
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switching the site-selectivity of C–H activation in aryl sulfonamides containing strongly coordinating N-heterocycles†
Yi
Dong‡
 ab,
XuePeng
Zhang‡
c,
Jiajing
Chen
ab,
Wenxing
Zou
ab,
Songwen
Lin
ab and
Heng
Xu
*ab
ab,
XuePeng
Zhang‡
c,
Jiajing
Chen
ab,
Wenxing
Zou
ab,
Songwen
Lin
ab and
Heng
Xu
*ab
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. E-mail: xuheng@imm.ac.cn
bBeijing Key Laboratory of Active Substances Discovery and Druggability Evaluation, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China
cLab of Computational and Drug Design, Peking University Shenzhen Graduate School, Shenzhen 518055, China
First published on 12th August 2019
Abstract
The limitations of arene C–H functionalization of aryl sulfonamides containing strongly coordinating N-heterocycles were overcome using a Rh(III) catalyst. The site-selectivity of C–H carbenoid functionalization at the ortho position relative to either the sulfonamide or N-heterocycle directing groups was elegantly switched using solvents of different polarities and different additive concentrations. Importantly, sulfonamide-group-directed ortho-C–H carbenoid functionalization tolerated strongly coordinating N-heterocycles, including pyridine, pyrrole, thiazole, pyrimidine, and pyrazine. Density functional theory (DFT) calculations were performed to rationalize the reaction mechanisms and the influence of reaction polarity.
Introduction
Transition-metal-catalyzed C–H functionalization is an important and efficient synthetic tool in organic synthesis owing to its straightforward and economical features.1–6 Although directing-group-oriented aryl C–H functionalization has developed rapidly in the past decade, some limitations have remained difficult to overcome.7–9 For example, C–H activation often selectively occurs at the ortho position relative to the directing group.10–12 However, when two or more directing groups coexist in one molecule, C–H activation directed by weakly coordinating groups is difficult to achieve owing to competing strongly coordinating directing groups, such as N-heterocycles.13–21 These groups often preferentially bind with transition metals and interfere with C–H functionalization through the desired directing group, which hinders late-stage modification in drug discovery.22–24 Recently, Dai, Yu, and Ackermann et al. reported pioneering examples of palladium-,25 copper-,26 and cobalt-catalyzed27 site-selective C–H functionalizations of amides and imidates that tolerated strongly coordinating N-heterocycles (Fig. 1A–C). However, switching the C–H functionalization selectivity of substrates containing two directing groups remains challenging (Fig. 1D).28–38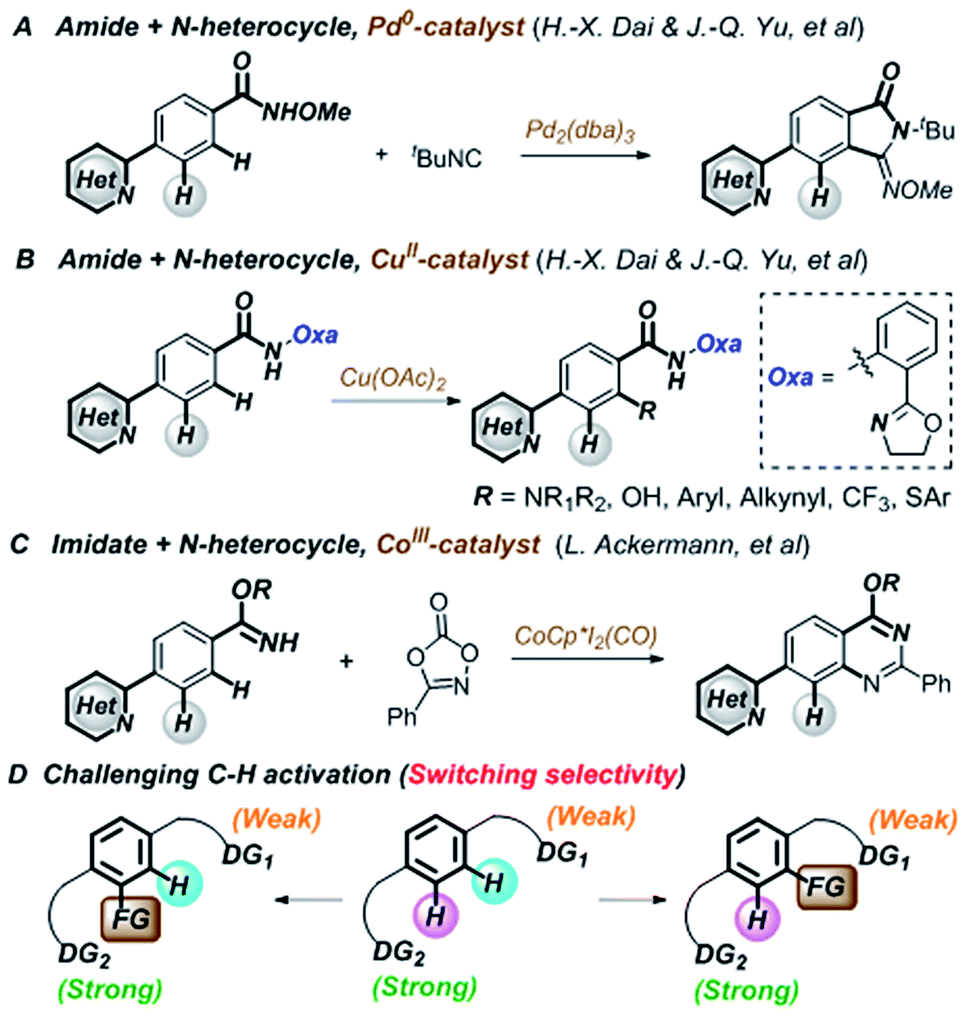 | ||
