusters continues to fascinate researchers worldwide.1–5 Neutral white phosphorus (P4) is the archetype of this class and a commodity chemical. Even nitrogen was shown to exist shortly as N4 in a plasma and has raised significant theoretical interest.6 However, the heavier homologous yellow arsenic (As4) is difficult to handle due to its high sensitivity to light. Recent years yielded several new representatives, such as AsP3 (Scheme 1) and As2P2, As3P and SbP3.2,7 In addition, a focus was put on the development of chemical tools to facilitate the handling of As4 with reagents such as Ag(As4)2[Al(ORF)4] or As4 intercalated in porous carbon that acts as a storage form of yellow arsenic and yields concentrated solutions of As4 that are light stable for several hours.4,8 These molecules continue to offer interesting follow-up chemistry that is not exhausted yet.9 For example, the side-on protonation of P4, [HP4]+ has only been realized very recently.10 In addition to these neutral molecules, the syntheses and structures of a multitude of Zintl-type anionic tetrahedra shown in Scheme 1 are known: [Tt4]4− (Tt = Si, Ge, Sn, Pb) or binary anions such as [Tt2Pn2]2− (Pn = P, As, Sb, Bi), [TrPn3]2− (Tr = Ga, In, Tl) and [TlSn3]5−.11 These are often accessible by solid-state reactions of the corresponding elements with alkaline metals. Several of these long known tetrahedra were also observed in polar media like liquid ammonia and/or with the presence of cryptands and crown ethers as sequestering reagents.12 Recent additions detected by NMR spectroscopy include the dissolved Zintl-tetrahedra [Si4]4−, [Sn4]4− and the protonation product ion [HSi4]3−.13,14 However, the only reports on any cationic tetrahedra are those on [SP3]+ and [SeP3]+ as well as the arsenic homologues that were observed by MS in the gas phase as fragmentation products of Pn4E3 (Pn = P, As; E = S, Se); the first observation dating back to 1971.15 Tellurium analogues have not been described in any phase.16 If turning to coordination chemistry, also no clear evidence for such cations is published. Only the related complexes [(triphos)Co(Pn2E)][BF4] (Pn = P, As; E = S, Se, Te); triphos = 1,1,1-tris(diphenylphosphinomethylethane) are known. Tentatively, these were formulated as coordinated [P2E]+ and [As2E]+ cations stemming from the fragmentation of the elusive [EP3]+ cations.17 Thus, to the best of our knowledge there is no proven condensed phase evidence for such binary tetrahedral main group cations in any composition. | ||
| Scheme 1 Exemplary overview of commonly known and structurally characterized tetrahedron shaped molecules in the condensed phase. The cations characterized in this work – albeit without crystal structure – are included for comparison. | ||
Despite a large number of binary pnictogen–chalcogen cations of nitrogen, arsenic, antimony and bismuth has been published,18,19 the first binary phosphorus–chalcogen cation isolated in the condensed phase, [P3Se4]+, was only published in 2015 by three independent groups via three different routes.20 Binary phosphorus–sulfur and phosphorus–tellurium cations remain unknown.21 The [P3Se4]+ cation adopts a nortricyclane-like structure similar to the heavier analogues [As3S4]+ and [As3Se4]+ known since 1981.19 Attempts to synthesize binary phosphorus–selenium cations by reaction of liquid phosphine PH3 with SeCl3[AlCl4] yielded insoluble polymeric materials22 tentatively assigned as “SeP[AlCl4]n”.23 Moreover, most known [ECl3]X salts (X = suitable anion) are poorly soluble in solvents compatible with white phosphorus. Therefore, any follow-up chemistry of [ECl3]X salts is scarce and, to our knowledge, in particular no reactions of any trihalochalcogenonium ion with P4 or any other elemental pnictogen was hitherto published. The synthesis of well soluble [ECl3]X salts in common solvents is a prerequisite for such investigations and was the starting point for our studies on the tetrahedral [EP3]+ cations (E = S, Se and Te).
Results and discussion
First attempts towards the synthesis of the [EP3]+ cations were performed in analogy to the published synthesis2 of AsP3 from Na[Nb(ODipp)3(P3)] and AsCl3. Thus, AsCl3 was replaced by isoelectronic ECl3[WCA] (WCA = weakly coordinating anion). Therefore, we briefly describe the synthesis of a series of well soluble ECl3[WCA] salts before turning to the investigated routes towards the [EP3]+ cations.Synthesis of well-soluble ECl3[WCA] salts as starting materials
