
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective ring opening of fully-substituted cyclopropanes via intramolecular Friedel–Crafts alkylation†
Veeranjaneyulu
Lanke
,
Fa-Guang
Zhang
 ,
Alexander
Kaushansky
and
Ilan
Marek
*
,
Alexander
Kaushansky
and
Ilan
Marek
*
Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City 3200009, Haifa, Israel. E-mail: chilanm@technion.ac.il
First published on 27th August 2019
Abstract
We herein disclose a diastereoselective ring opening of non-donor–acceptor cyclopropanes via an intramolecular Friedel–Crafts alkylation en route to functionalized dihydronaphthalene scaffolds possessing quaternary carbon stereocentres. The transformation proceeds through a selective bond breaking at the most alkylated carbon centre with a pure retention of configuration. Mechanistic investigations and computational studies revealed that alkoxy functionality is the key for selective bond breaking leading to a complete retention of configuration.
Cyclopropenes have always been a valuable molecular scaffold in organic synthesis as they could selectively be functionalized into more densely substituted three-membered rings.1 In the case of nucleophilic addition on the strained double bond, the resulting cyclopropane can undergo selective ring opening2 producing highly functionalized cyclic, acyclic or heterocyclic building blocks that are essential for the synthesis of natural products and biological active compounds.3,4 Among all possible strategies for ring-opening of strained-rings, donor–acceptor cyclopropanes5 have attracted a particular attention because of the simplicity, reliability, atom-economy and notable chemo-, regio-, and stereoselectivities of the reaction although the formation of acyclic quaternary stereocentre remains one challenging problem (Scheme 1a).6 In the context of selective ring-opening, and based on the catalytic Nazarov cyclization,7 the formal homo-Nazarov process (Scheme 1b),8 transforms an activated cyclopropane into a six-membered ring carbocycle. Despite the high reactivity of cyclopropanes, this formal homo-Nazarov reaction proceed efficiently only when additional activating groups are present either by (i) introducing a second electron-withdrawing group α- to the carbonyl group, (ii) introducing a heteroatom at the α′-position to the ketone or (iii) increasing the donor–acceptor properties of the cyclopropane core by using heteroatoms as donor partners in a γ-position (Scheme 1b).9 Mechanistically, the reaction usually involves cyclopropane ring-opening in the presence of a Lewis acid catalyst, followed by an intramolecular Friedel–Crafts alkylation.10 In these lines, Nishii and coworkers has utilized the homo-Nazarov strategy to synthesize (±)-cyclogalgravin and (+)-podophyllic aldehydes.10d,e
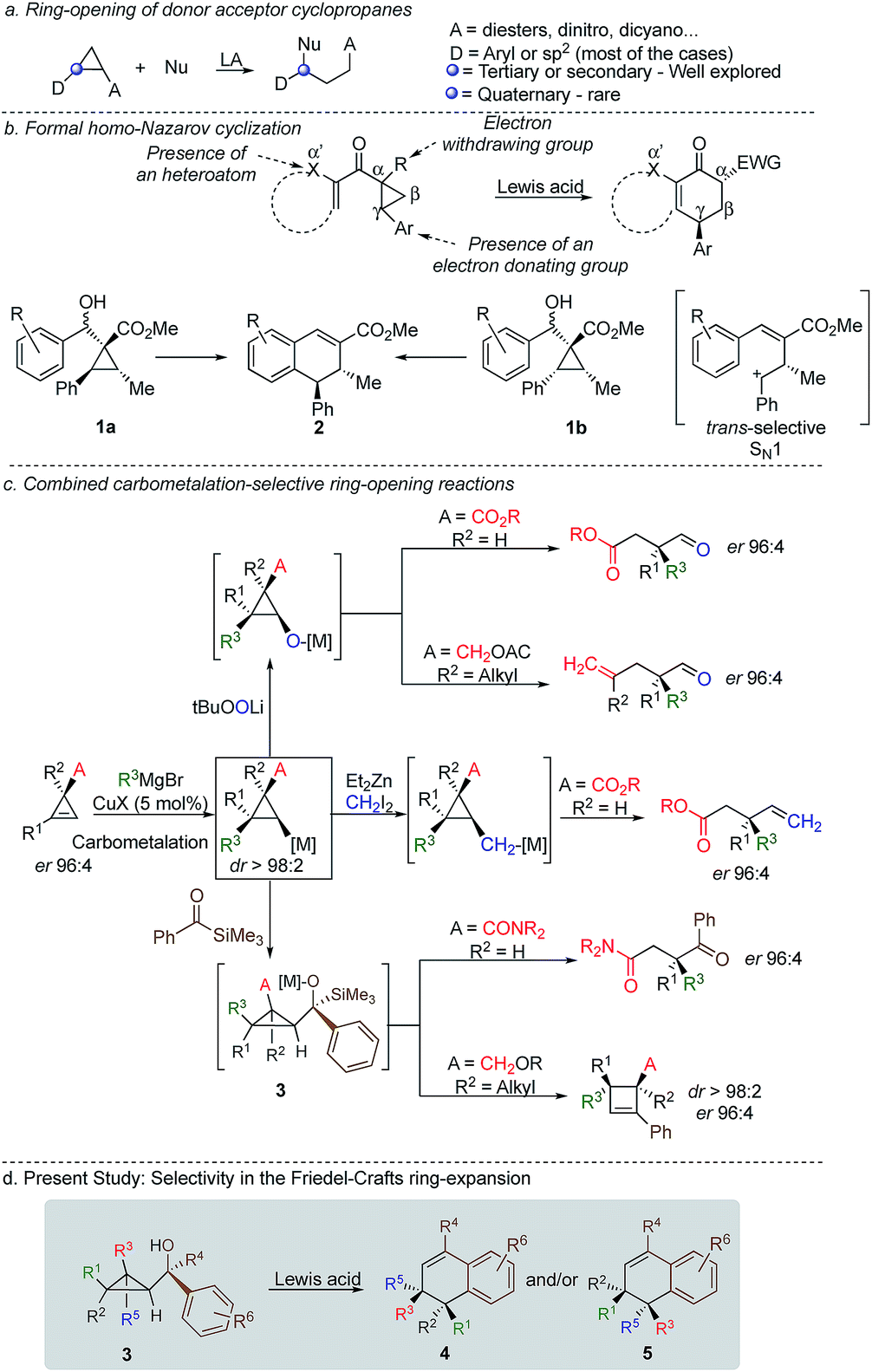 | ||
| Scheme 1 Selectivity in the ring-opening of three-membered rings. | ||
To further validate the mechanism of the cyclization reaction, the same group has synthesized the two independent diastereoisomers10a1a and 1b and under the same experimental condition, both diastereoisomers provided the same product 2. Although not a formal homo-Nazarov process, the trans stereochemistry between the two stereogenic centres supports a stepwise Friedel–Crafts type mechanism (SN1) where the benzylic stereocentre epimerizes into the less sterically shielded stereoface (anti to the substituent in the β-position). As a consequence of this stepwise Friedel–Crafts type mechanism (SN1), no example was reported for the formation of quaternary stereocentre as the two diastereoisomers should be formed in equivalent amount.