| Fig. 1 Overcoming the limitation of directed C–H functionalization. | ||
Both N-heterocycles and sulfonamides are ubiquitous in biologically active molecules owing to their hydrogen bond forming abilities and favorable drug-like properties.39–43 Considering their dual pharmacological effects, switching the selectivity of the C–H functionalization of aryl sulfonamides containing strongly coordinating N-heterocycles would be invaluable in the late-stage modification of potential drug molecules (Fig. 2A).44–46 In this study, we successfully switched the selectivity of the C–H carbenoid functionalization47–66 of sulfonamides67–80 containing N-heterocycles81 using a rhodium catalyst. The selectivity was precisely controlled by changing the reaction medium polarity and additive concentration, affording C–H activation at the ortho position relative to the N-heterocycle (charge-neutral forms, denoted as L-type ligands) or sulfonamide (easily deprotonated to charge-negative forms, denoted as X-type ligands) directing groups.
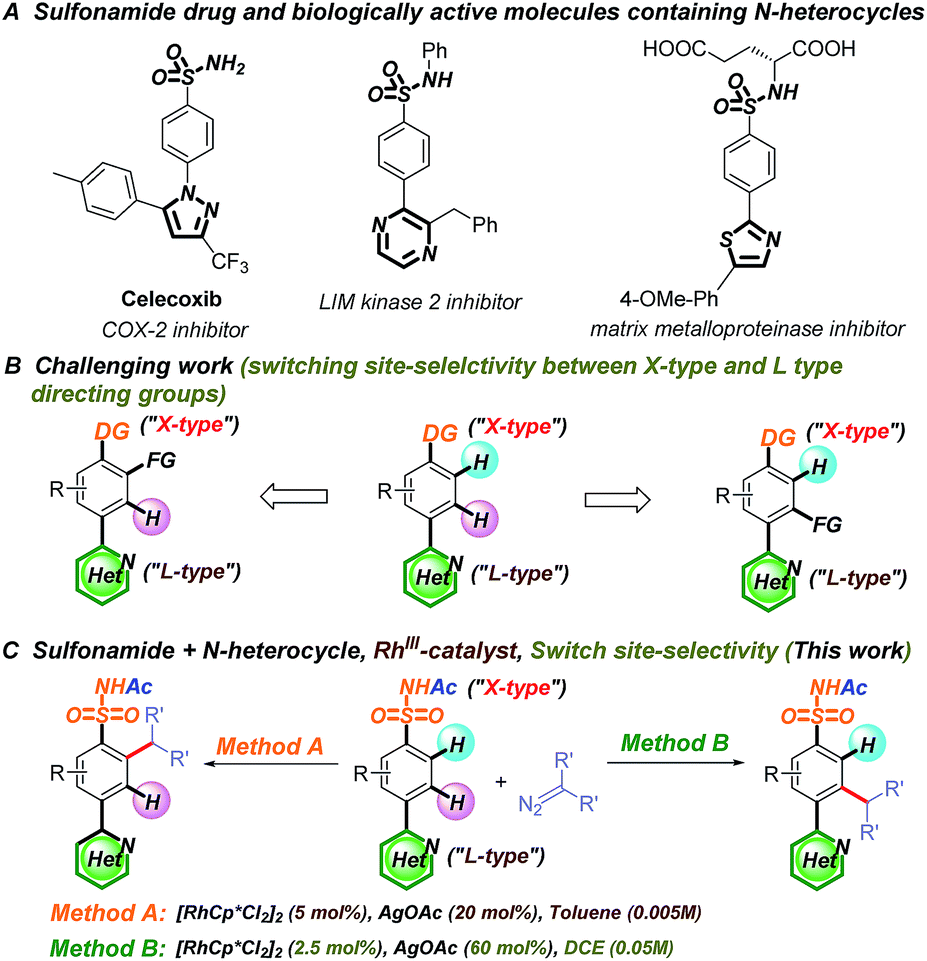 | ||
| Fig. 2 Site-selective C–H functionalization of sulfonamides containing N-heterocycles. | ||
Results and discussion
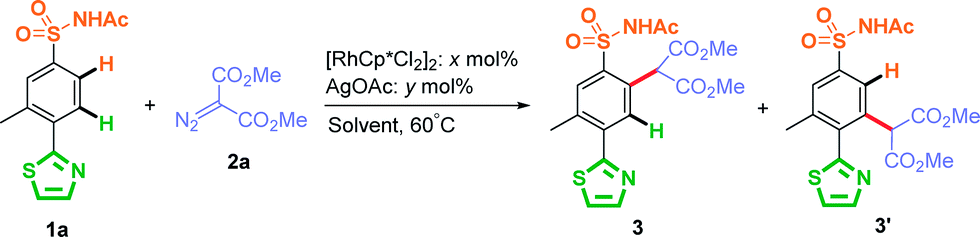
Initially, this Rh(III)-catalyzed site-selective C–H carbenoid functionalization was investigated for solvent effects (0.05 M concentration) by using AgOAc as an additive at 60 °C, with the results summarized in Table 1 (entries 1 to 5). When 3-methyl-4-thiazole-N-acetyl sulfonamide (1a) was used as a model substrate, C–H carbenoid functionalization product 3′ at the ortho-position of thiazole was only observed and isolated in 79% yield when MeOH (a polar protonic solvent) was used as the reaction solvent (Table 1, entry 1). Other tested polar solvents, such as DMF and MeCN, also favored generation of the C–H activated product at the ortho-position of the thiazole group (Table 1, entries 2 and 3). When non-polar solvents were used, such as DCE and toluene, the thiazole-directed C–H activated product was decreased significantly (Table 1, entries 4 and 5). It was notable that the amount of sulfonamide directed C–H activated product 3 was close to thiazole directed C–H activated product 3′ (3/3′ ratio, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6) when less-polar toluene was used as the reaction solvent (Table 1, entry 5). These results suggested that the polarity of the solvent strongly determined the site-selectivity of this rhodium catalyzed C–H carbenoid functionalization: the less polar the solvent, the more favorable was the sulfonamide-directed C–H activation product. Importantly, this phenomenon was more obvious at low concentrations (Table 1, entries 6–8), and when the reaction concentration was reduced to 0.01 M in DCE or