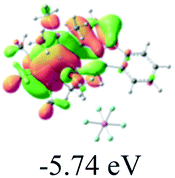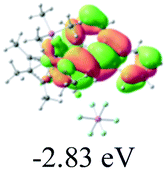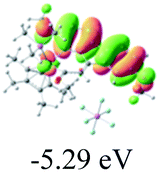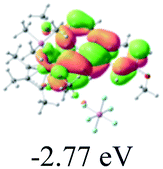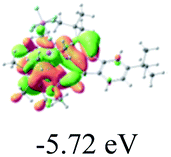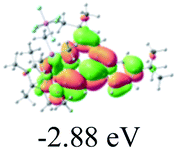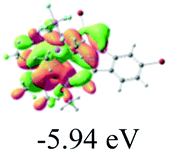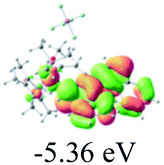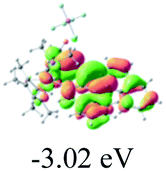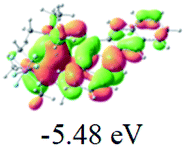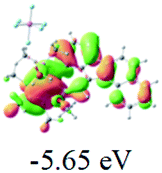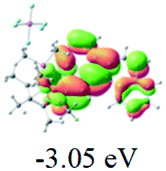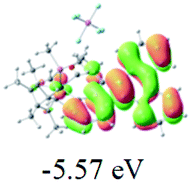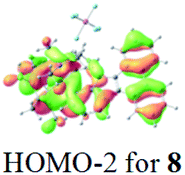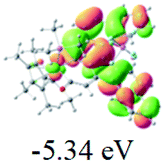DOI:
10.1039/C9SC03914G
(Edge Article)
Chem. Sci., 2019,
10, 10894-10899
One-pot syntheses of irida-polycyclic aromatic hydrocarbons†
Received
6th August 2019
, Accepted 12th October 2019
First published on 14th October 2019
Abstract
Metalla-analogues of polycyclic aromatic hydrocarbons (PAHs) have captivated chemists with their fascinating structures and unique electronic properties. To date, metallabenzene, metallanaphthalene and metallaanthracene have been reported. Metalla-analogues with more complicated fused rings have rarely been reported. Herein, we have successfully synthesized a series of new iridafluoranthenes and fused-ring iridafluoranthenes ranging from pentacyclic to heptacyclic metallaaromatic hydrocarbons in high yields under mild reaction conditions for the first time. Their photophysical and redox properties were also explored using UV-vis spectroscopy and electrochemistry combined with TD-DFT calculations. The present work may offer an important guideline for the design and construction of new polycyclic metallaaromatic hydrocarbons and metalla-nanographenes.
Introduction
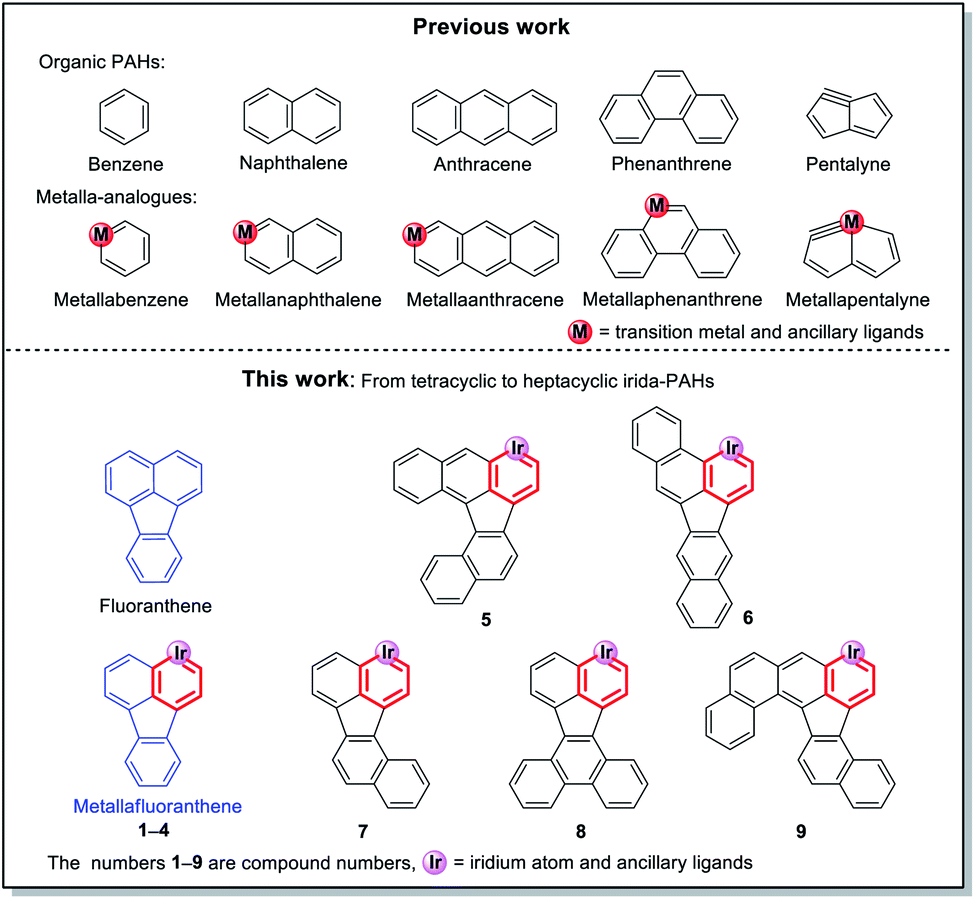
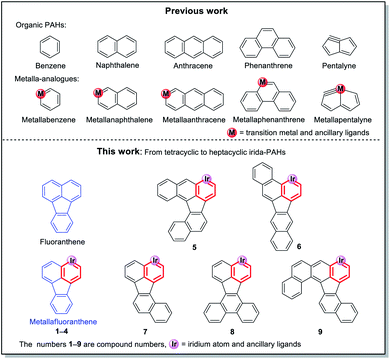
Polycyclic aromatic hydrocarbons (PAHs), as important components in the field of organic chemistry, have attracted a significant amount of attention due to their wide range of applications in organic light-emitting diodes,1 field-effect transistors2 and organic photovoltaic cells.3 Since the first metallabenzene (Scheme 1) was reported by Roper et al. in 1982,4 scientists have devoted enormous efforts to developing transition metal-containing metalla-aromatics.5 However, only very limited systems, including metallanaphthalene,6 metallaanthracene,7 metallaphenanthrenes,8 metallabenzynes,9 metallanaphthalynes,10 metallaanthracynes, metallaphenanthrynes11 and metallapentalenes,12 have been reported over the past few decades. Moreover, metalla-analogues of PAHs containing multiple fused rings have rarely been reported until now. Among these developed systems, only a handful of examples involve irida-PAHs.