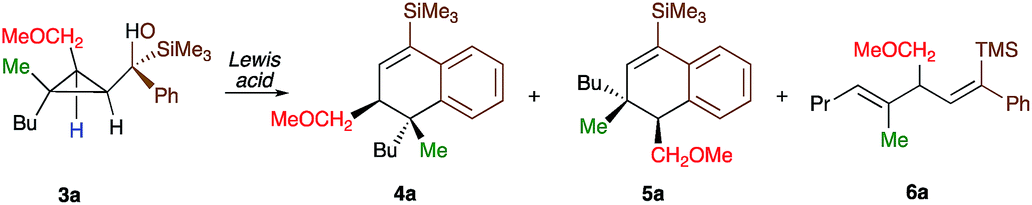
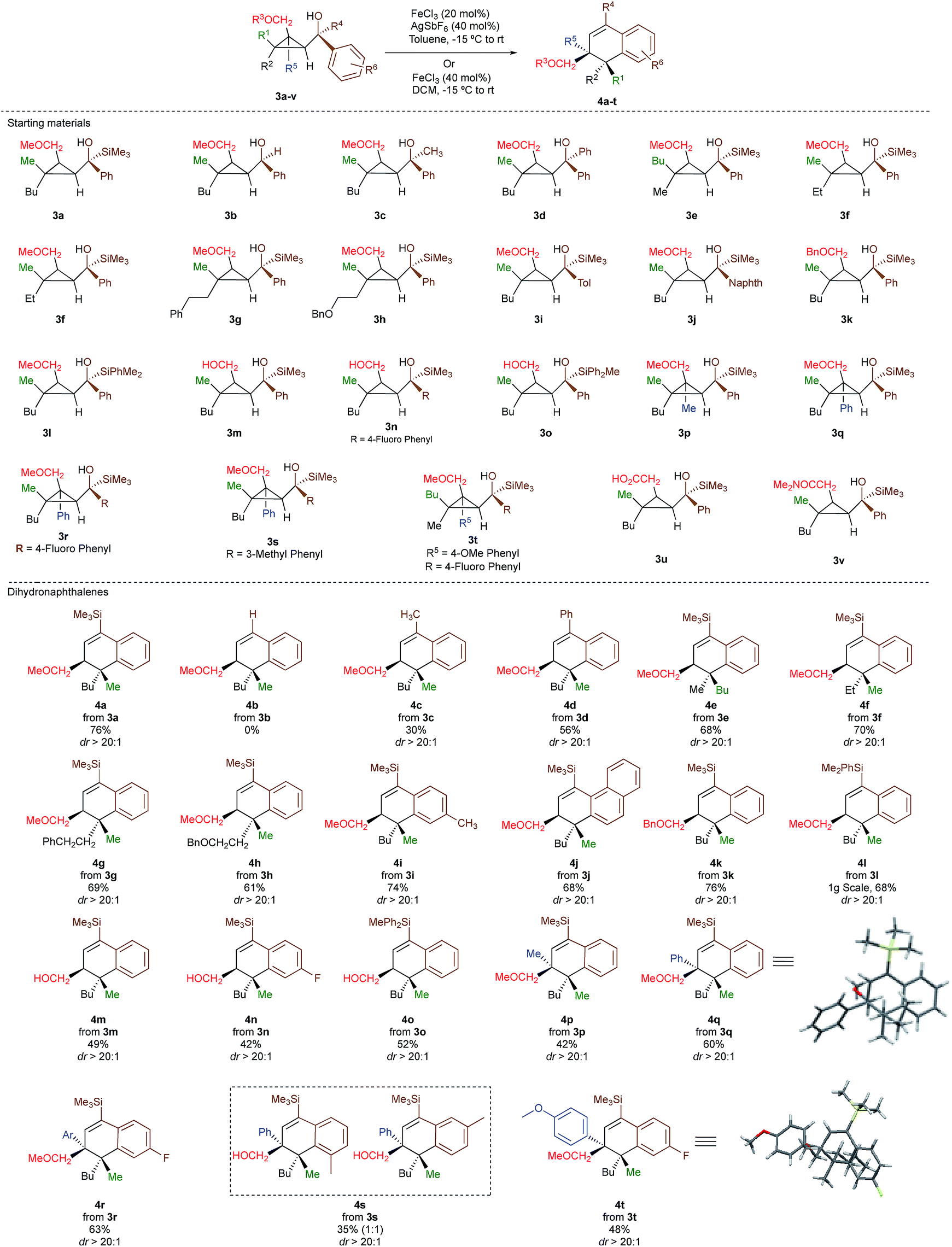
As one of the research areas of our group concern with the stereoselective preparation of quaternary stereocentres, we have reported the preparation of these latter11 by the in situ formation of donor–acceptor cyclopropanes. Those strategies relied on the combined diastereoselective carbometalation reactions of cyclopropenes12 followed by reaction of the generated cyclopropyl metal species with ambiphilic electrophiles such as carbenoids,13 oxenoids14 or acylsilanes15 promoting subsequent selective ring-opening reactions (Scheme 1c). In all of these above-mentioned transformations, the quaternary stereocentre generated during the carbometalation reaction was at no risk of epimerization except for the ring-expansion of 3 into cyclobutene (Scheme 1c).15b In this case, a complete preservation of the stereochemistry was observed during the C–C bond migration. Although mechanistically not related, we were nevertheless intrigued by the discrepancy between this complete preservation of the stereochemistry during the ring-expansion of 3 into polysubstituted cyclobutenes (Scheme 1c)15b with the SN1 character of the ring expansion of 1a and 1b (Scheme 1b).10a We were therefore wondering what would be the stereochemistry of the Lewis-acid catalysed Friedel–Crafts ring-expansion products of 3 possessing the previously unreported quaternary sterocentres (Scheme 1d). When our model substrate 3a (R1 = Me, R2 = Bu, R3 = CH2OMe, R4 = SiMe3, R5 = R6 = H) was treated with FeCl3 (20 mol%) as Lewis acid, the desired Friedel–Crafts alkylation product (4a) was obtained in 50% or 58% yield at respectively 60 °C or room temperature along with intramolecular fragmented product 6a (Table 1, entries 1 and 2) but without any traces of the rearranged product 5a. Remarkably, 4a was obtained as a single diastereoisomer. Screening various different Lewis or Brønsted acids did not improve yield of 4a (Table 1, entries 3–7). When the reaction was performed at lower temperature, 4a could be isolated in 72% yield with a minimum amount of 6a (Table 1, entry 8). After additional screening (see ESI†), we were delighted to find two additional optimal conditions (Table 1, entries 9 and 10). In the first case, FeCl3 (20 mol%) was combined with AgSbF6 as cocatalyst (40 mol%) in toluene (0.1 M) at −15 °C to room temperature to provide the desired product in 76% yield. In the second case, the quantity of FeCl3 was increased to 40 mol% without cocatalyst and the product 4a was isolated in 81% yield as single diastereoisomer without any detectable amount of 5a. Having in hand the best experimental conditions, we first briefly evaluated the role of the TMS group in the formation of 4 and subsequently explored the scope of the reaction by varying the nature of all substituents R1 to R5 on the cyclopropanes (Scheme 2). For the effect of the TMS group, four different substrates 3a–d were prepared using our conventional strategy (see the ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | T (°C)/time (h) | 4a/5a/6ab |