toluene, an increasing amount of sulfonamide-directed activation product 3 was observed. It was worth noting that product 3 was isolated as the major product and the 3/3′ ratio could reach 33:1 when the reaction was carried out in toluene at 0.01 M concentration (Table 1, entry 7). The result given by a solvent mixture of DCE and toluene (v/v = 1:1) more clearly reflected the solvent polarity effect on this Rh(III)-catalyzed site-selective C–H carbenoid functionalization (Table 1, entry 8). Further decreasing the concentration to 0.005 M resulted in an excellent site-selectivity of 3 (3/3′ ratio, >99:1, Table 1, entry 9). Reducing the amount of [RhCp*Cl2]2 to 2.5 mol% resulted in a decreased 3:3′ ratio of 28:1, which showed that 5 mol% of the catalyst loading amount was necessary for this selective sulfonamide directed C–H carbenoid functionalization (entry 10). In contrast, increasing the reaction concentration led to increase of the thiazole-directed product 3′, which also well reflected the significant solvent effect of this site-selective C–H carbenoid functionalization (Table 1, entries 11 and 12). On the other hand, the effect of the amount of AgOAc on site-selectivity was also considered and systematically investigated in DCE (Table 1, entries 13 to 16). Increasing the amount of AgOAc resulted in a lower 3/3′ ratio (entries 13 and 14), which reached <1:99 when the amount of AgOAc was 8- and 12-fold that of [RhCp*Cl2]2 respectively. It is worth noting that 2.5 mol% [RhCp*Cl2]2 and 60 mol% AgOAc loading amount could guarantee an excellent site-selectivity of 3′ (3/3′ ratio, <1:99, entry 15). These results showed that increasing the amount of AgOAc favored C–H functionalization at the thiazole ortho-position. It may be possible that changing the AgOAc concentration affected the polarity of the reaction environment, influencing the site-selectivity. It is also possible that AgOAc plays the role of a Lewis acid which could bind to an N-heterocycle or diazo compound and affect the site-selectivity.82 When a more economic acetate salt tetrabutylammonium acetate was used as an additive instead of AgOAc, similar yields were obtained. However, the site-selectivities of the C–H carbenoid functionalization were significantly reduced (entries 17 and 18). Therefore, two optimal conditions to switch site-selectivity at the ortho-position of sulfonamide and N-heterocycle were chosen respectively (entries 9 and 15). These two methods not only gave good yields, but more importantly excellent site-selectivities.