 |
| | Scheme 1 Organic PAHs and metalla-analogues. | |
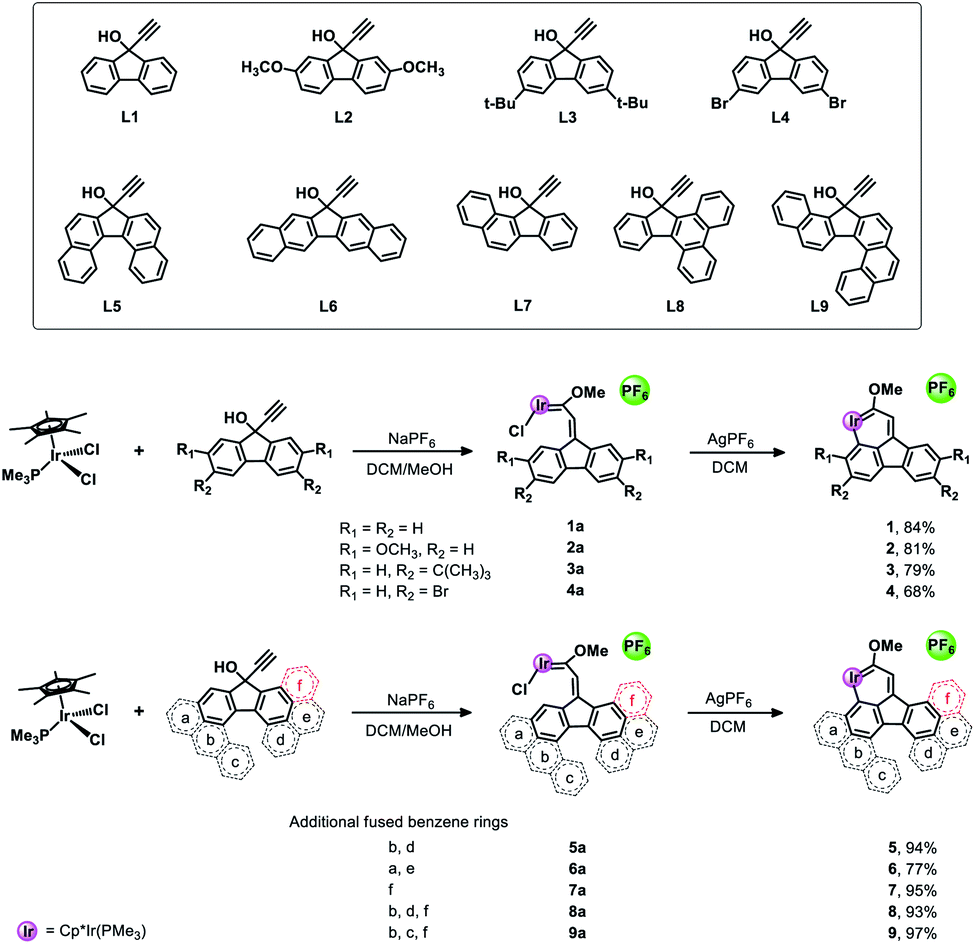
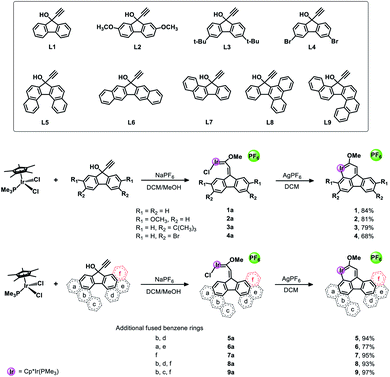
In 2017, Wright et al. successfully synthesized the first iridaanthracene complex.7 In another recent breakthrough, Bolaño et al. reported a novel iridanaphthalene compound in a high yield of 74% from its corresponding methoxy(alkenyl)carbeneiridium compound by using an intramolecular C–H activation reaction.6b In 2018, they further developed iridaphenanthrene, iridanaphthalene and iridaanthracene from their corresponding methoxy(alkenyl)carbeneiridium intermediates via reactions of [IrCp*Cl(NCMe) (PMe3)]PF6 with diarylpropargyl alcohols.8 In the present work, we have proposed a one-pot synthetic route to obtain a series of pure iridafluoranthenes in high yields (70–87%) by using IrCp*Cl2(PMe3) and 9-ethynyl-9-fluorenol derivatives as reagents (Scheme 2), all of which were successfully embedded into a fused five-membered ring to achieve higher conjugation. In the solid state all the new compounds are stable in air and can be stored in contact with the atmosphere for half a year without any signs of decomposition. To the best of our knowledge, the present work is the first case involving synthesis of metallafluoranthenes to date. Specifically, compounds L5–L9 were used to produce complexes 5–9 with the most abundant fused rings and high conjugation, which did not involve any isomerization and thus maintained high yield and high purity. In addition, diverse polycyclic metallaarenes, including four tetracyclic complexes (1–4) and pentacyclic (7), hexacyclic (5, 6 and 8) and heptacyclic (9) complexes, have been presented in this work. Although there are several reports on nitrogen- or oxygen-containing polycyclic metallaaromatics,13 this series of new structures that contain more than four fused rings in the polycyclic skeletons consisting of a metal atom and carbon atoms only, is unprecedented.