| a Condition: 3a (0.3 mmol), solvent (3 mL). b NMR yield using p-methoxy acetophenone as internal standard. c AgSbF6 (40 mol%) used as an additive. d Yield of isolated product after purification by column chromatography. | ||||
| 1 | FeCl3 (20) | Toluene | 60/3 | 50/0/12 |
| 2 | FeCl3 (20) | Toluene | rt/3 | 58/0/10 |
| 3 | FeCl2 (20) | Toluene | rt/12 | 0/0/0 |
| 4 | Bi(OTf)3 (20) | Toluene | rt/12 | 40/0/18 |
| 5 | Cu(OTf)2 (20) | Toluene | rt/12 | 22/0/8 |
| 6 | BF3·OEt2 (20) | Toluene | rt/4 | 48/0/0 |
| 7 | TsOH (100) | Toluene | rt/0.5 | 32/0/11 |
| 8 | FeCl3 (20) | CH2Cl2 | −15 to rt/3 | 72/0/10 |
| 9c | FeCl3 (20) | Toluene | −15 to rt/3 | 76d/0/7 |
| 10 | FeCl3 (40) | CH2Cl2 | −15 to rt/3 | 81d/0/9 |
 | ||
| Scheme 2 Preparation of dihydronaphthalenes. | ||
In contrast to 3a, where the corresponding dihydronaphthalene 4a was obtained in good yield (Scheme 2), secondary alcohol 3b (R4 = H) led quantitatively to the product of fragmentation without any trace of 4b (Scheme 2). This result is in good agreement with previous studies underlining the need of either a donor–acceptor cyclopropane or a substrate possessing a benzylic stereocentre.12–15 On the other hand, benzylic tertiary alcohols (3c R4 = Me and 3d R4 = Ph) provided the desired product 4c, 4d in moderate yields along the fragmented products (Scheme 2, respectively). It should be noted again that in all these cases, the products 4a, 4c and 4d were formed as a single diastereoisomer. These results suggest that the reaction doesn't proceed anymore through a stepwise Friedel–Crafts (SN1) type mechanism as a mixture of two diastereoisomer would be expected at the quaternary carbon centre. To further check this mechanistic assumption, the nature of the substituents R1 and R2 were permuted and the opposite diastereoisomer at the quaternary carbon stereocentre was formed with a similar diastereoselectivity underlying that the reaction is stereospecific (Scheme 2a, compare 4a and 4e from 3a and 3e). Various products possessing differently substituted quaternary carbon stereocentres were prepared with similar efficiencies and yields (Scheme 2, 4f–h). Structural variation of the aromatic ring doesn't impede the reaction to proceed (Scheme 2, 4i, 4j, 4n). Changing the coordinating group from methoxy (4a) to benzyloxy (4k) or changing TMS group (4a) to DMPS (4l), didn't alter the transformation. The presence of a free alcohol seriously decreased the overall yield (4m–4o), whereas the presence of a carboxylic acid or amide as a coordinating group failed to furnish the expected products under all tested reaction conditions (Scheme 2, 3u, 3v).17 Finally, five additional interesting examples show that products possessing two quaternary carbon stereocentres can be prepared underlining the unique selectivity of the carbon–carbon bond cleavage in the process (Scheme 2, 4p–4t). The configuration of 4p was established by NMR studies (see ESI†) and by X-ray crystallographic analysis for compounds 4q and 4t confirming that the rearrangement proceeds with a pure overall retention of configuration at the quaternary stereocentre.16 The configuration of all other products were assigned by analogy.