:1.6) when less-polar toluene was used as the reaction solvent (Table 1, entry 5). These results suggested that the polarity of the solvent strongly determined the site-selectivity of this rhodium catalyzed C–H carbenoid functionalization: the less polar the solvent, the more favorable was the sulfonamide-directed C–H activation product. Importantly, this phenomenon was more obvious at low concentrations (Table 1, entries 6–8), and when the reaction concentration was reduced to 0.01 M in DCE or toluene, an increasing amount of sulfonamide-directed activation product 3 was observed. It was worth noting that product 3 was isolated as the major product and the 3/3′ ratio could reach 33:1 when the reaction was carried out in toluene at 0.01 M concentration (Table 1, entry 7). The result given by a solvent mixture of DCE and toluene (v/v = 1:1) more clearly reflected the solvent polarity effect on this Rh(III)-catalyzed site-selective C–H carbenoid functionalization (Table 1, entry 8). Further decreasing the concentration to 0.005 M resulted in an excellent site-selectivity of 3 (3/3′ ratio, >99:1, Table 1, entry 9). Reducing the amount of [RhCp*Cl2]2 to 2.5 mol% resulted in a decreased 3:3′ ratio of 28:1, which showed that 5 mol% of the catalyst loading amount was necessary for this selective sulfonamide directed C–H carbenoid functionalization (entry 10). In contrast, increasing the reaction concentration led to increase of the thiazole-directed product 3′, which also well reflected the significant solvent effect of this site-selective C–H carbenoid functionalization (Table 1, entries 11 and 12). On the other hand, the effect of the amount of AgOAc on site-selectivity was also considered and systematically investigated in DCE (Table 1, entries 13 to 16). Increasing the amount of AgOAc resulted in a lower 3/3′ ratio (entries 13 and 14), which reached <1:99 when the amount of AgOAc was 8- and 12-fold that of [RhCp*Cl2]2 respectively. It is worth noting that 2.5 mol% [RhCp*Cl2]2 and 60 mol% AgOAc loading amount could guarantee an excellent site-selectivity of 3′ (3/3′ ratio, <1:99, entry 15). These results showed that increasing the amount of AgOAc favored C–H functionalization at the thiazole ortho-position. It may be possible that changing the AgOAc concentration affected the polarity of the reaction environment, influencing the site-selectivity. It is also possible that AgOAc plays the role of a Lewis acid which could bind to an N-heterocycle or diazo compound and affect the site-selectivity.82 When a more economic acetate salt tetrabutylammonium acetate was used as an additive instead of AgOAc, similar yields were obtained. However, the site-selectivities of the C–H carbenoid functionalization were significantly reduced (entries 17 and 18). Therefore, two optimal conditions to switch site-selectivity at the ortho-position of sulfonamide and N-heterocycle were chosen respectively (entries 9 and 15). These two methods not only gave good yields, but more importantly excellent site-selectivities.