 |
| | Scheme 2 Synthesis of target complexes 1–9. | |
Results and discussion
As depicted in Scheme 2, the target complexes (1–9) were synthesized by a one-pot method in high yield (70–87%) under mild reaction conditions. Specifically, as shown in Scheme S1,† a solution of Cp*IrCl2(PMe3), NaPF6 and a pro-ligand (L1–L9, respectively) in methanol was stirred at room temperature overnight, and then methanol was removed under vacuum and the residue was dissolved in dichloromethane. Subsequently, AgPF6 was added and the mixture was further stirred for 5 minutes to obtain the corresponding target complex (1–9). Furthermore, we propose a possible reaction mechanism according to the previous report of Bolaño et al.14 by taking complex 1 as an example. As shown in Scheme S3,† initially, de-coordination of one chloride from Cp*IrCl2(PMe3) occurred, which is followed by further coordination of alkynol compound L1 to form an allenylidene intermediate (A). Subsequently, nucleophilic attack by the oxygen atom of methanol on the Cα of the allenylidene followed by proton transfer at Cβ gives the (methoxyalkenylcarbene)iridium intermediate 1a. Intramolecular C–H activation then occurred after treatment with AgPF6 to remove another chloride and to generate the ultimate product complex 1. Fortunately, we successfully isolated the intermediate 1a and obtained its single crystal (Fig. S2†) to further verify the reaction mechanism we proposed above. In addition, it is well documented in the literature that carbene complexes like 1a could easily be formed from a reaction between allenylidene intermediates and alkynol pro-ligands.15 Additionally, the structure of the fluorenyl ligand of 1a was symmetric, so pure products could be obtained in the subsequent ring-closing reaction due to the equivalent C–H activation sites (Ha and Hb in Scheme S3†) in 1a. Interestingly, when asymmetric compounds (L7–L9) reacted with Cp*IrCl2(PMe3), the corresponding pure complexes 7–9 were obtained in high yields above 90%. In addition, we also successfully isolated carbene intermediates 7a and 8a and obtained their structures by single crystal X-ray crystallography (Fig. S3†). The complexes 7–9 were obtained by CH metalation of the benzene ring that is directly fused to the five-membered ring and remote from the fused ring “f”. Metalation of this benzene ring results in the formation of a six-membered metallacyclic ring. Presumably this is favoured over CH metalation of the fused benzene ring “f” because metalation of this ring would give a seven-membered metallacycle, the formation of which is expected to be less favourable. After treating 8a with AgPF6, ring-closed product 8 was successfully produced in a high yield of 93%.