With a practical method to prepare dihydronaphthalenes possessing a quaternary and tertiary carbon stereocentres from simple cyclopropenes, substrate 3w (R1 = Me, R2 = Hex, R3 = Me, R4 = SiMe3, R5 = H) was readily synthesized from the corresponding cyclopropene with high enantioselectivity (er 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) through catalytic decomposition of diazo ester with 1-octyne.18 The copper-catalysed carbomagnesiation followed by addition of an acylsilane provide 3w as a single diastereoisomer with the same enantiomeric ratio than the starting material (Scheme 3, dr > 98:2; er 95:5). The subsequent Lewis acid-catalysed selective ring-opening and rearrangement afforded dihydronaphthalene 4w as a single diastereoisomer without any loss in enantiomeric purity (dr > 20:1, er 95:5, Scheme 3, see ESI†) confirming that the reaction proceeds with a complete retention of configuration at the migratory carbon centre. To determine whether the migratory aptitude is independent of the stereochemistry at the benzylic stereocentre in C4, a mixture of the two diastereoisomers of 3c were prepared and subjected to our experimental conditions. Although lower yields were observed as discussed previously when R4 = Me, the rearranged product 4c was generated with the same selectivity (dr > 20:1, Scheme 3) underlining that the rearrangement is unrelated to the stereochemistry at the benzylic alcohol.
:5) through catalytic decomposition of diazo ester with 1-octyne.18 The copper-catalysed carbomagnesiation followed by addition of an acylsilane provide 3w as a single diastereoisomer with the same enantiomeric ratio than the starting material (Scheme 3, dr > 98:2; er 95:5). The subsequent Lewis acid-catalysed selective ring-opening and rearrangement afforded dihydronaphthalene 4w as a single diastereoisomer without any loss in enantiomeric purity (dr > 20:1, er 95:5, Scheme 3, see ESI†) confirming that the reaction proceeds with a complete retention of configuration at the migratory carbon centre. To determine whether the migratory aptitude is independent of the stereochemistry at the benzylic stereocentre in C4, a mixture of the two diastereoisomers of 3c were prepared and subjected to our experimental conditions. Although lower yields were observed as discussed previously when R4 = Me, the rearranged product 4c was generated with the same selectivity (dr > 20:1, Scheme 3) underlining that the rearrangement is unrelated to the stereochemistry at the benzylic alcohol.
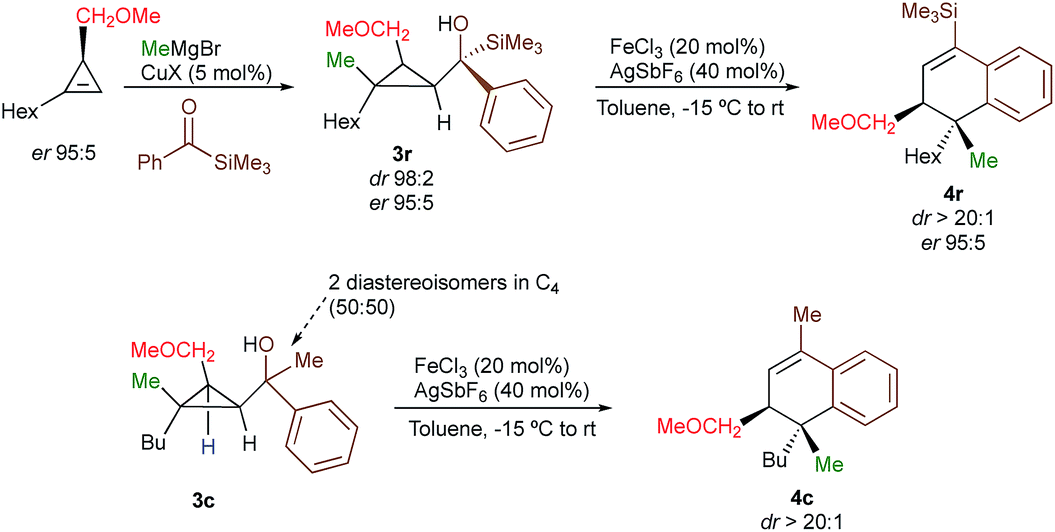 | ||
| Scheme 3 Diastereo- and enantioselective preparation of dihydronaphthalene. | ||
Since the dihydronaphthalene or tetralin cores represent a common scaffold in many biologically active compounds,10,19 we could showcase the selective reduction of the double bond (formation of 7) and deprotection of TMS group (formation of 4b, Scheme 4) with equal efficiency. It should be noted that the silicon moiety could be used for subsequent cross-coupling reactions20 but also as a surrogate of a hydrogen, leading to the product 4b that was not accessible directly from 3b (Scheme 2). All attempts to transform 4r into tetralone by oxidation of the corresponding vinylsilane failed.