|
|
|||||
|---|---|---|---|---|---|
| Entry | x/y | Solvent | Conc. | Yield/%b | 3/3′c |
| a Reaction conditions: [RhCp*Cl2]2 (x mol%), AgOAc (y mol%), 1a (0.125 mmol), 2a (1.1 equiv.), solvent, 60 °C. b Isolated yield. c Determined by 1H NMR analysis of the crude reaction mixture before separation. d Tetrabutylammonium acetate instead of AgOAc. | |||||
| 1 | 5/20 | MeOH | 0.05 M | 3′/79 | Only 3′ |
| 2 | 5/20 | DMF | 0.05 M | 3′/77 | 1:48 |
| 3 | 5/20 | MeCN | 0.05 M | 3′/78 | 1:11 |
| 4 | 5/20 | DCE | 0.05 M | 3′/65 | 1:4 |
| 5 | 5/20 | Toluene | 0.05 M | 3 + 3′/26 + 50 | 1:1.6 |
| 6 | 5/20 | DCE | 0.01 M | 3 + 3′/40 + 44 | 1:1 |
| 7 | 5/20 | Toluene | 0.01 M | 3/80 | 33:1 |
| 8 | 5/20 | Toluene/DCE = 1:1 |
0.01 M | 3/62 | 4:1 |
| 9 | 5/20 | Toluene | 0.005 M | 3 /81 |
>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 10 | 2.5/10 | Toluene | 0.005 M | 3/80 | 28:1 |
| 11 | 5/20 | DMF | 0.25 M | 3′/81 | <1:99 |
| 12 | 5/20 | MeCN | 0.25 M | 3′/80 | 1:47 |
| 13 | 5/40 | DCE | 0.05 M | 3′/88 | <1:99 |
| 14 | 5/60 | DCE | 0.05 M | 3′/87 | <1:99 |
| 15 | 2.5/60 | DCE | 0.05 M | 3′ /88 |
<1:99
|
| 16 | 2.5/20 | DCE | 0.05 M | 3′/83 | 1:30 |
| 17d | 5/20 | Toluene | 0.005 M | 3/79 | 26:1 |
| 18d | 2.5/60 | DCE | 0.05 M | 3 + 3′/29 + 46 | 1:1.5 |
With the two optimal conditions in hand, sulfonamide substrates containing various N-heterocycles in a competitive position were further investigated, including thiazole, pyrazole, imidazole, pyridine, pyrimidine, and pyrazine substrates (Scheme 1). As expected, sulfonamide-group-directed ortho-C–H carbenoid functionalization proceeded in the presence of 5.0 mol% [RhCp*Cl2]2 catalyst and 20 mol% AgOAc as the additive in less-polar toluene (0.005 M) at 60 °C, affording excellent site-selectivity (Scheme 1A, Method A, 4–14). In addition to phenyl sulfonamides, thienyl sulfonamides also showed excellent site-selectivity (15 and 16), while substrates containing phosphate ester and sulfone ester diazo groups instead of dimethyl 2-diazomalonate also gave satisfactory results (17 and 18). Notably, C–H di-activation at the ortho-positions relative to the sulfonamide group afforded the major product when para-heterocycle-substituted benzenesulfonamide was used (19–22). As shown in Scheme 1B, when the reaction was conducted in DCE (0.05 M), the N-heterocycle-directed C–H carbenoid functionalization of this difunctional compound performed well in the presence of 2.5 mol% [RhCp*Cl2]2 catalyst and 60 mol% AgOAc as the additive at 60 °C, with good yields and excellent site-selectivity (Method B, 23–30). A 1,2-disubstituted substrate was also tried, but no desired carbenoid functionalization product was obtained, which may be the result of a bidentate coordination, impeding the free coordination site for cyclometallation.