The structures of complexes 1–9 were verified by using nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS). Typically, for complex 1, the proton signals corresponding to the pentamethylcyclopentadienyl group can be observed at δ = 1.84 ppm and the 13C (CH3) signals are located at δ = 100.0 and 9.7 ppm, respectively. The characteristic signals of group P(CH3)3 are located at δ = 1.21 ppm in the 1H NMR spectrum and at δ = 13.8 ppm (d, 1JC–P = 41.6 Hz) in the 13C NMR spectrum, respectively. The 31P NMR spectrum also shows two signals at −35.3 ppm (s, P(CH3)3) and −144.3 ppm (hept, 1JP–F = 712.3 Hz, PF6), respectively. Additionally, the downfield signal observed at δ = 250.5 ppm in the 13C NMR spectrum corresponds to C1, which is a typical metal-bonded carbon atom. Similar spectra were obtained for complexes 2–9 and their NMR data were consistent with those reported in the literature.6b,c,8
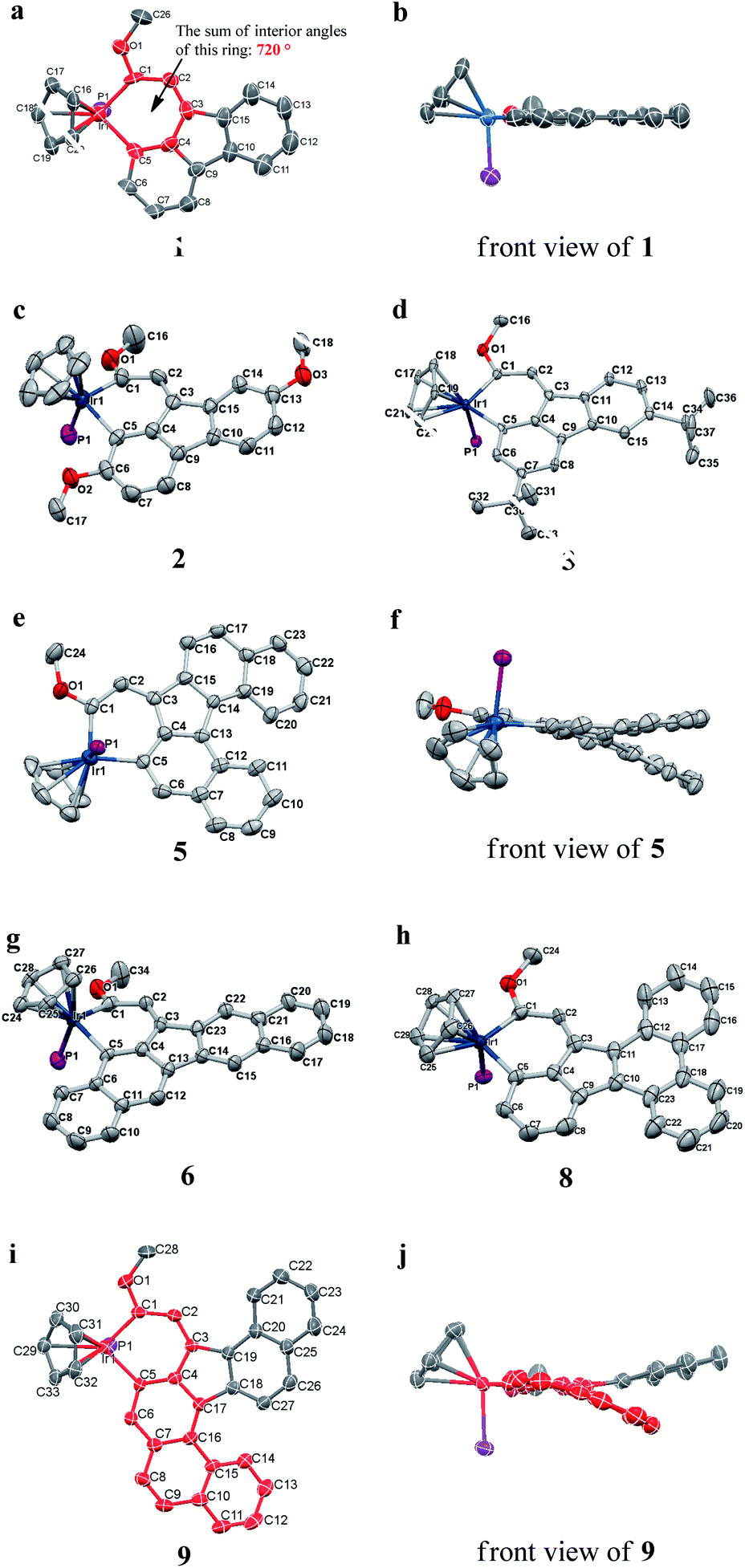
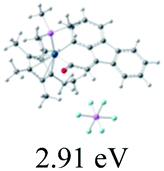
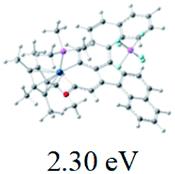
We successfully obtained single crystals of 1–3, 5, 6, 8 and 9 to further confirm their structures. As shown in Fig. 1a and b, the tetracyclic skeleton of complex 1 exhibited good planarity. The sum of the angles in the iridium-containing six-membered ring was 720.0°, which is consistent with the ideal value of a planar six-membered ring. The bond lengths of Ir1–C1 and Ir1–C5 were 1.984 and 2.056 Å, respectively, while the corresponding bond lengths in complexes 2, 3, 5, 6, 8 and 9 were 1.954–1.992 Å and 2.034–2.071 Å, respectively. These bond lengths are also comparable to the values of the previously reported systems including iridanaphthalene,6 iridaphenanthrene and iridaanthracene.7,8 In addition, as an iridafluoranthene derivative, complex 1 can be considered to be composed of an iridanaphthalene moiety and a benzene ring linked by a fused five-membered ring. In the crystal structure of 2 (Fig. 1c), the sum of the angles in the iridium-containing six-membered ring was 711.4°, indicative of a slightly lower planarity compared with 1. This may be due to the steric hindrance between the methoxy group (bonded to C6 and C13) and the pentamethylcyclopentadienyl group, which makes the iridium atom stand slightly out of the plane formed by C1–C2–C3–C4–C5. The iridium-containing polycyclic skeletons observed in the crystal structures of 3, 6 and 8 also show excellent planarity (Fig. 1d, g and h), while the polycyclic frameworks of 5 and 9 exhibit some distortion (Fig. 1e, f, i and j), which is presumably caused by the intramolecular repulsion between the hydrogen atoms connected with C12 and C20 in 5 and C14 and C27 in 9.