The large majority of the many known salts [EX3]A (E = S, Se, Te, X = F, Cl, Br, I) feature halometallate counterions A like [AlCl4]−, [MF6]− (M = Sb, As) and [AuCl4]−.24 They were synthesized from melts or in oxidizing media like SOCl2 or SO2Cl2.24 Typically, they do exhibit poor solubility in common organic solvents. In addition, many of the media employed for synthesis, would be highly reactive towards reductants such as P4. Therefore, salts EX3[WCA] (E = S, Se, Te, X = F, Cl, Br, I) soluble in common, non-reactive solvents were needed. Since the WCAs [Al(ORF)4]− and [F(Al(ORF)3)2]− often lend high solubility to salts of the respective cations, the salts ECl3[WCA] were prepared by halide abstraction of the chalcogen tetrachlorides with M[Al(ORF)4] or M[F(Al(ORF)3)2] (M = Ag, Tl for E = Se; M = Ag for E = Te) in CH2Cl2 or liquid SO2. Eqn (1) is related to the known synthesis of TeCl3[Al(ORF)4] (2a).25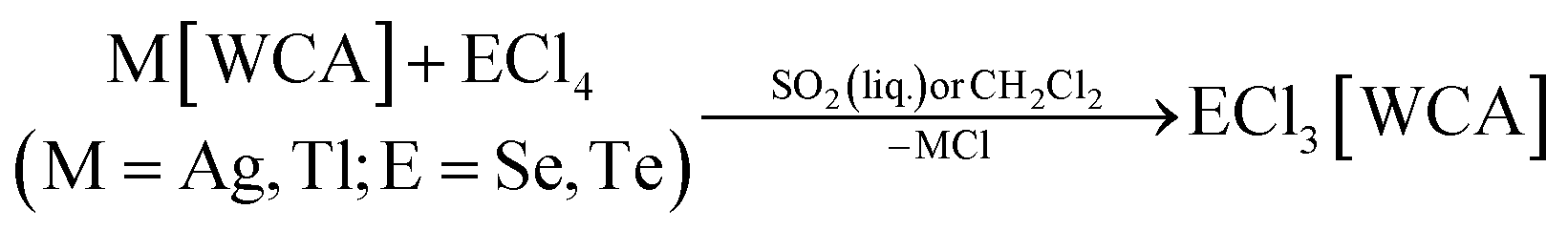 | (1) |
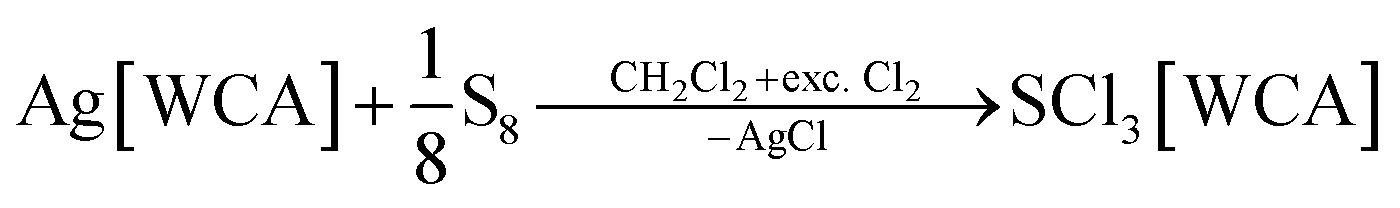 | (2) |
The compounds SeCl3[Al(ORF)4] (1a), SeCl3[F(Al(ORF)3)2] (1b), SeCl3[FAl(ORF)3] (1c) and TeCl3[F(Al(ORF)3)2] (2b) are well accessible and soluble in organic media like CH2Cl2 and 1,2-F2C6H4 (o-dfb). By contrast, [SCl3]+ salts turned out to be very reactive. Due to the instability of SCl4, it was prepared as an intermediate by the reaction of sulfur with Cl2 in CH2Cl2 and slowly warming the reaction mixture from −196 °C to room temperature. Reaction of this SCl4 intermediate with Ag[Al(ORF)4] according to eqn (2) was accompanied by anion decomposition and yielded predominantly SCl3[F(Al(ORF)3)2] (3). A selective way to 3 in good yields (>90%) simply substitutes [Al(ORF)4]− with the more stable26–29 anion [F(Al(ORF)3)2]−. Care needs to be taken, to ensure a complete reaction during the synthesis of the ECl3[WCA] salts. If there is residual Ag[WCA] present in ECl3[WCA], Ag(P4)2[Al(ORF)4] or Ag(P4)2[F(Al(ORF)3)2] tend to crystallize from the reaction mixtures in subsequent reactions. This could be overcome by using Tl[WCA] as a starting material.30 Attempts to synthesize [SeCl3]+ salts from Li[Al(ORF)4] and SeCl4 or through metathesis reaction with SeCl3[AlCl4] only led to the re-isolation of the starting materials (see ESI Section 3.5†). Since it appeared useful to also test a [EF3]+ source as starting material, we investigated the preparation of SF3[Al(ORF)4] from Li[Al(ORF)4] and SF4, but all efforts of its synthesis failed (see ESI Section 3.5†). Attempts to synthesize 1b by reacting SeCl4 with two equivalents of the strong Lewis acid Me3Si–F–Al(ORF)3 and trying to skip the synthesis of Ag[F(Al(ORF)3)2], only led to methylation and the isolation of SeMeCl2[F(Al(ORF)3)2].31 Yet, to all prepared salts ECl3[WCA], optimized routes were found and the products were fully characterized (yields, NMR, IR, Raman; crystal structures of 1a, 1b, 1c and 3). As a side, the crystal structure of Tl[Al(ORF)4] was determined and deposited with the CCDC. Due to the well-known properties of the long-known [ECl3]+ cations, the characterization and discussion of the novel salts ECl3[WCA] is found in the ESI.†
Syntheses directed towards the ion [EP3]+
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[P5Cl2]+ = 0.3 (E = S), 1.1 (E = Se), 0.3 (E = Te)).
:[P5Cl2]+ = 0.3 (E = S), 1.1 (E = Se), 0.3 (E = Te)).
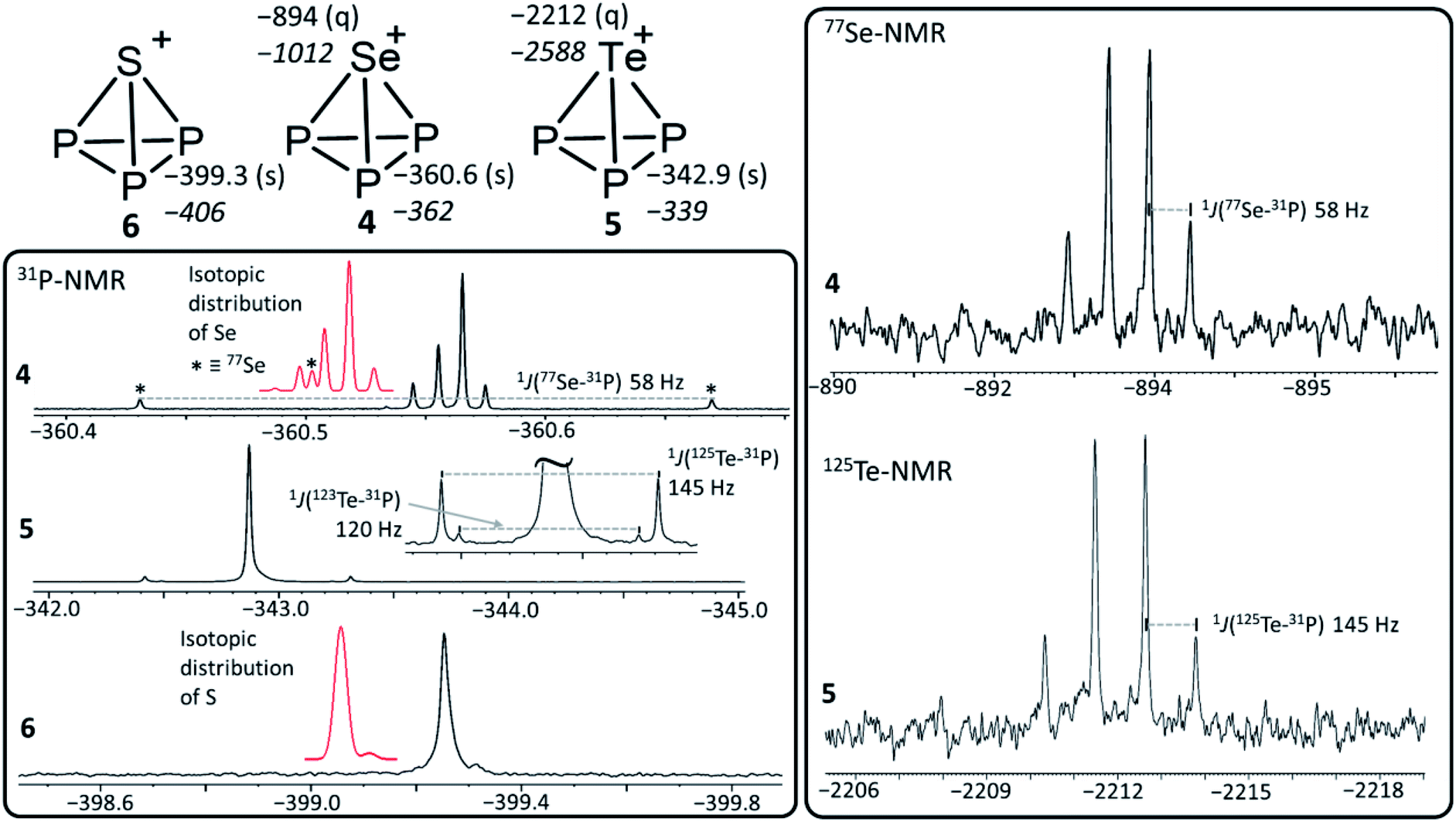 | ||
| Fig. 1 Summary of the experimental and calculated (in italics) NMR data of 4, 5 and 6 (top left) including a graphical representation of the structures of [EP3]+ (E = S, Se, Te). 77Se (top right) and 125Te (bottom right) spectra of 4 and 5 as well as the 31P spectra of 4, 5 and 6 (bottom left). The red traces in the 31P-NMR box indicate the isotopic distributions of Se and S calculated with a standard mass spectrometry program. Of all noticeable S and Se isotopes, only 77Se is NMR active (33S has an abundancy of only 0.7%). Thus, only the NMR signal of [77Se31P3]+, labelled by an asterisk in the isotopic distribution, is visibly splitted to a doublet in the NMR trace of 4. Apparently, the 31P-chemical shift of [EP3]+ follows the weight of the isotopes for the lighter elements S and Se, with higher isotope weights leading to incrementally slightly higher field shifted 31P-NMR resonances. However, for Te, where the relative weight differences between isotopomers are smallest among the three cations, the high-field shift in the 31P-NMR is so small that the signals due to all Te-isotopes fall onto one visible line (with the exception of the two NMR active nuclei 123Te and 125Te that couple and give doublets). | ||
Multiple efforts testing both, the [Al(ORF)4]− and the [F(Al(ORF)3)2]− anion, to crystallize the obtained product ions 4, 5 and 6 as pure salts from the complex reaction mixtures were unsuccessful in many solvents. Since the crystal structure of 1c did show no superstructure and no disorder, as contrasted by the other [SeCl3]+ salts, 1c was tested for the synthesis of 4. This was also futile (see ESI Section 3.4.2†): the [FAl(ORF)3]− anion is not able to withstand the fluorophilic nature of phosphorus cations, and decomposes with formation of several P–F bond containing compounds in the 31P-NMR (PF4(ORF), dynamic PF3 complexes, unknown PF2X, PFX2).