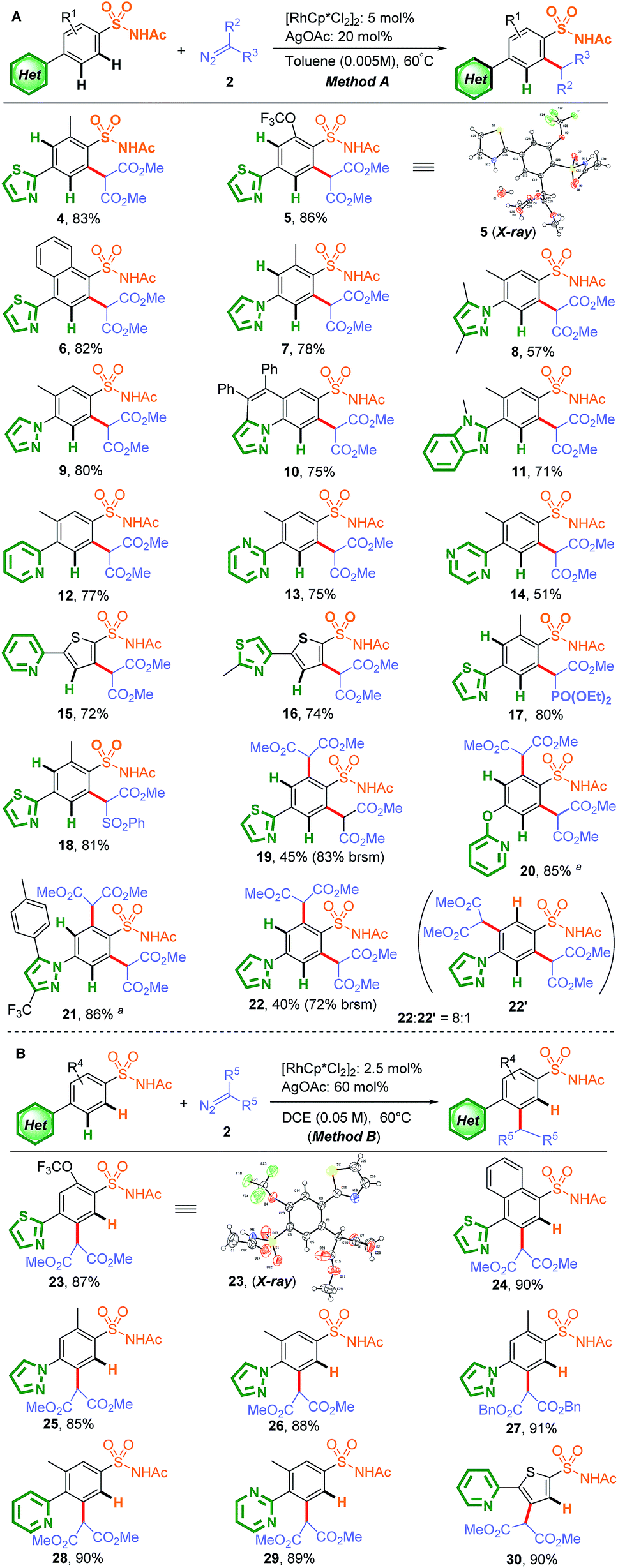 | ||
| Scheme 1 Site-selective ortho C–H carbenoid functionalization of sulfonamides containing N-heterocycles. (A) Method A: [RhCp*Cl2]2 (5 mol%), AgOAc (20 mol%), sulfonamides containing N-heterocycles (0.125 mmol), 2a (1.1 equiv.), toluene (25 mL), 60 °C. brsm = based on the recovered starting material. a2.1 equiv. of 2a was used. (B) Method B: [RhCp*Cl2]2 (2.5 mol%), AgOAc (60 mol%), sulfonamides containing N-heterocycles (0.125 mmol), 2a (1.1 equiv.), DCE (2.5 mL), 60 °C. | ||
Intermolecular competition experiments were conducted to explore the unique chemoselectivity of difunctional sulfonamides containing N-heterocycle substrates (Scheme 2). Notably, X-type sulfonamides outcompeted strongly coordinating L-type thiazole, pyrazole, and pyridine substrates using method A. In contrast, when the competition reaction was conducted using method B, N-heterocycle-oriented C–H carbenoid functionalization was the major pathway. These intermolecular competition results implied that changing the C–H carbenoid functionalization reaction conditions could affect the competitive reactivity between X-type and L-type directing groups. More importantly, this strategy could prevent interference from L-type N-heterocycles to afford X-type sulfonamide-oriented C–H activation, despite N-heterocycles possessing strongly coordinating properties with metal catalysts.