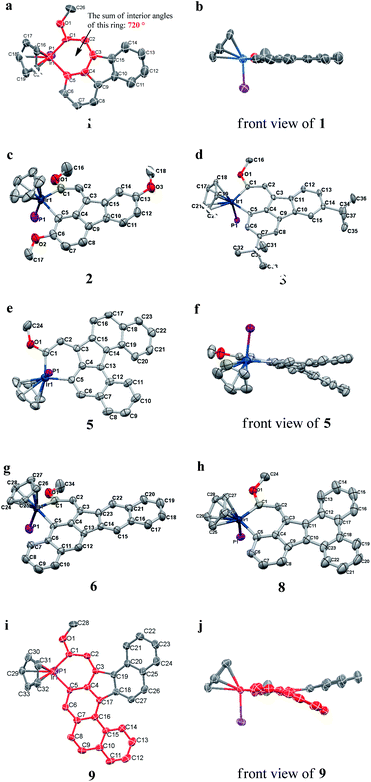 |
| | Fig. 1 Molecular structure of 1 (a and b), 2 (c), 3 (d), 5 (e and f), 6 (g), 8 (h) and 9 (i and j) (thermal ellipsoids set at 50% probability). Hydrogen atoms and the methyl group in Cp* and PMe3 are omitted for clarity. | |
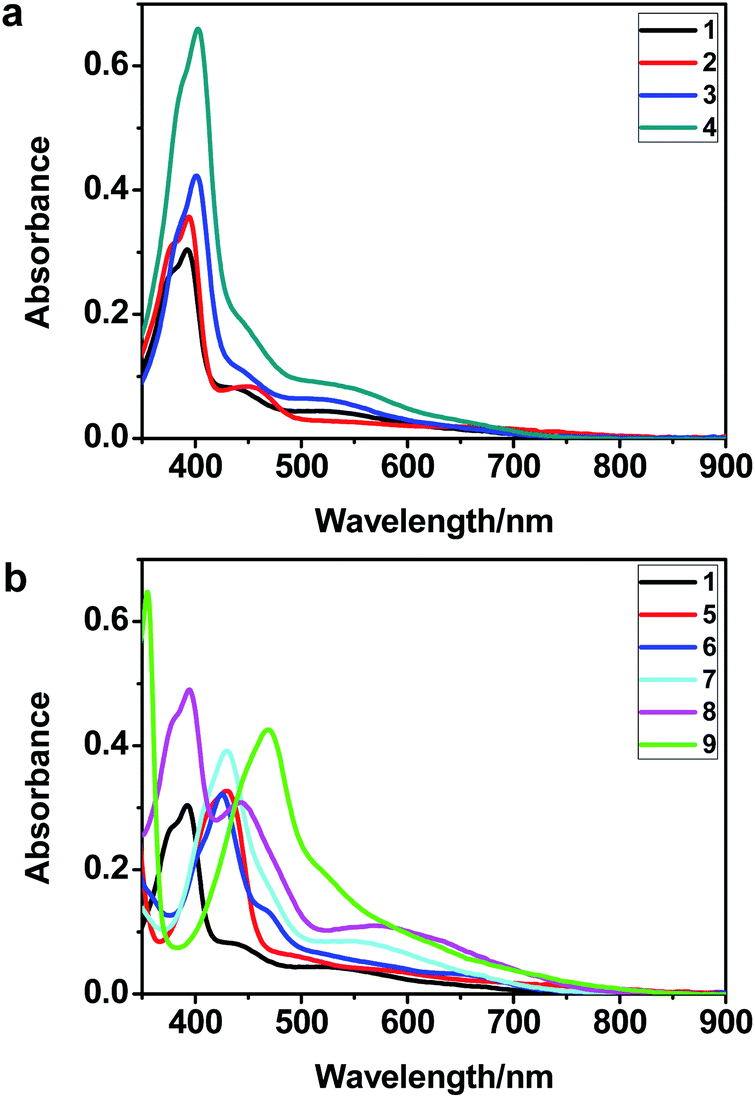
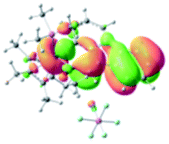
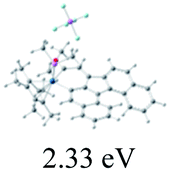
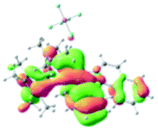
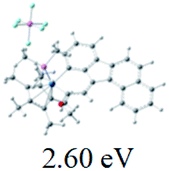
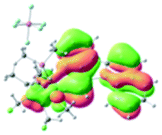
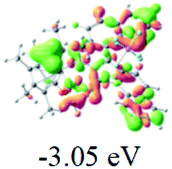
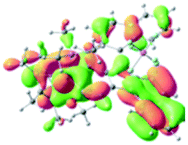
The UV-vis spectra of complexes 1–9 are illustrated in Fig. 2. Complexes 1–4 displayed similar absorption profiles with the maximum peak at around 400 nm. Comparatively, the absorption bands of complexes 5–9 were located at 430–470 nm with an obvious red shift compared with complex 1. These results clearly indicate that increasing the conjugation degree of metallaaromatic rings can effectively increase their corresponding absorption wavelength, while introducing different substituent groups has no significant influence on their absorption. In addition, tails were observed for complexes 1–9 similar to those observed for pure organic fluoranthene derivatives,16 while these tails were broader and their positions were significantly red-shifted compared with those for pure fluoranthene derivatives due to the presence of iridium atoms. Therefore, TD-DFT calculations were performed to confirm the above observations and the pertinent data are collected in Table 1 and Table 2. The simulated absorption spectra of complexes 1–9 are well consistent with the experimental data. According to the TD-DFT results, the absorptions of all complexes are attributed to the transitions from HOMO−4 or HOMO−2 (8) to the LUMO, which were mainly involved in the intra-ligand and metal (Ir) to ligand transitions, further verifying the significant role of the metal atoms.