Towards optimized reaction conditions. The ratio of [EP3]+ to [P5Cl2]+ in the reaction is highest for [SeCl3]+ amongst the [ECl3]+ starting materials. Equal amounts of both cations were obtained in the reaction with [SCl3]+ and with [TeCl3]+. Keeping the reaction temperature at −40 °C leads to [SeP3]+ being around 48% (compared to 28% with the starting conditions) of the total intensity of the cation signals in the 31P spectrum (23% including unremoved PCl3, see ESI Table S7†) and increases the ratio [SeP3]+
:[P5Cl2]+ from 1.1 to 1.9:1 (Fig. 2).32 Slow anion decomposition was observed in the ambient temperature 19F-NMR spectra. This can be avoided by keeping the reaction at −40 °C. Screenings of the reaction in different solvents did show no significant improvement for the selectivity of the reaction and led to further side reactions (see ESI Section 3.4.1†). In agreement with this, the calculated Gibbs reaction energies in solvents of different relative dielectric permittivity (CH2Cl2, o-dfb, SO2) using the COSMO model revealed no significant solvent dependency (discussion below, Table 2). Experiments using CS2 to enhance the solubility of P4 have shown no formation of 4 at all. Raman spectra obtained from reactions on a small scale have only shown fluorescence. Only bulk material obtained from experiments on a larger scale did yield Raman spectra. The vibrational bands can be assigned to the [Al(ORF)4]− anion and tentatively to the overlapping vibrational bands of the [SeP3]+, [P5Cl2]+ and [P3Se4]+ cations, as well as some Se2Cl2 showing its most intense band33 at 361 cm−1 (ESI, Section 3.4.3.2†). An exact assignment of all vibrational bands is not possible due to the unavoidable occurrence of [P5Cl2]+ having strong overlaps with the predicted bands of [P3Se]+. Electro-spray-MS spectra of solutions containing 4 showed the presence of the [SeP3]+ (m/z = 172.83) and the [P5Cl2]+ (m/z = 225.0) cation (ESI, Section 3.4.3.2†).
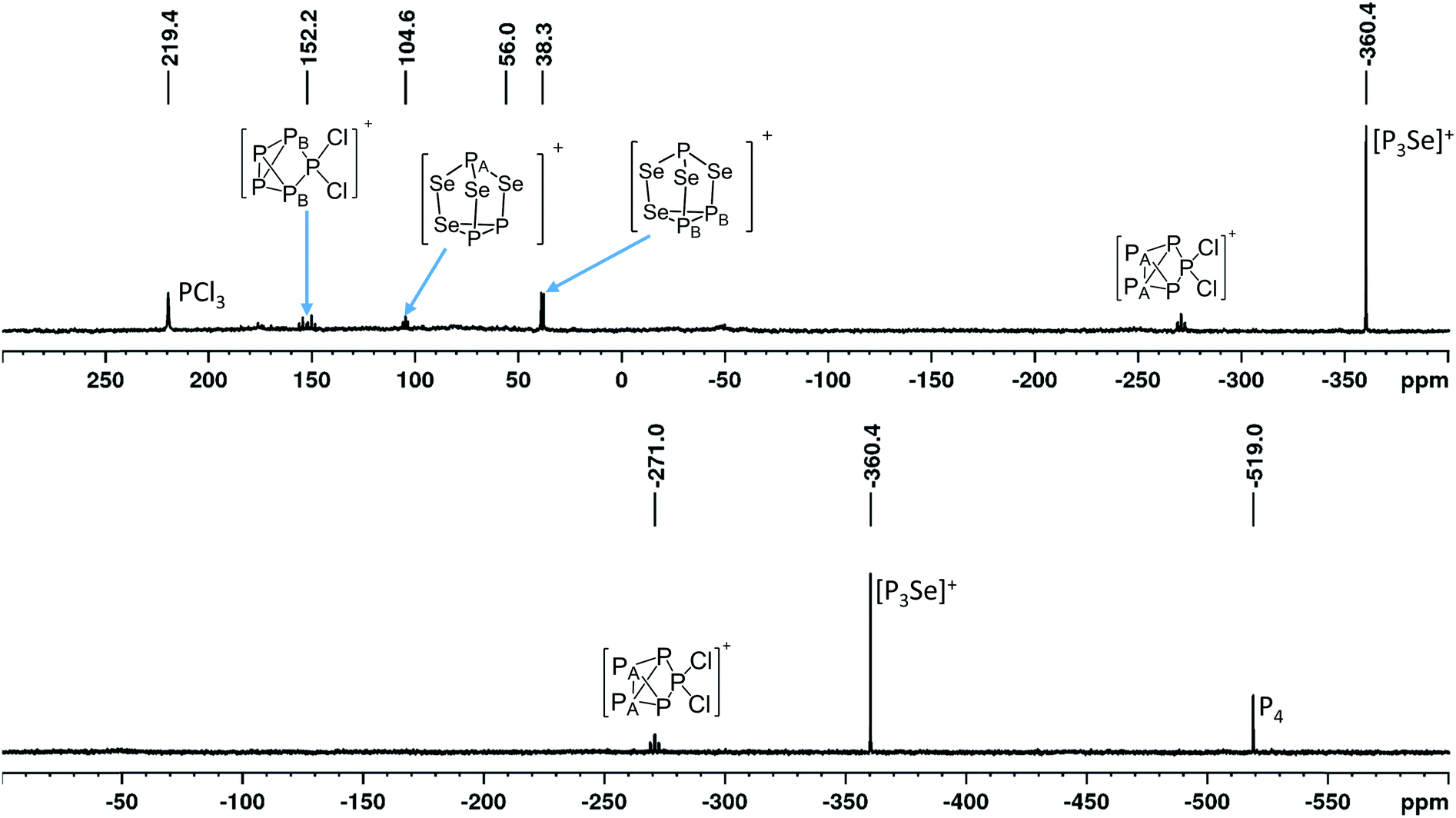 | ||
| Fig. 2 Product ion mixture obtained from the larger scale reaction of 1a with P4 at −40 °C. Combined views of the measured range of the 31P-NMR spectra (81.0 MHz, CD2Cl2, RT) from 300 to −400 ppm (top) and 0 to −600 ppm (bottom). | ||
| Compound | δ(31P)a | NICSb | NICS/eb | d(P–E) [pm] | d(P–P) [pm] | δ(E)exp.c | 1 J(P, E) |
|---|---|---|---|---|---|---|---|
| a 31P chemical shifts were referenced to the chemical shift of P4 in CH2Cl2. b NICS values are calculated at the cage center. NICS/e denotes the NICS per valence electron. c 77Se and 125Te chemical shifts were referenced against the chemical shifts of SePMe3/TePMe3 more details on the referencing of the chemical shifts is given in the ESI in Section 4.2. | |||||||
| P4 | −520 −520 | −61.9 | −3.10 | — | 219.94(3)37219.3 | — | — |
| AsP3 | −484 (ref. 2) −484 | −61.3 | −3.07 | 230.4(1)37230.4 | 219.5(3)37219.4 | — | — |
| SbP3 | −462 (ref. 2) −452 | −58.6 | −2.93 | 250.0 | 219.8 | — | — |
| [SP3]+ | −399 −406 | −60.6 | −3.03 | 214.8 | 220.2 | — | — |
| [SeP3]+ | −361 −362 | −60.3 | −3.02 | 228.1 | 220.5 | −894 −1012 | 1 J(31P, 77Se) = 58 Hz |
| [TeP3]+ | −342 −339 | −59.4 | −2.97 | 246.9 | 220.9 | −2212 −2588 | 1 J(31P, 125Te) = 145 Hz, 1J(31P, 123Te) = 120 Hz |
| Reaction | E = | ΔrH298 (g) | ΔrG298 (g) | ΔrG298 (CH2Cl2)b | ΔrG298 (o-dfb)b | ΔrG298 (SO2)b | Cation | FIA | E = |
|---|---|---|---|---|---|---|---|---|---|
| a Efforts to synthesize [SI3]+ previously yielded [S2I4]2+ instead.43 b The free reaction energies in solvents were calculated at the (RIJ)BP86/def2-TZVPP level using the COSMO model. | |||||||||