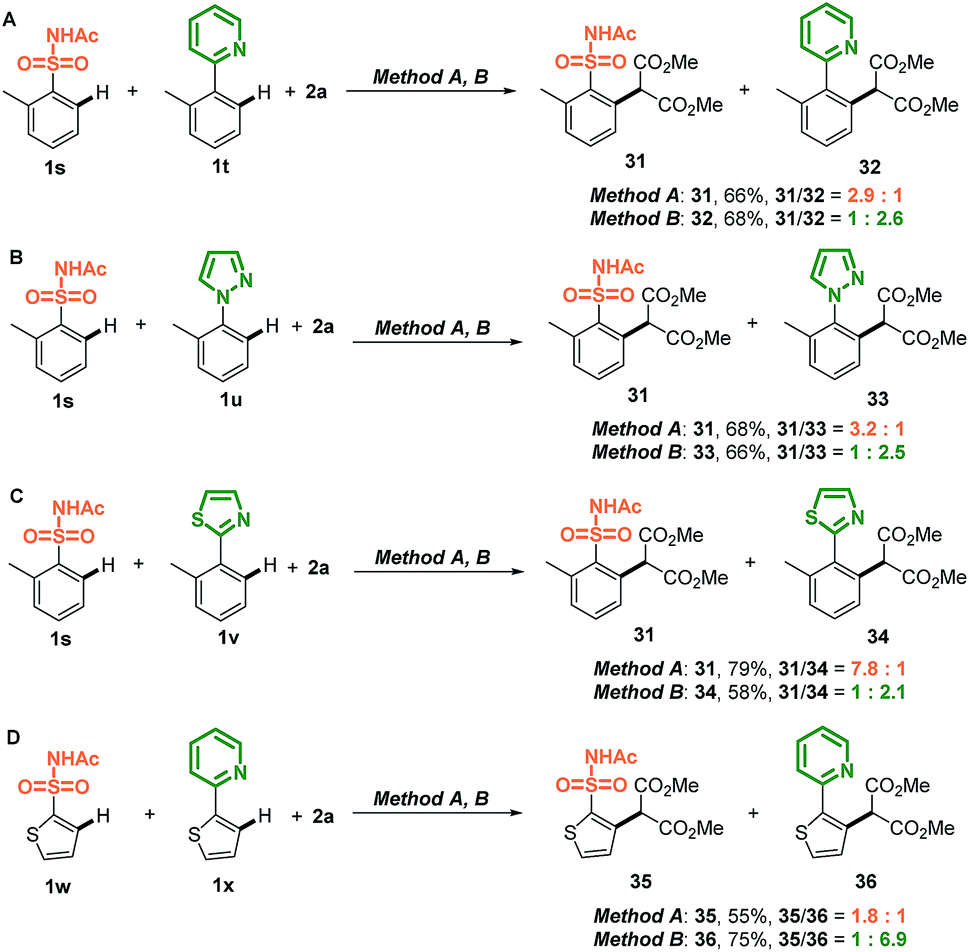 | ||
| Scheme 2 Intermolecular competition experiments between X-type sulfonamides and L-type N-heterocycles. | ||
The mechanism of the site-selective aryl C–H carbenoid functionalization between sulfonamide and N-heterocycles was further investigated by experimental studies and theoretical calculations. The mechanism of directing group oriented aryl C–H carbenoid functionalization has been illustrated in the previous literature, and three major steps including C–H activation, metal–carbene formation and final C–C bond formation process were proposed to take place.50 Firstly, as shown in Scheme 3, isotope-labelling experiments of 3-methyl-4-thiazole-N-acetyl sulfonamide (1a) by method A and B in the presence of [D4]-MeOD showed that reversible processes for sulfonamide and thiazole directed C–H activation were involved under method A and B conditions.83–85 Notably, there was a relatively slight H/D exchange (5%) at the ortho position of the thiazole group using [D4]-MeOD as a co-solvent in the method A catalytic system, implying weak reversible properties. As shown in Scheme 4, kinetic isotope effect (KIE) experimental results showed a relatively small value, indicating that C–H bond cleavage was likely not the rate-determining step in the catalytic cycle.86,87 These results suggested that the selectivity does not appear in the first C–H activation step, but in the subsequent steps possibly.