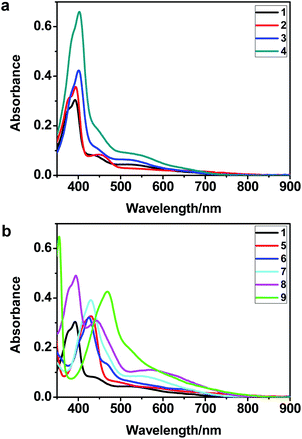 |
| | Fig. 2 UV/vis absorption spectra of complexes 1–9 (2.0 × 10−5 M) measured in CH2Cl2 at room temperature. | |
Table 1 Detailed photophysical data and calculation results
| Compound |
λ
abs/nm calc. (exp.) |
Excitation |
f
|
Percentage |
1stEredvs. Fc+/0 (V) |
1stEoxivs. Fc+/0 (V) |
|
1
|
379 (393) |
HOMO−4 → LUMO |
0.4074 |
91% |
−1.21 |
0.98 |
|
2
|
378 (394) |
HOMO−4 → LUMO |
0.3799 |
94% |
−1.24 |
0.72 |
|
3
|
388 (401) |
HOMO−4 → LUMO |
0.4428 |
80% |
−1.27 |
0.98 |
|
4
|
398 (402) |
HOMO−4 → LUMO |
0.4158 |
70% |
−1.05 |
1.09 |
|
5
|
420 (429) |
HOMO−4 → LUMO |
0.1776 |
65% |
−0.99 |
0.83 |
|
6
|
422 (425) |
HOMO−4 → LUMO |
0.3625 |
78% |
−1.20 |
0.97 |
|
7
|
428 (429) |
HOMO−4 → LUMO |
0.2863 |
95% |
−1.07 |
0.96 |
|
8
|
490 (443) |
HOMO−2 → LUMO |
0.1554 |
91% |
−1.06 |
0.96 |
| 377 (394) |
HOMO−5 → LUMO |
0.3277 |
76% |
|
9
|
482 (468) |
HOMO−4 → LUMO |
0.2385 |
64% |
−0.97 |
0.94 |
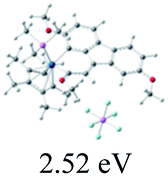
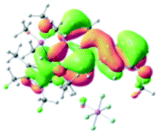
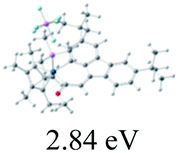
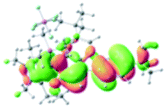
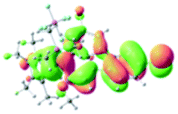
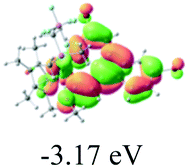
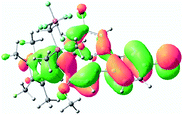
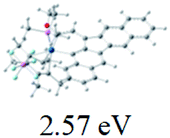
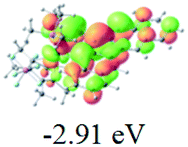
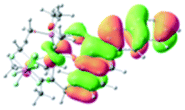
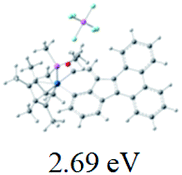
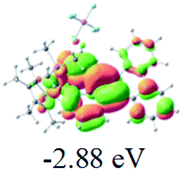
Table 2 Selected molecular orbitals and energy levels of complexes 1–9
| Compound |
Optimized structure (ΔEHOMO/LUMO) |
HOMO |
LUMO |
HOMO−4 |
|
1
|

|
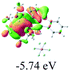
|
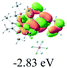
|
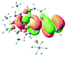
|
|
2
|

|
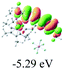
|
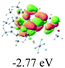
|

|
|
3
|

|
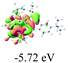
|
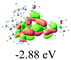
|
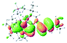
|
|
4
|
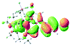
|
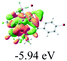
|
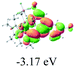
|
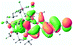
|
|
5
|

|
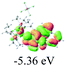
|
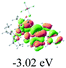
|
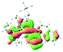
|
|
6
|

|

|
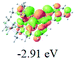
|
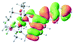
|
|
7
|

|
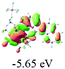
|
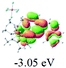
|

|
|
8
|
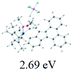
|

|

|
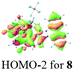
|
|
9
|

|
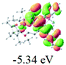
|
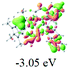
|

|
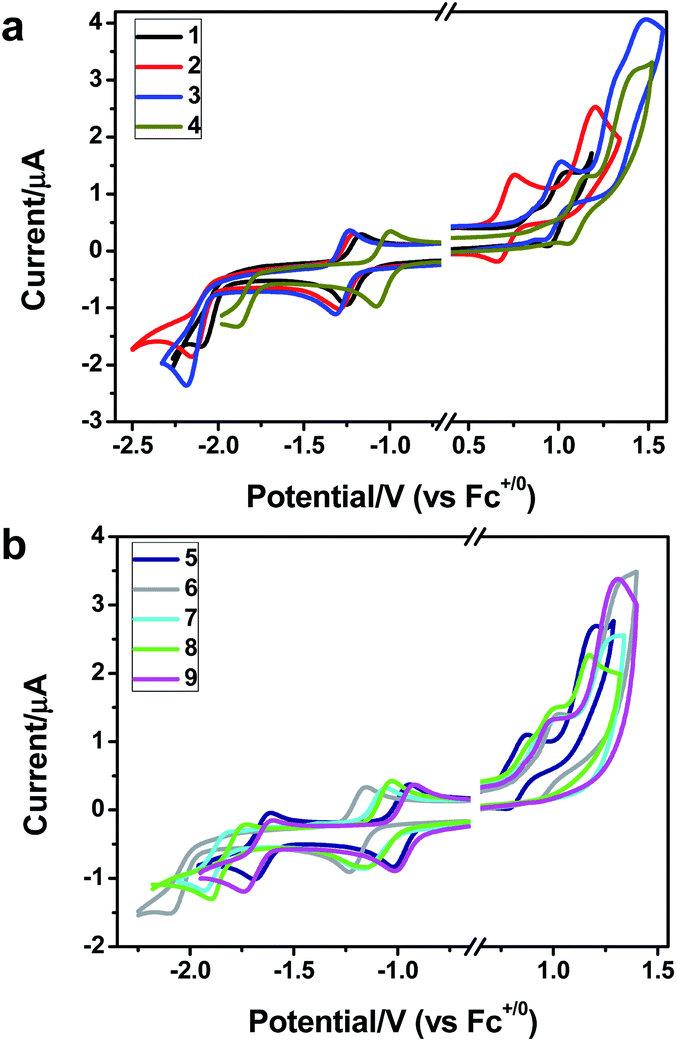
To gain insight into the electronic properties of these complexes, cyclic voltammetry (CV) was performed in dichloromethane using a platinum disk as the working electrode, a platinum wire as the counter electrode and an Ag wire as the reference electrode. All complexes showed multi-electron redox processes (Fig. 3) and only their first reduction processes were reversible (Fig. S1†). As presented in Table 1, the introduction of electron-donating –OCH3 and t-Bu groups in complexes 2 and 