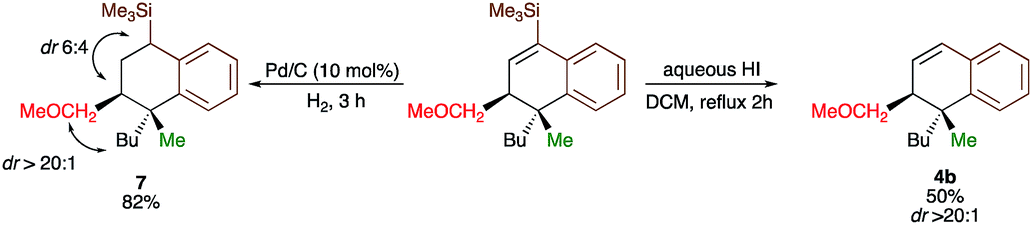 | ||
| Scheme 4 Selective manipulation of dihydronaphthalene. | ||
To shed additional light on the selectivity of the ring-expansion proceeding with a pure retention of configuration at the migratory carbon centre, we compared the behaviour of several starting materials (Schemes 2 and 5a) in this Lewis acid-catalysed Friedel–Crafts rearrangement. In all cases, the C–C bond cleavage is exceptionally selective with a pure retention of configuration always leading either to the fragmentation at the most-substituted carbon centre or with a unique selectivity when two quaternary stereocentres are present in the cyclopropyl ring. Even when a benzylic centre is concerned (R5 = Ph, formation of 4q), the selectivity is unique and the migrating group is always the one that do not possess the alkoxy-functionality. However, when the methoxy group is absent, the ring expansion still proceeds but without any selectivity (formation of 4x and 5x in a 2 to 1 ratio as inseparable mixture of products, Scheme 5b). Based on these control experiments, we can conclude that the selectivity of the ring-expansion is controlled by the presence of the alkoxy group allowing the Friedel–Crafts reaction to proceed stereospecifically. It should be added that the effect of the substituent R4 on the rearrangement (compare 4a to 4d, Scheme 2), only α-hydroxysilane provides good yield of the rearranged products, suggesting that the silicon atom add an extra stabilization in the process.
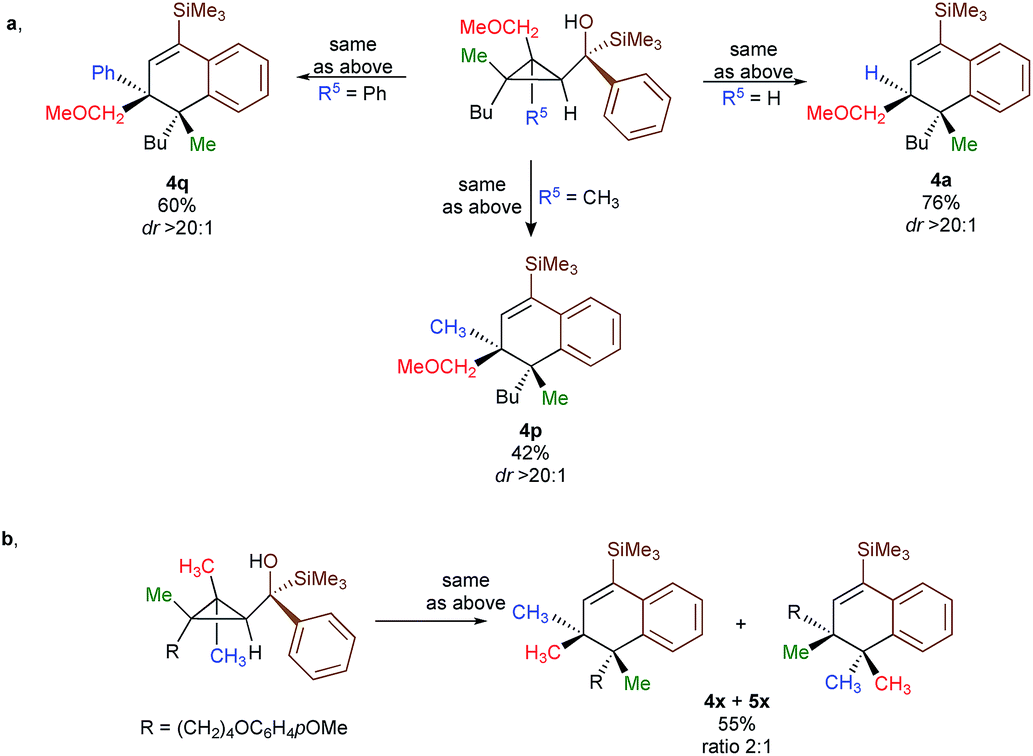 | ||
| Scheme 5 Mechanistic insights. | ||
Computational studies
To better understand the origin of the selectivity for the ring-opening as well as the stereospecificity of the process, the reaction mechanism was investigated by density functional theory calculations21 at M062X/6-311+g(d,p) level of theory in dichloromethane. The overall process is essentially exothermic with enthalpy and Gibbs free energy −31.1 and −33.7 kcal mol−1 respectively (see ESI for all details†). We first studied the selectivity of the ring-opening of the reaction and we found that the stability of the intermediate carbocation dictates the selectivity of the ring-opening. Although the selectivity was obvious for the transformation of 3a to 3o into 4a to 4o, where the formation of a tertiary carbocation is preferred over the formation of a secondary carbocation (Scheme 6a), it became much less evident when two tertiary carbocations (3p–3t into 4p–4t) are concerned. Although our initial hypothesis was that an anchimeric effect was controlling the selectivity of the ring-opening as well as the stereospecificity, calculations show that the negative inductive effect of the methoxy group is the key-controlling element by slightly destabilizing the formation of the carbocation A, generated through the cleavage of the C1–C2 bond, as compared to the formation of the carbocation B that would be generated by the cleavage of the C1–C3 bond (Scheme 6b). Full details of the results of the calculations are given in the ESI† material including the non-productive approach through anchimeric effect, where the 4-membered oxonium could not undergo the subsequent Friedel–Crafts cyclization. To corroborate this hypothesis, the isodesmic reaction represented in Scheme 6c has been calculated and indeed shows that carbocation C is less stabilized than carbocation D. If the methoxy group is not present as described in Scheme 5d, two isomers are formed and the final ratio depends on the stability of the carbocation intermediates.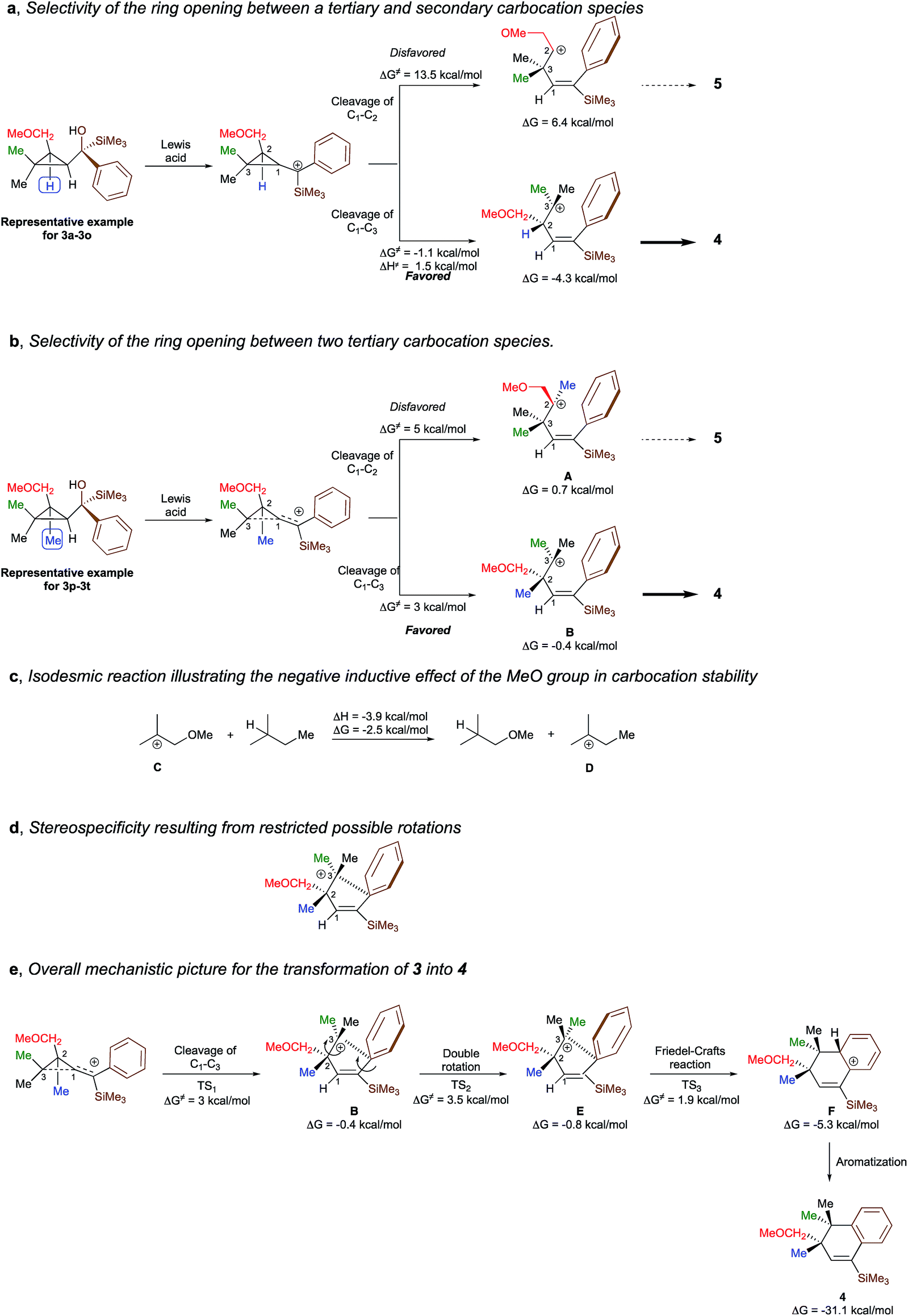 | ||
| Scheme 6 Rationalization for the proposed mechanism. | ||
Having now a good understanding on the selectivity of the ring-opening, we then focus our attention to the stereospecificity of the reaction that was particularly puzzling in a process proceeding through carbocationic intermediates. Calculations shows that for the carbocation B to epimerize, a rotation around the C2–C3 bond has to occur. However, the system is geometrically blocked by the proximity of the aromatic ring (d[C3–Cipso] = 2.86 A) impeding all epimerization (Scheme 6d). One could also consider that the C1–C2 rotation precedes the C2–C3 rotation but is much higher in energy (ΔG# = 8.6 kcal mol−1) than the Friedel–Crafts reaction described in Scheme 6e. Interestingly, the aromatization step (F into 4) is exergonic (ΔG = −25.9 kcal mol−1) and is almost barrierless.
The overall picture of the reaction mechanism is then described in Scheme 6e where product 4 is selectively formed from the corresponding cyclopropane 3 by combining the selective ring-opening reaction controlled by the inductive effect of the alkoxy functionality and the stereospecific cyclization controlled by a restricted conformation of the carbocation.