| P4 + [EF3]+ → [EP3]+ + PF3 | S | −550 | −554 | −477 | −475 | −474 | [EF3]+ | 909 | S |
| (Se) | (−560) | (−563) | (−468) | (−464) | (−463) | (941) | (Se) | ||
| [Te] | [−407] | [−408] | [−319] | [−315] | [−314] | [962] | [Te] | ||
| P4 + [ECl3]+ → [EP3]+ + PCl3 | S | −339 | −343 | −324 | −325 | −325 | [ECl3]+ | 775 | S |
| (Se) | (−291) | (−295) | (−269) | (−268) | (−268) | (803) | (Se) | ||
| [Te] | [−167] | [−170] | [−143] | [−143] | [−142] | [844] | [Te] | ||
| P4 + [EBr3]+ → [EP3]+ + PBr3 | S | −263 | −267 | −271 | −272 | −273 | [EBr3]+ | 735 | S |
| (Se) | (−206) | (−210) | (−209) | (−210) | (−210) | (762) | (Se) | ||
| [Te] | [−97] | [−99] | [−96] | [−97] | [−97] | [805] | [Te] | ||
| P4 + [EI3]+ → [EP3]+ + PI3 | S | −165 | −168 | −184 | −186 | −187 | [EI3]+a | 675a | S |
| (Se) | (−115) | (−118) | (−127) | (−129) | (−129) | (704) | (Se) | ||
| [Te] | [−29] | [−32] | [−39] | [−41] | [−41] | [753] | [Te] | ||
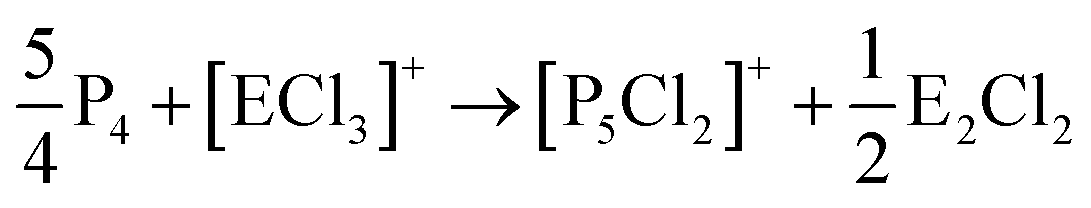
|
S | −297 | −267 | −227 | −225 | −225 | [EP3]+ (F bound via P) | 828 | S |
| (Se) | (−256) | (−226) | (−182) | (−181) | (−180) | (819) | (Se) | ||
| [Te] | [−152] | [−121] | [−84] | [−82] | [−82] | [798] | [Te] | ||
On the occurrence of [P5Cl2]+. The formation of 4, 5 and 6 is necessarily accompanied by the formation of PCl3 (eqn (3)). From NMR investigations it became clear that the [EP3]+ cations are unreactive towards PCl3. On the other hand, the formation of [P5L2]+ (L = organic residue or halide) cations is typically ascribed to the insertion of an intermediate carbene-like [PL2]+ into P4.1,5,27,34 However, this intermediate [PCl2]+ was never observed in the condensed phase, but assumed to be a complex [(S)Ag–Cl–PCl2]+ (S = solvent) delivering [PCl2]+in situ.35 To completely rule out the [P5Cl2]+ presence as a byproduct ion resulting from the reaction of residual Ag+ with PCl3 and P4, ion 4 was synthesized from a silver-free SeCl3[Al(ORF)4] prepared from SeCl4via the thallium salt36 Tl[Al(ORF)4] according to eqn (1). Yet, these experiments revealed a similar quantity of [P5Cl2]+ as side product ion. Thus, the formation of [P5Cl2]+ does not occur due to silver impurities of the starting material and may arise by reaction of P4 with [ECl3]+ as in eqn (4).
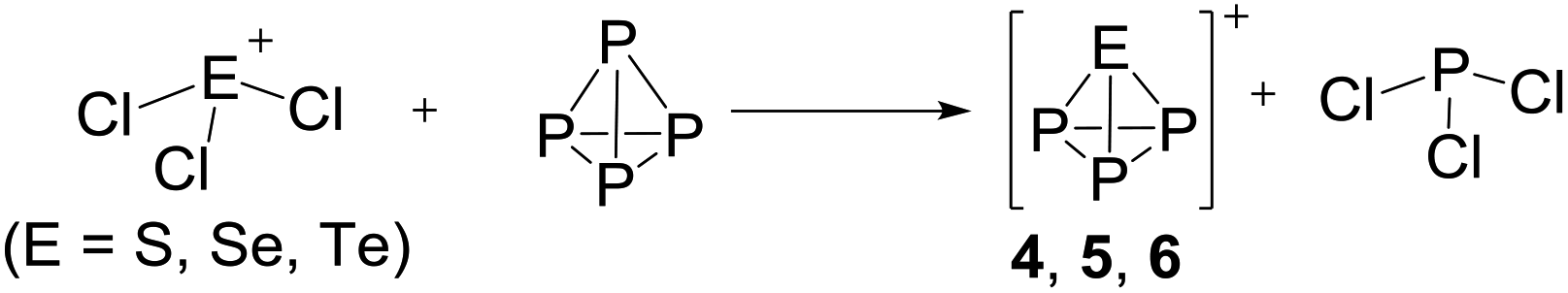 | (3) |
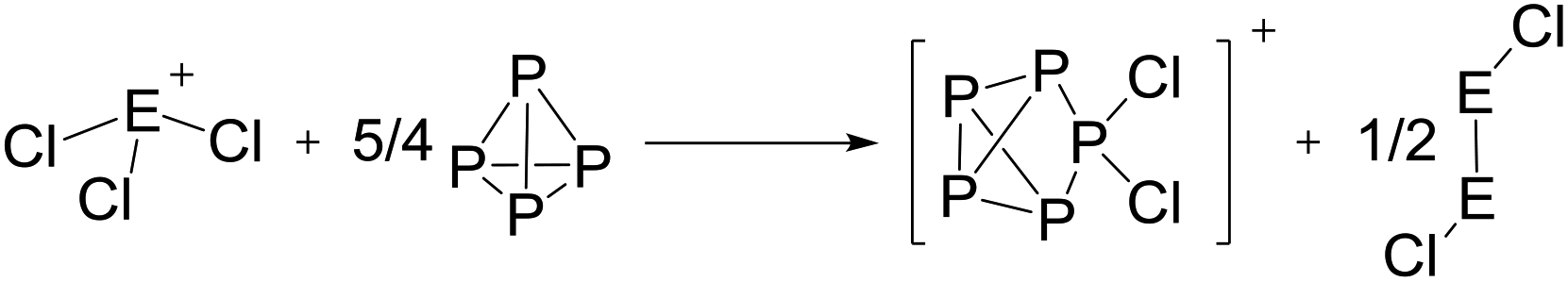 | (4) |
The reaction course according to eqn (4) is further supported by the observation of a yellow volatile material (Se2Cl2) present in the traps of the vacuum line after pumping on the mixtures as well as the Se2Cl2 Raman band observed in solid product mixtures at 361 cm−1 (ESI, Section 3.1†). Efforts to suppress this side reaction by slowly adding P4 to the reaction at low temperatures showed no significant improvement towards a clean reaction (ESI, Table S7†).
Further side product ions. The known20 [P3Se4]+ additionally occurs as a side product ion. Performing the reaction at −40 °C lowers the amount of obtained [P3Se4]+, but does not allow to completely get rid of this side product ion. Depending on the solvent, the higher chlorinated oxidized [PCl4]+ was also observed (ESI, Section 3.4.1†). An unknown side product/product ion including five chemically equivalent phosphorus atoms and three chemically equivalent tellurium atoms occurs during the synthesis of 5. Analogously, in the synthesis of 6, side-products of unknown origin occurred (ESI, Section 3.4.3†).