3, respectively, caused a cathodic shift in the reduction potential compared with that in 1, while the electron-withdrawing bromine atom in complex 4 led to an anodic shift. For complexes 5–9, the extension of conjugated fused rings resulted in anode-shifted reduction potentials and cathode-shifted oxidation potentials, and especially the first reversible reduction potentials of 5 and 9 reached −0.99 V and −0.97 V, respectively. As presented in Table 2, the calculated HOMO and LUMO levels also matched well with the changing tendencies of the first oxidation and reduction potentials, respectively. Taken together, introducing electron-donating groups increased both the HOMO and LUMO levels, while electron-withdrawing units resulted in an opposite effect; enlarging the π-skeletons decreased the LUMO level and increased the HOMO level.17
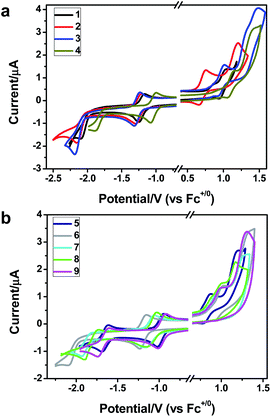 |
| | Fig. 3 Cyclic voltammograms of complexes 1–9 (2.0 × 10−3 M) measured in CH2Cl2 at room temperature (0.1 M, n-Bu4NPF6). | |
Conclusions
In summary, a series of novel iridafluoranthene derivatives as metalla-analogues of PAHs were synthesized in high yields under mild conditions by a one-pot synthetic route. In addition, fused rings ranging from four to seven in number were successfully obtained for the first time. Among them, metalla-PAHs containing seven fused rings is a new record. Meanwhile, a sterically-oriented synthetic strategy was revealed and this new method may provide a feasible approach to prepare larger conjugated polycyclic metallaaromatic hydrocarbons or even multinuclear metalla-analogues of PAHs in high yields. In addition, introducing different substituent groups and enlarging the conjugation of the π-skeletons can effectively tune the electronic properties of metallaaromatics. Electron-donating groups can elevate both the HOMO and LUMO levels, while electron-withdrawing units will lead to an opposite effect. Enlarging the π-skeletons will decrease the LUMO level and increase the HOMO level. Therefore, our present results open up a new avenue for the design and synthesis of new polycyclic metallaaromatic hydrocarbons and metalla-nanographenes.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21772054 and 21676113) and the 111 Project (B17019).
Notes and references
- Q. Yan, Y. Zhou, B.-B. Ni, Y. Ma, J. Wang, J. Pei and Y. Cao, J. Org. Chem., 2008, 73, 5328–5339 CrossRef CAS PubMed; R. C. Chiechi, R. J. Tseng, F. Marchioni, Y. Yang and F. Wudl, Adv. Mater., 2006, 18, 325–328 CrossRef; J. Dong, X. Li, K. Zhang, Y. D. Yuan, J. Jiang and D. Zhao, J. Am. Chem. Soc., 2018, 140, 4035–4046 CrossRef PubMed; G. M. Paternò, Q. Chen, X.-Y. Wang, K. Müllen, A. Narrita and F. Scotognella, Angew. Chem., Int. Ed., 2017, 56, 6753–6757 CrossRef PubMed.
-
(a) S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed;
(b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed;
(c) M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS PubMed.
-
(a) J. Farinhas, D. Molina, A. Olcina, C. Costa, F. Fernández-Lázaro, Á. Sastre-Santos and A. Charas, Dyes Pigm., 2019, 161, 188–196 CrossRef CAS;
(b) H. Wu, L. Yang, Y. Li, M. Zhang, Y. Guo and P. Wang, J. Mater. Chem. A, 2016, 4, 519–528 RSC.