Conclusions
In conclusion, a protocol for the diastereoselective preparation of functionalized dihydronaphthalene backbones, useful precursor of diterpenoids, was reported from easily accessible cyclopropanes in three catalytic steps from commercially available alkynes. The versatility of the transformation was illustrated by the complete selectivity of the ring-opening and by the stereospecificity during the migration. Mechanistic control experiments and computational studies reveal the key inductive effect of the alkoxy functionality in the selectivity of the carbon–carbon bond cleavage and the π-stabilization of the carbocationic intermediates leading to an overall stereospecific process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreement 786976-ERC and by the Ministry of Science and Technology (grant No. 330/17).Notes and references
- (a) L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415 CrossRef CAS PubMed; (b) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651 CrossRef CAS PubMed; (c) X. Liu and J. M. Fox, J. Am. Chem. Soc., 2006, 128, 5600 CrossRef CAS PubMed; (d) V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382 CrossRef CAS PubMed; (e) M. L. Deem, Synthesis, 1972, 675 CrossRef CAS; (f) A. B. Charette and A. Beauchemin, Org. React., 2001, 58, 1 CAS; (g) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (h) M. Rubin, M. Rubina and V. Gevorgyan, Synthesis, 2006, 8, 1221 Search PubMed; (i) M. Rubina and V. Gevorgyan, Tetrahedron, 2004, 60, 3129 CrossRef CAS.
- (a) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CrossRef CAS PubMed; (b) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (c) M. E. Van Der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (d) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (e) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (f) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (g) K. Ruhland, Eur. J. Org. Chem., 2012, 2683 CrossRef CAS.
- (a) S. Danishefsky, Acc. Chem. Res., 1979, 12, 66 CrossRef CAS; (b) O. G. Kulinkovich, Russ. Chem. Rev., 1993, 62, 839 CrossRef; (c) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (d) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625 CrossRef CAS PubMed; (e) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (f) W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS; (g) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (h) A. de Meijere, Cyclopropanes, Transformations, Houben-Weyl Methods of Organic Chemistry, Thieme, Stuttgart, Germany, 1997, vol. E17c Search PubMed; (i) Topics in Current Chemistry, ed. A. de Meijere, Springer, Berlin, 2000, vol. 207, DOI:10.1007/3-540-48255-5.
- (a) P. Tang and Y. Qin, Synthesis, 2012, 44, 2969 CrossRef CAS; (b) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS PubMed; (c) D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC.
- Latest reviews on functionalization of donor–acceptor cyclopropane: (a) S. Liao, X. Sun and Y. Tang, Acc. Chem. Res., 2014, 47, 2260 CrossRef CAS PubMed; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (c) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC; (d) K. L. Ivanov, E. V. Villemson, E. M. Budynina, O. A. Ivanova, I. V. Trushkov and M. Y. Melnikov, Chem.–Eur. J., 2015, 21, 4975 CrossRef CAS PubMed; (e) A. K. Pandey, A. Ghosh and P. Banerjee, Isr. J. Chem., 2016, 56, 512 CrossRef CAS; (f) S. Racine, J. Vuilleumier and J. Waser, Isr. J. Chem., 2016, 56, 566 CrossRef CAS; (g) E. M. Budynina, K. L. Ivanov, I. D. Sorokin and M. Y. Melnikov, Synthesis, 2017, 49, 3035 CrossRef CAS.
- Selected examples of donor–acceptor cyclopropane reactions: (a) Y.-Y. Zhou, L.-J. Wang, J. Li, X.-L. Sun and Y. Tang, J. Am. Chem. Soc., 2012, 134, 9066 CrossRef CAS PubMed; (b) J. Kaschel, T. F. Schneider, D. Kratzert, D. Stalke and D. B. Werz, Angew. Chem., Int. Ed., 2012, 51, 11153 CrossRef CAS PubMed; (c) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239 CrossRef CAS PubMed; (d) Q.-K. Kang, L. Wang, Q.-J. Liu, J.-F. Li and Y. Tang, J. Am. Chem. Soc., 2015, 137, 14594 CrossRef CAS PubMed; (e) T. Kaicharla, T. Roy, M. Thangaraj, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2016, 55, 10061 CrossRef CAS PubMed; (f) A. U. Augustin, M. Sensse, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2017, 56, 14293 CrossRef CAS PubMed; (g) A. Lücht, L. J. Patalag, A. U. Augustin, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2017, 56, 10587 CrossRef PubMed; (h) R. A. Novikov, D. D. Borisov, A. V. Tarasova, Y. V. Tkachev and Y. V. Tomilov, Angew. Chem., Int. Ed., 2018, 130, 10450 CrossRef; (i) A. Ortega, R. Manzano, U. Uria, L. Carrillo, E. Reyes, T. Tejero, P. Merino and J. L. Vicario, Angew. Chem., Int. Ed., 2018, 57, 8225 CrossRef CAS PubMed; (j) A. Reding, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 7266 CrossRef CAS PubMed.
- (a) I. N. Nazarov and I. I. Zaretskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1941, 211 CAS. For reviews, see: (b) K. L. Habermas, S. E. Denmark and T. K. Jones, Org. React., 1994, 45, 1 CAS; (c) M. A. Tius, Eur. J. Org. Chem., 2005, 2193 CrossRef CAS; (d) A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577 CrossRef CAS; (e) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676 RSC; (f) W. Nakanishi and F. G. West, Curr. Opin. Drug Discovery Dev., 2009, 12, 732 CAS; (g) T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531 CrossRef CAS.
- (a) W. S. Murphy and S. Wattanasin, Tetrahedron Lett., 1980, 21, 1887 CrossRef CAS; (b) W. S. Murphy and S. Wattanasin, J. Chem. Soc., Perkin Trans. 1, 1981, 2920 RSC; (c) W. S. Murphy and S. Wattanasin, J. Chem. Soc., Perkin Trans. 1, 1982, 271 RSC; (d) W. S. Murphy and S. Wattanasin, J. Chem. Soc., Perkin Trans. 1, 1982, 1029 RSC; (e) O. Tsuge, S. Kanemasa, T. Otsuka and T. Suzuki, Bull. Chem. Soc. Jpn., 1988, 61, 2897 CrossRef CAS; (f) L. Greiner-Bechert, T. Sprang and H.-H. Otto, Monatsh. Chem., 2005, 136, 635 CrossRef CAS; (g) V. K. Yadav and N. V. Kumar, Chem. Commun., 2008, 3774 RSC.