Quantum chemical calculations on [EP3]+
Since it proved impossible to crystallize any [EP3]+ salt, we turned to quantum chemical calculations, to shed some light on energetics, structure, properties and bonding.Molecular structures. The molecular structures of the title tetrahedra were calculated at the (RIJ)MP2/def2-QZVPP level, which gives very good agreement with experimental data for P4 and PnP3 (Pn = As, Sb). Apparently, the calculated [EP3]+ bond lengths dPP are slightly elongated, if compared to the neutral tetrahedra and increase from S to Se to Te.
Calculated NMR-shifts. The calculated NMR-shifts of 4, 5 and 6, as well as that of the neutral molecules AsP3 and SbP3 for comparison, were obtained using a fully decontracted basis set with (RIJ)B3-LYP(DKH2)/cc-pVQZ single point calculations, using the (RIJ)MP2/def2-QZVPP structures. They were referenced against P4 (Table 1). The calculated 31P chemical shifts of all [EP3]+ ions deviate by less than 7 ppm from the experimental values. This is similar to the deviation of the calculated NMR shifts from the known AsP3 and SbP3 shifts (less than 10 ppm).2 However, the calculated 77Se and 125Te chemical shifts show larger deviations from the experimental values. Yet, the trends in chemical shifts are well reproduced, as investigated on a larger test set of Se- and or Te-containing molecules spanning a range of 2300 ppm (Se) and even 5000 ppm (Te) (ESI, Section 4.2†). With such a broad range of chemical shifts, the choice of the reference compound is crucial, although the calculated absolute shielding tensors remain the same. Yet, no truly close relative to [EP3]+ is known experimentally to serve as a reference to tie to the calculated shift. The best compromise was to use EPMe3 (E = Se, Te). We note, that the observed resonances of 4 and 5 underlie an extreme high-field shift to untypical regions for inorganic Se(IV) and Te(IV) compounds compared to their [ECl3]+ counterparts. This raises the question, whether the chalcogen atoms in the [EP3]+ cations can be formally rationalized as deriving from an E4+ cation being exposed to the ring current of a 6π Hückel-aromatic [P3]3− ring or if the chalcogen atoms exhibit a lower formal oxidation state (see next section).
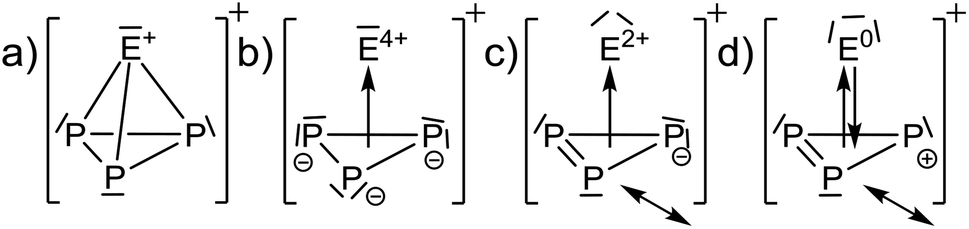 | ||
| Scheme 2 Extreme, limiting sketches that might contribute to the [EP3]+ structures. (a) Formally electron precise structure, (b) cluster bonding as described by π-donation from a 6π Hückel-aromatic P33− unit. (c) Similar, but π-donation from a 4π antiaromatic P3− and (d) synergistic π-donation from a 2π Hückel-aromatic P3+ unit to an E0 and back bonding from p-type AOs of E0 to π*-MOs of P3+. | ||
Thus, with the cluster-like description as in Scheme 2b and c, one would expect that the E atoms experience a large high-field shift due to the proximity to an aromatic ring current. Thus, it first needed to be investigated, if diamagnetic ring currents are present.
NICS values. Calculated nuclear independent chemical shift (NICS) values at the cage centers of the [EP3]+ cations suggest pronounced diamagnetic ring currents with a very slightly decreased order of magnitude, if compared with P4 and AsP3 (Table 1). This suggests a delocalized cluster-like bonding.
Population analyses and MO diagram. Calculated NBO, PABOON and AIM charges (Fig. 3) only agree that the positive charge on the E atom of the [EP3]+ cluster increases in going from S to Se and Te and that on P decreases in the same direction. The similarity of the calculated MO diagrams to the known7 MO diagrams of P4 and AsP3 clearly shows that the [EP3]+ cations can be understood as clusters related to AsP3 and P4. This is in accordance with the generally at very high field observed NMR shifts as well as the NICS values in Table 1, and similar to the known tetrahedra P4, AsP3, Si44− and Sn44−.2,14 However, closer inspection reveals some differences. First, due to the cationic nature, all orbital energies are lowered by 6 to 7 eV in comparison to the neutral tetrahedra. Second, the lowering of the symmetry from Td to C3v in AsP3 splits the t2-MOs of P4 giving a pair of e and a1 MOs. The sequence of orbital energies for this pair of e (higher E) and a1 MOs (lower E) in AsP3 matches those in [EP3]+ only for Se and Te. For the [SP3]+ system, these orbitals are reversed in energy and the a1 MO is now higher in energy than the e MO (Fig. 3).
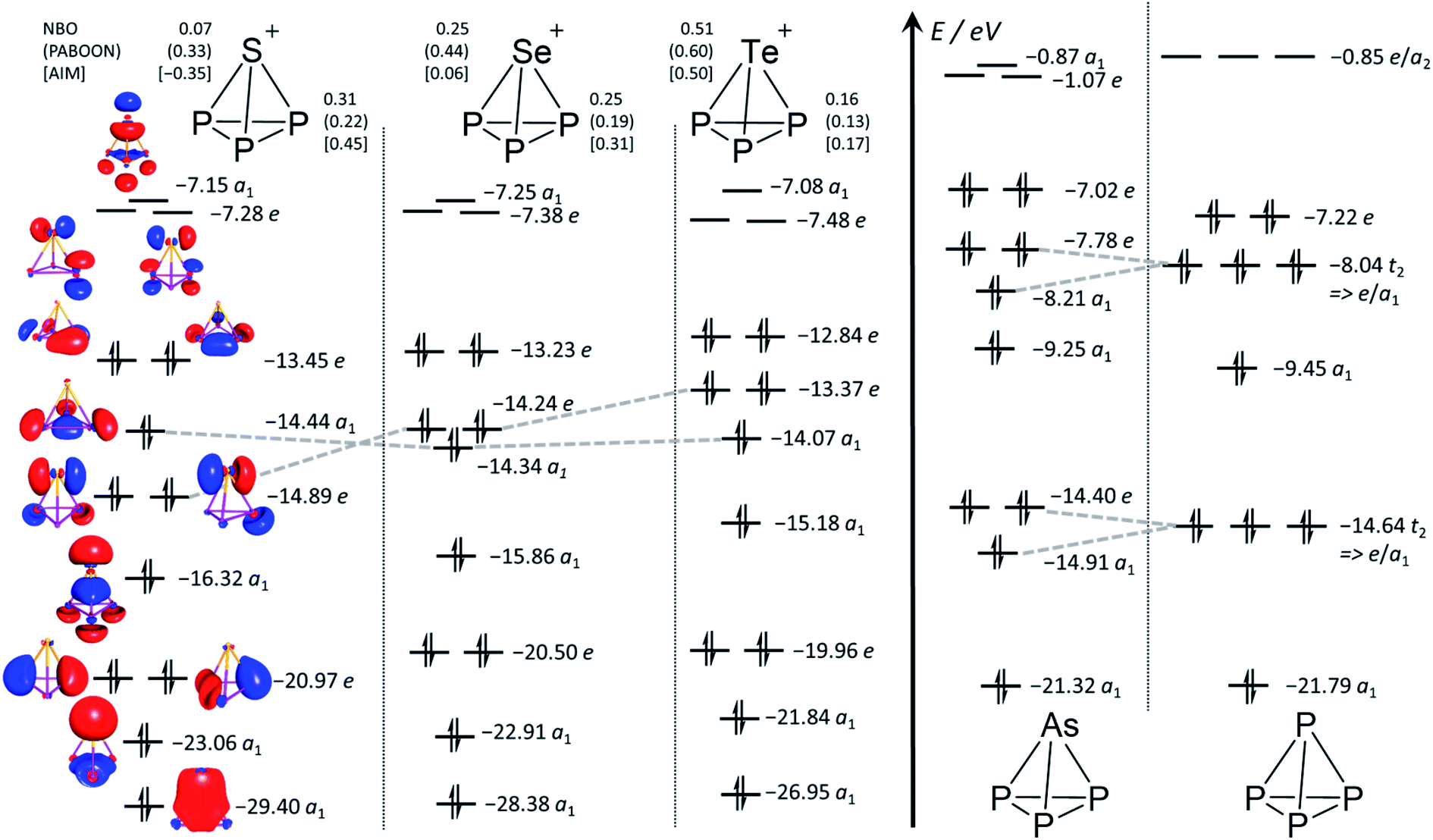 | ||
| Fig. 3 Calculated MO Diagrams and NBO,38 PABOON and AIM39 charges of [EP3]+ ((RIJ)B3-LYP/def2-TZVPP). The corresponding MO diagrams of P4 and AsP3 were also generated and are in accordance with the previously published MO diagrams (cf. also ESI Section 4.1†).7 The grey broken lines delineate the switch of the a1 and e MO energies in going from E = S to Se and Te as well as the splitting of the t2 MOs in P4 upon symmetry lowering in AsP3. | ||
The HOMO LUMO gaps decrease in the order [SP3]+ (6.2 eV) > [SeP3]+ (5.9 eV) > [TeP3]+ (5.4 eV) and are comparable to P4 and AsP3 (6.4 eV and 6.0 eV with the same method). This speaks for a stable structure and bonding within [EP3]+. Closer inspection of the MOs in view of the descriptions in Scheme 2 would be in agreement with the suggestion that the back bonding component from p-type AOs of E0 to π*-MOs of P3+ (Scheme 2d), which might be assigned to the e MO at −14.89 (S)/−14.24 (Se)/−13.37 (Te) eV in Fig. 3, is inferior for the heavier elements. Thus, apparently the sizes of the heavier p-type AOs do not fit similarly well as that of sulfur and the respective MO energies increase from S to Te.