- G. P. Elliott, W. R. Roper and J. M. Waters, J. Chem. Soc., Chem. Commun., 1982, 811–813 RSC.
-
(a) X.-Y. Cao, Q. Zhao, Z. Lin and H. Xia, Acc. Chem. Res., 2014, 47, 341–354 CrossRef CAS PubMed;
(b) A. F. Dalebrook and L. J. Wright, Adv. Organomet. Chem., 2012, 60, 93–177 CrossRef CAS;
(c) B. J. Frogly and L. J. Wright, Chem.–Eur. J., 2018, 24, 2025–2038 CrossRef PubMed.
-
(a) M. Paneque, C. M. Posadas, M. L. Poveda, N. Rendón, V. Salazar, E. Oñate and K. Mereiter, J. Am. Chem. Soc., 2003, 125, 9898–9899 CrossRef CAS PubMed;
(b) M. Talavera, S. Bolaño, J. Bravo, J. Castro, S. García-Fontán and J. M. Hermida-Ramón, Organometallics, 2013, 32, 4058–4060 CrossRef CAS;
(c) M. Talavera, J. Bravo, J. Castro, S. García-Fontán, J. M. Hermida-Ramón and S. Bolaño, Dalton Trans., 2014, 43, 17366–17374 RSC.
- B. J. Frogly and L. J. Wright, Angew. Chem., Int. Ed., 2017, 56, 143–147 CrossRef PubMed.
- M. Talavera, A. Peña-Gallego, J. L. Alonso-Gómez and S. Bolaño, Chem. Commun., 2018, 54, 10974–10976 RSC.
-
(a) J. Chen, C. Shi, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Angew. Chem., Int. Ed., 2011, 50, 7295–7299 CrossRef CAS PubMed;
(b) J. Chen and G. Jia, Coord. Chem. Rev., 2013, 257, 2491–2521 CrossRef CAS.
-
(a) G. He, J. Zhu, T. B. Wen, I. D. Williams, Z. Lin and G. Jia, Angew. Chem., Int. Ed., 2007, 46, 9065–9068 CrossRef CAS PubMed;
(b) B. Liu, H. Xie, Q. Zhao, J. Chen, T. B. Wen, Z. Cao and H. Xia, Angew. Chem., Int. Ed., 2009, 48, 5461–5464 CrossRef CAS PubMed;
(c) M.-X. Zhang, J. Zhang, X. Jin, X. Sun, J. Yin, F. Hartl and S. H. Liu, Chem.–Eur. J., 2018, 24, 18998–19009 CrossRef CAS PubMed.
-
(a) M.-X. Zhang, Z. Xu, T. Lu, J. Yin and S. H. Liu, Chem.–Eur. J., 2018, 24, 14891–14895 CrossRef CAS PubMed;
(b) W. Ruan, T.-F. Leung, C. Shi, I. D. Williams, Z. Lin and G. Jia, Chem. Sci., 2018, 9, 5994–5998 RSC.
-
(a) C. Zhu, M. Luo, Q. Zhu, J. Zhu, P. R. Schleyer, J. I.-C. Wu, X. Lu and H. Xia, Nat. Commun., 2014, 5, 3265–3271 CrossRef PubMed;
(b) C. Zhu, X. Zhou, H. Xing, K. An, J. Zhu and H. Xia, Angew. Chem., Int. Ed., 2015, 54, 3102–3106 CrossRef CAS PubMed;
(c) C. Zhu, J. Zhu, X. Zhou, Q. Zhu, T. B. Wen and H. Xia, Angew. Chem., Int. Ed., 2018, 57, 3154–3157 CrossRef CAS PubMed.
-
(a) R. Li, Z. Lu, Y. Cai, F. Jiang, W. Hong and H. Xia, J. Am. Chem. Soc., 2017, 139, 14344–14347 CrossRef CAS PubMed;
(b) C. Zhu, Q. Zhu, J. Zhu, X. He, X.-Y. Cao and H. Xia, Angew. Chem., Int. Ed., 2014, 53, 6232–6236 CrossRef CAS PubMed.
- M. Talavera, S. Bolaño, J. Bravo, J. Castro, S. García-Fontán and J. M. Hermida-Ramón, Organometallics, 2013, 32, 4402–4408 CrossRef CAS.
-
(a) M. A. Esteruelas, A. V. Gómez, A. M. López, E. Oñate and N. Ruiz, Organometallics, 1998, 17, 2297–2306 CrossRef CAS;
(b) M. A. Esteruelas, A. V. Gómez, A. M. López, M. Oliván, E. Oñate and N. Ruiz, Organometallics, 2000, 19, 4–14 CrossRef CAS;
(c) B. Weberndörfer and H. Werner, J. Chem. Soc., Dalton Trans., 2002, 1479–1486 RSC;
(d) A. Asensio, M. L. Buil, M. A. Esteruelas and E. Oñate, Organometallics, 2004, 23, 5787–5798 CrossRef CAS.
- G. S. Mohammad-Pour, R. T. Ly, D. C. Fairchild, A. Burnstine-Townley, J. K. Harper and F. J. Uribe-Romo, J. Org. Chem., 2018, 83, 8036–8053 CrossRef CAS PubMed.
-
(a) C. Yan, S. Barlow, Z. Wang, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS;
(b) A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763–13772 RSC;
(c) J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Haiping
Xia
*a,
Haiping
Xia