- (a) M. C. Martin, R. Shenje and S. France, Isr. J. Chem., 2016, 56, 499 CrossRef CAS; (b) C. W. Williams, R. Shenje and S. France, J. Org. Chem., 2016, 81, 8253 CrossRef CAS PubMed; (c) M. J. Sandridge and S. France, Org. Lett., 2016, 18, 4218 CrossRef CAS PubMed; (d) F. De Simone, T. Saget, F. Benfatti, S. Almeida and J. Waser, Chem.–Eur. J., 2011, 17, 14527 CrossRef CAS PubMed; (e) F. De Simone, J. Andres, R. Torosantucci and J. Waser, Org. Lett., 2009, 11, 1023 CrossRef CAS PubMed.
- (a) W. Wu, Z. Lin and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315 RSC; (b) K. Sasazawa, S. Takada, T. Yubune, N. Takaki, R. Ota and Y. Nishii, Chem. Lett., 2017, 46, 524 CrossRef CAS; (c) S. Takada, N. Takaki, K. Yamada and Y. Nishii, Org. Biomol. Chem., 2017, 15, 2443 RSC; (d) E. Yoshida, K. Nishida, K. Toriyabe, R. Taguchi, J. Motoyoshiya and Y. Nishii, Chem. Lett., 2010, 39, 194 CrossRef CAS; (e) J. Ito, D. Sakuma and Y. Nishii, Chem. Lett., 2015, 44, 297 CrossRef CAS; (f) D. Sakuma, J. Ito, R. Sakai, R. Taguchi and Y. Nishii, Chem. Lett., 2014, 43, 610 CrossRef CAS.
- (a) G. Sklute, D. Amsallem, A. Shibli, J. P. Varghese and I. Marek, J. Am. Chem. Soc., 2003, 125, 11776 CrossRef CAS PubMed; (b) G. Sklute and I. Marek, J. Am. Chem. Soc., 2006, 128, 4642 CrossRef CAS PubMed; (c) G. Sklute and I. Marek, Chem. Commun., 2007, 1683 Search PubMed; (d) G. Kolodney, G. Sklute, S. Perrone, P. Knochel and I. Marek, Angew. Chem., Int. Ed., 2007, 46, 9291 CrossRef CAS PubMed; (e) B. Dutta, N. Gilboa and I. Marek, J. Am. Chem. Soc., 2010, 132, 5588 CrossRef CAS PubMed; (f) T. Mejuch, B. Dutta, M. Botoshansky and I. Marek, Org. Biomol. Chem., 2011, 10, 5803 RSC; (g) Y. Minko, M. Pasco, M. Botoshansky and I. Marek, Nature, 2012, 490, 522 CrossRef CAS PubMed; (h) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed.
- (a) I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed; (b) D. Didier, P.-O. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem.–Eur. J., 2014, 20, 1038 CrossRef CAS PubMed; (c) D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414 CrossRef PubMed; (d) D. S. Müller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552 RSC; (e) M. Preshel-Zlatsin, F.-G. Zhang, G. Eppe and I. Marek, Synthesis, 2016, 48, 3279 CrossRef CAS; (f) L. Dian, D. S. Muller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783 CrossRef CAS PubMed; (g) L. Dian and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 3682 CrossRef CAS PubMed; (h) H. Sommer and I. Marek, Chem. Sci., 2018, 9, 6503 RSC; (i) J. Bruffaerts, D. Pierrot and I. Marek, Nat. Chem., 2018, 10, 1164 CrossRef CAS.
- S. R. Roy, D. Didier, A. Kleiner and I. Marek, Chem. Sci., 2016, 7, 5989 RSC.
- (a) M. Simaan, P.-O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345 CrossRef CAS PubMed; (b) M. Simaan and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 1543 CrossRef CAS PubMed.
- (a) F. G. Zhang, G. Eppe and I. Marek, Angew. Chem., Int. Ed., 2016, 55, 714 CrossRef CAS PubMed; (b) F.-G. Zhang and I. Marek, J. Am. Chem. Soc., 2017, 139, 8364 CrossRef CAS PubMed; (c) C. Tugny, F. Zhang and I. Marek, Chem.–Eur. J., 2018, 25, 205 CrossRef PubMed.
- Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, accession number 4q: 1888210 and 4t: 1937437.†.
- Although the focus of this research was on bond breaking and bond making even at quaternary centre possessing aliphatic groups with non-donor substituents (the most challenging substrates), a remark by a reviewer led us also to consider the stereochemical outcome of a more donor–acceptor substrate 3 with a benzylic stereocenter (Scheme 1d with R1 = Bu, R2 = H, R3 = Me, R4 = SiMe3, R5 = Ph and R6 = H). In this case, the product 5 was obtained with a diastereoisomer ratio of 95:05 indicating that even in this case, the stereochemistry is preserved.
- Y. Lou, M. Horikawa, R. A. Kloster, N. A. Hawryluk and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 8916 CrossRef CAS PubMed.
- (a) J. D. Sellars and P. G. Steel, Eur. J. Org. Chem., 2007, 3815 CrossRef CAS; (b) P. Finkbeiner, K. Murai, M. Röpke and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 11349 CrossRef CAS.
- A. Reding, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2018, 57, 10610 CrossRef CAS PubMed.
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1994 Search PubMed; (b) W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, 2001 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1888210 and 1937437. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03832a |
| This journal is © The Royal Society of Chemistry 2019 |