Further AIM analyses. Contour plots of the Laplacian of the electron density (Section 4.2, ESI†) show a similar situation for all [EP3]+ cations. They are closely related to the neutral molecules AsP3 and P4, with [P3S]+ being similar to P4 and [P3Se]+ and [P3Te]+ being more similar to AsP3. Exohedral lone pairs of the clusters are absent for any of the tetrahedra, which confirms the nature of the tetrahedra as delocalized clusters rather than electron precise molecules. The electron densities (ρXCP) residing at the critical points XCP (X = R (ring), C (cluster), B (bond)) of the [EP3]+ cations have similar values to P4 and AsP3 and are included with the ESI.† It is worth noting that the calculated increasing bond ellipticities for E = Se and Te would be in agreement with a description as in Scheme 2d and suggest a larger electronic separation of the ‘E’- and ‘P3’-parts for the heavier elements and its interpretation as resulting from π-interactions.
[SF3]+ with a FIA of 915 kJ mol−1, similar to the one of [CCl3]+ (904 kJ mol−1)44 that is stable as [Al(ORF)4]− salt and in solution at temperatures of −20 °C,45 might be accessible with the aluminate counterions. Therefore, we have tried to prepare the salt SF3[Al(ORF)4], but all efforts failed and no [EF3]+ salts could be tested. However, this might be a reaction path that could be pursued in future; given the use of a suitable WCA tolerant towards extremely Lewis acidic cations – for example a halogenated carborane anion. As to whether the strongly exergonic Gibbs energy for the formation of the [EP3]+ cations from [EF3]+ salts and P4 would guarantee a selective reaction, remains to be tested. However, we do not expect that fluorometallates [MF6]− (M = P, As, Sb), with considerably lower FIA values of MF5 (389 to 495 kJ mol−1) than Al(ORF)3 (543 kJ mol−1),41 would stabilize salts of [EP3]+, but rather form fluoro-phosphorus compounds and MF5.
Conclusion
Investigations on the existence of tetrahedral [EP3]+ (E = S, Se, Te) in condensed phases required the here reported synthesis and characterization of well-soluble salts ECl3[WCA]. We note, that these cations are very strong Lewis acids with FIA values of 775 (S); 803 (Se) and 844 (Te) kJ mol−1 that could be relevant in view of the renewed interest46 in strong47 (also cationic48) Lewis acids.49 They should be compared to classical phosphenium ions [PR2]+ with FIA values of 721 (R = NMe2), 789 (R = Mes) and 828 (R = Ph).50 The easily and in high yield accessible salts ECl3[WCA] were tested for their reaction with the known P33− precursor Na[Nb(P3)(ODipp)3], related to the synthesis of AsP3 from AsCl3. Only a clean generation of P4 occurred. This is due to the strongly chlorinating nature of the [ECl3]+ cations that immediately reacted with the niobate to give PCl3. Subsequently, the PCl3 was transformed by the P3-niobate to P4. Contrary, the reaction of the salts ECl3[WCA] with P4, led to reaction mixtures containing mainly the [EP3]+ cations, PCl3 and [P5Cl2]+ as well as other byproducts/byproduct ions. Even under optimized reaction conditions, the clean formation of [EP3]+ and PCl3 from [ECl3]+ and P4 is rivaled by side reactions. High level quantum chemical calculations show that the formation of [EP3]+ and of [P5Cl2]+ are both strongly exergonic and independent of the solvent choice. Apparently, the similar activation energies of the reactions prevent the separation of both pathways by all tested reaction conditions. Experimental 31P-, 77Se- and 125Te-NMR spectra as well as calculations of the chemical shifts prove the presence of the first cationic tetrahedra in the condensed phase. The selenium analogue [SeP3]+ was also unambiguously detected by ESI-MS of the obtained samples, while IR- and Raman spectra show the expected mixtures of cations in agreement with the optimized NMR conditions. The [EP3]+ ions are the first tetrahedral cations stabilized in the condensed phase, contrasted by the many anionic and the long known neutral tetrahedra. These cationic tetrahedra are also the first clear evidence for binary P–S and P–Te cations in the condensed phase, with the 2015 published [P3Se4]+ being the first phosphorus chalcogen cation at all. Theoretical investigations show, that the [EP3]+ tetrahedra can be understood as clusters similar to P4 and AsP3 as indicated by their MO diagrams, AIM analyses and calculated NICS values. The unusually high-field shifted 77Se- and 125Te-NMR shifts may arise from a synergistic π-interaction between formal E0 and P3+ fragments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg and by the DFG in the Normalverfahren. We would like to thank Fadime Bitgül and Dr Arthur Martens for the measurement of NMR spectra and Dr Daniel Himmel for valuable discussions concerning the DFT and ab initio calculations.References
- J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295 CrossRef CAS PubMed.
- B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS PubMed.
- (a) L. C. Forfar, T. J. Clark, M. Green, S. M. Mansell, C. A. Russell, R. A. Sanguramath and J. M. Slattery, Chem. Commun., 2012, 48, 1970 RSC; (b) S. Heinl, E. V. Peresypkina, A. Y. Timoshkin, P. Mastrorilli, V. Gallo and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 10887 CrossRef CAS PubMed; (c) F. Hennersdorf, J. Frötschel and J. J. Weigand, J. Am. Chem. Soc., 2017, 139, 14592 CrossRef CAS PubMed; (d) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 CrossRef CAS PubMed; (e) M. Ichinohe, M. Toyoshima, R. Kinjo and A. Sekiguchi, J. Am. Chem. Soc., 2003, 125, 13328 CrossRef CAS PubMed; (f) I. Krossing and L. van Wüllen, Chem.–Eur. J., 2002, 8, 700 CrossRef CAS PubMed; (g) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (h) T. Mennekes, P. Paetzold, R. Boese and D. Bläser, Angew. Chem., Int. Ed., 1991, 30, 173 CrossRef; (i) F. Meyer-Wegner, S. Scholz, I. Sänger, F. Schödel, M. Bolte, M. Wagner and H.-W. Lerner, Organometallics, 2009, 28, 6835 CrossRef CAS; (j) S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend and S. Dehnen, Nat. Commun., 2016, 7, 10480 CrossRef CAS PubMed; (k) C. Schwarzmaier, A. Schindler, C. Heindl, S. Scheuermayer, E. V. Peresypkina, A. V. Virovets, M. Neumeier, R. Gschwind and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 10896 CrossRef CAS PubMed; (l) F. Spitzer, M. Sierka, M. Latronico, P. Mastrorilli, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 4392 CrossRef CAS PubMed; (m) M. Waibel and T. F. Fässler, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 732 CAS; (n) J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2014, 53, 12836 CrossRef CAS PubMed; (o) I. de los Rios, J.-R. Hamon, P. Hamon, C. Lapinte, L. Toupet, A. Romerosa and M. Peruzzini, Angew. Chem., Int. Ed., 2001, 40, 3910 CrossRef CAS.
- C. Schwarzmaier, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 858 CrossRef CAS PubMed.
- M. H. Holthausen, K.-O. Feldmann, S. Schulz, A. Hepp and J. J. Weigand, Inorg. Chem., 2012, 51, 3374 CrossRef CAS PubMed.
- (a) J. P. Zheng, J. Waluk, J. Spanget-Larsen, D. M. Blake and J. G. Radziszewski, Chem. Phys. Lett., 2000, 328, 227 CrossRef CAS; (b) P. Jerabek and G. Frenking, Theor. Chem. Acc., 2014, 133, 1447 Search PubMed; (c) E. E. Rennie and P. M. Mayer, J. Chem. Phys., 2004, 120, 10561 CrossRef CAS PubMed.
- B. M. Cossairt and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 15501 CrossRef CAS PubMed.
- A. E. Seitz, F. Hippauf, W. Kremer, S. Kaskel and M. Scheer, Nat. Commun., 2018, 9, 361 CrossRef PubMed.
- T. A. Engesser, W. J. Transue, P. Weis, C. C. Cummins and I. Krossing, Eur. J. Inorg. Chem., 2019, 2019, 2607 CrossRef CAS.
- A. Wiesner, S. Steinhauer, H. Beckers, C. Müller and S. Riedel, Chem. Sci., 2018, 9, 7169 RSC.
- (a) M. Baitinger, Y. Grin, R. Kniep and H. G. von Schnering, Z. Kristallogr., 1999, 214 Search PubMed; (b) M. Baitinger, K. Peters, M. Somer, W. Carrillo-Cabrera, Y. Grin, R. Kniep and H. G. von Schnering, Z. Kristallogr., 1999, 214 Search PubMed; (c) A. Betz, H. Schäfer, A. Weiss and R. Wulf, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1968, 23, 878 CAS; (d) S. Bobev and S. C. Sevov, Polyhedron, 2002, 21, 641 CrossRef CAS; (e) E. Busmann, Z. Anorg. Allg. Chem., 1961, 313, 90 CrossRef CAS; (f) J. Curda, W. Carrillo-Cabrera, A. Schmeding, K. Peters, M. Somer and H. G. von Schnering, Z. Anorg. Allg. Chem., 1997, 623, 929 CrossRef CAS; (g) I. F. Hewaidy, E. Busmann and W. Klemm, Z. Anorg. Allg. Chem., 1964, 328, 283 CrossRef CAS; (h) C. Hoch and C. Röhr, Z. Anorg. Allg. Chem., 2002, 628, 1541 CrossRef CAS; (i) C. Hoch, M. Wendorff and C. Röhr, Z. Anorg. Allg. Chem., 2003, 629, 2391 CrossRef CAS; (j) C. Hoch, M. Wendorff and C. Röhr, J. Alloys Compd., 2003, 361, 206 CrossRef CAS; (k) K. H. Janzon, H. Schäfer and A. Weiss, Z. Anorg. Allg. Chem., 1970, 372, 87 CrossRef CAS; (l) J. Llanos, R. Nesper and H. G. von Schnering, Angew. Chem., 1983, 95, 1026 CrossRef CAS; (m) R. E. Marsh and D. P. Shoemaker, Acta Crystallogr., 1953, 6, 197 CrossRef CAS; (n) V. Quéneau, E. Todorov and S. C. Sevov, J. Am. Chem. Soc., 1998, 120, 3263 CrossRef; (o) C. Röhr, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 802 Search PubMed; (p) R. Schäfer and W. Klemm, Z. Anorg. Allg. Chem., 1961, 312, 214 CrossRef; (q) H. G. von Schnering, M. Baitinger, U. Bolle, W. Carrillo-Cabrera, J. Curda, Y. Grin, F. Heinemann, J. Llanos, K. Peters, A. Schmeding and M. Somer, Z. Anorg. Allg. Chem., 1997, 623, 1037 CrossRef CAS; (r) H. G. von Schnering, M. Schwarz and R. Nesper, Angew. Chem., Int. Ed., 1986, 25, 566 CrossRef; (s) J. Witte, H. G. Schnering and W. Klemm, Z. Anorg. Allg. Chem., 1964, 327, 260 CrossRef CAS; (t) G. Cordier and W. Blase, Z. Kristallogr., 1991, 196, 207 CrossRef.
- (a) R. Ababei, J. Heine, M. Hołyńska, G. Thiele, B. Weinert, X. Xie, F. Weigend and S. Dehnen, Chem. Commun., 2012, 48, 11295 RSC; (b) S. C. Critchlow and J. D. Corbett, Inorg. Chem., 1982, 21, 3286 CrossRef CAS; (c) S. C. Critchlow and J. D. Corbett, Inorg. Chem., 1985, 24, 979 CrossRef CAS; (d) F. Lips, M. Raupach, W. Massa and S. Dehnen, Z. Anorg. Allg. Chem., 2011, 637, 859 CrossRef CAS; (e) F. Lips, I. Schellenberg, R. Pöttgen and S. Dehnen, Chem.–Eur. J., 2009, 15, 12968 CrossRef CAS PubMed; (f) S. Mitzinger, J. Bandemehr, K. Reiter, J. Scott McIndoe, X. Xie, F. Weigend, J. F. Corrigan and S. Dehnen, Chem. Commun., 2018, 54, 1421 RSC; (g) L. Xu and S. C. Sevov, Inorg. Chem., 2000, 39, 5383 CrossRef CAS PubMed; (h) U. Friedrich, M. Neumeier, C. Koch and N. Korber, Chem. Commun., 2012, 48, 10544 RSC.
- (a) C. B. Benda, T. Henneberger, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2017, 643, 146 CrossRef CAS; (b) F. Hastreiter, C. Lorenz, J. Hioe, S. Gärtner, N. Lokesh, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2019, 3133–3137 CrossRef CAS PubMed; (c) C. Lorenz, S. Gärtner and N. Korber, Z. Anorg. Allg. Chem., 2017, 643, 141 CrossRef CAS.
- M. Neumeier, F. Fendt, S. Gärtner, C. Koch, T. Gärtner, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2013, 52, 4483 CrossRef CAS PubMed.
- (a) G. J. Penney and G. M. Sheldrick, J. Chem. Soc. A, 1971, 243 RSC; (b) L. Operti, G. A. Vaglio, M. Peruzzini and P. Stoppioni, Inorg. Chim. Acta, 1985, 96, 43 CrossRef CAS.
- D. A. Northrop, Mater. Res. Bull., 1972, 7, 147 CrossRef CAS.
- (a) M. Di Vaira, F. Mani, S. Moneti, M. Peruzzini, L. Sacconi and P. Stoppioni, Inorg. Chem., 1985, 24, 2230 CrossRef CAS; (b) M. Di Vaira, M. Peruzzini and P. Stoppioni, J. Chem. Soc., Chem. Commun., 1982, 894 RSC; (c) M. Di Vaira, M. Peruzzini and P. Stoppioni, Polyhedron, 1986, 5, 945 CrossRef CAS.
- (a) M. F. Groh, A. Wolff, M. A. Grasser and M. Ruck, Int. J. Mol. Sci., 2016, 17 Search PubMed; (b) T. Chivers and I. Manners, Inorganic rings and polymers of the p-block elements. From fundamentals to applications, Royal Society of Chemistry, Cambridge, 2009 Search PubMed; (c) J. Beck, S. Schlüter and N. Zotov, Z. Anorg. Allg. Chem., 2005, 631, 2450 CrossRef CAS; (d) A. Eich, S. Schlüter, G. Schnakenburg and J. Beck, Z. Anorg. Allg. Chem., 2013, 639, 375 CrossRef CAS; (e) J. Beck, S. Schlüter and N. Zotov, Z. Anorg. Allg. Chem., 2004, 630, 2512 CrossRef CAS; (f) J. Beck, M. Dolg and S. Schlüter, Angew. Chem., Int. Ed., 2001, 40, 2287 CrossRef CAS.
- B. H. Christian, R. J. Gillespie and J. F. Sawyer, Inorg. Chem., 1981, 20, 3410 CrossRef CAS.
- K.-O. Feldmann, T. Wiegand, J. Ren, H. Eckert, J. Breternitz, M. F. Groh, U. Müller, M. Ruck, B. Maryasin, C. Ochsenfeld, O. Schön, K. Karaghiosoff and J. J. Weigand, Chem.–Eur. J., 2015, 21, 9697 CrossRef CAS PubMed.
- M. Donath, F. Hennersdorf and J. J. Weigand, Chem. Soc. Rev., 2016, 45, 1145 RSC.
- T. M. Klapötke and C. M. Riencker, Z. Anorg. Allg. Chem., 1994, 620, 2104 CrossRef.
- A structural characterization was not possible due to its insolubility in SO2 and CFCl3. A monomeric [SeP]+ was disproven by vibrational spectra. Dissolution in HMPT, DMF, DMSO or MeOH led to spontaneous combustion and mechanical stress led towards explosion. Using other [SeCl3]+ salts such as SeCl3[SbCl6] or SeCl3[AsF6] failed due to the high affinity of phosphorus to fluorine or led to an even more sensitive material. Using solvents other than liquid PH3 failed due to the insolubility of SeCl3[AlCl4] in common organic solvents or the tendency to sidereactions due to the oxophilic nature of phosphorus in inorganic solvents such as SO2.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC.
- T. A. Engesser, P. Hrobárik, N. Trapp, P. Eiden, H. Scherer, M. Kaupp and I. Krossing, ChemPlusChem, 2012, 77, 643 CrossRef CAS.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 45, 789 Search PubMed.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem.–Eur. J., 2004, 10, 5041 CrossRef CAS PubMed.
- (a) I. Krossing and I. Raabe, Chem.–Eur. J., 2004, 10, 5017 CrossRef CAS PubMed; (b) A. Martens, M. Kreuzer, A. Ripp, M. Schneider, D. Himmel, H. Scherer and I. Krossing, Chem. Sci., 2019, 10, 2821 RSC.
- J. Bohnenberger, W. Feuerstein, D. Himmel, M. Daub, F. Breher and I. Krossing, Nat. Commun., 2019, 10, 624 CrossRef PubMed.
- Tl[Al(ORF)4] forms no complex with P4 which is beneficial for the removal of residual P4 (see ESI Section 3.5†).
- P. Weis, H. Scherer and I. Krossing, Z. Anorg. Allg. Chem., 2019, 645, 64 CrossRef CAS.
- The integral ratios are given in the ESI in Tables S7, S9 and S10.†.
- S. G. Frankiss, J. Mol. Struct., 1968, 2, 271 CrossRef CAS.
- (a) M. H. Holthausen and J. J. Weigand, Dalton Trans., 2016, 45, 1953 RSC; (b) M. H. Holthausen, A. Hepp and J. J. Weigand, Chem.–Eur. J., 2013, 19, 9895 CrossRef CAS PubMed; (c) M. H. Holthausen, C. Richter, A. Hepp and J. J. Weigand, Chem. Commun., 2010, 46, 6921 RSC; (d) M. H. Holthausen, C. Sala and J. J. Weigand, Eur. J. Inorg. Chem., 2016, 2016, 667 CrossRef CAS; (e) M. H. Holthausen and J. J. Weigand, J. Am. Chem. Soc., 2009, 131, 14210 CrossRef CAS PubMed; (f) M. H. Holthausen and J. J. Weigand, Z. Anorg. Allg. Chem., 2012, 638, 1103 CrossRef CAS; (g) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406 CrossRef CAS.
- (a) M. Gonsior, I. Krossing and E. Matern, Chem.–Eur. J., 2006, 12, 1703 CrossRef CAS PubMed; (b) N. Burford, C. A. Dyker and A. Decken, Angew. Chem., Int. Ed., 2005, 44, 2364 CrossRef CAS PubMed; (c) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404 CrossRef CAS PubMed.
- Reactions of Tl[Al(ORF)4] with PCl3 have shown that no halide abstraction occurs with Tl+ contrary to Ag+ (see ESI Section 3.5†).
- B. M. Cossairt, C. C. Cummins, A. R. Head, D. L. Lichtenberger, R. J. F. Berger, S. A. Hayes, N. W. Mitzel and G. Wu, J. Am. Chem. Soc., 2010, 132, 8459 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS.
- (a) S. C. Meier, A. Holz, A. Schmidt, D. Kratzert, D. Himmel and I. Krossing, Chem.–Eur. J., 2017, 23, 14658 CrossRef CAS PubMed; (b) M. M. Schwab, D. Himmel, S. Kacprzak, Z. Yassine, D. Kratzert, C. Felbek, S. Weber and I. Krossing, Eur. J. Inorg. Chem., 2019, 2019, 3309 CrossRef CAS; (c) S. C. Meier, A. Holz, J. Kulenkampff, A. Schmidt, D. Kratzert, D. Himmel, D. Schmitz, E.-W. Scheidt, W. Scherer, C. Bülow, M. Timm, R. Lindblad, S. T. Akin, V. Zamudio-Bayer, B. von Issendorff, M. A. Duncan, J. T. Lau and I. Krossing, Angew. Chem., Int. Ed. Engl., 2018, 57, 9310 CrossRef CAS PubMed.
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489 RSC.
- M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chemistry, 2014, 20, 1218 CrossRef CAS PubMed.
- S. Brownridge, T. S. Cameron, H. Du, C. Knapp, R. Koppe, J. Passmore, J. M. Rautiainen and H. Schnöckel, Inorg. Chem., 2005, 44, 1660 CrossRef CAS PubMed.
- I. Krossing, A. Bihlmeier, I. Raabe and N. Trapp, Angew. Chem., Int. Ed., 2003, 42, 1531 CrossRef CAS PubMed.
- A. J. Lehner, N. Trapp, H. Scherer and I. Krossing, Dalton Trans., 2011, 40, 1448 RSC.
- L. Greb, Chem.–Eur. J., 2018, 24, 17881 CrossRef CAS PubMed.
- J. F. Kögel, D. A. Sorokin, A. Khvorost, M. Scott, K. Harms, D. Himmel, I. Krossing and J. Sundermeyer, Chem. Sci., 2018, 9, 245 RSC.
- H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984 CrossRef CAS PubMed.
- A. R. Jupp, T. C. Johnstone and D. W. Stephan, Inorg. Chem., 2018, 57, 14764 CrossRef CAS PubMed.
- J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1860640, 1898379, 1898380, 1898381 and 1898382. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03915e |
| This journal is © The Royal Society of Chemistry 2019 |