 | ||
| Scheme 3 Isotope-labelling experiments. | ||
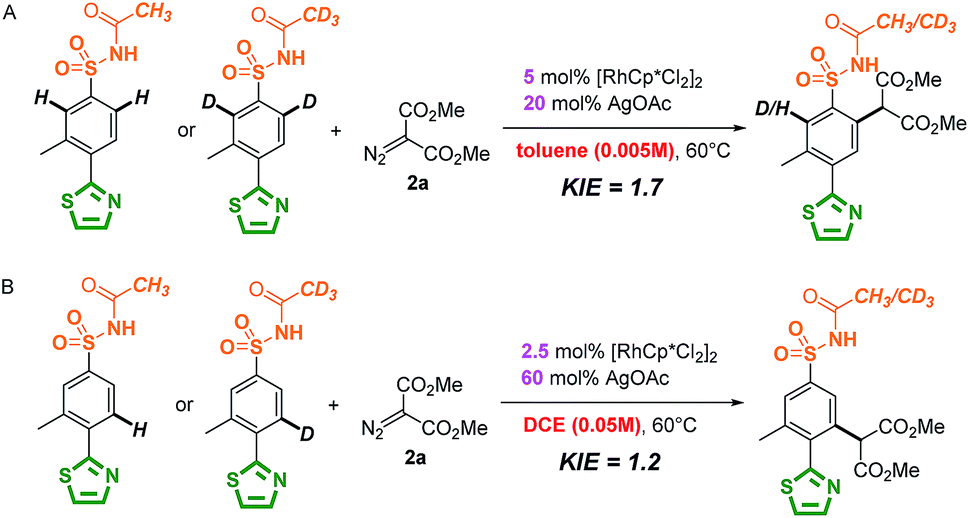 | ||
| Scheme 4 Kinetic isotope effect experiments. | ||
In order to delve into our conjecture about the mechanism on selectivity, density functional theory (DFT) calculations were performed. The RhCp*(OAc)2 complex was commonly considered as the active catalyst under the conditions of [RhCp*Cl2]2 with an abundant AgOAc additive.51,54,57 As implied in our experiments, the results of site-selective C–H carbenoid functionalization catalyzed by RhCp*(OAc)2 were similar to those catalyzed by RhCp*Cl2/AgOAc, which proved that RhCp*(OAc)2 was an active catalyst for the C–H carbenoid functionalization (details in the ESI†). An easier N–H deprotonation of the acetyl-substituted sulfonamide was predicted, and the active catalyst RhCp*(OAc)2 would firstly dissociate one OAc− ligand to coordinate with the deprotonated sulfonamide substrate to form the reactant complex RC. Similar to previous reports, domino C–H activation, metal–carbene formation, and finally C–C bond formation processes were predicted to take place.88–90 The deprotonated sulfonamide moiety or para-thiazole group would act as a directing group to locate the Rh(III) center adjacent to the ortho-C–H bond of either the sulfonamide or thiazole group, which guaranteed facilitation of the ortho-C–H activation processes in the transition state TS1 or TS1′. Both aryl C–H deprotonation to the mono-coordinated OAc− ligand and Rh–C bond formation processes were observed in the concerted-metalation–deprotonation (CMD)91–93 type C–H activation transition state TS1 or TS1′. Subsequently, the Rh(III)-bound neutral HOAc molecule would be easily replaced by the diazo compound 2a, and metal–carbene formation through clear nitrogen extrusion processes was observed in both TS2 and TS2′. The active metal–carbene would attack an adjacent aryl carbon center to generate C–C bond formation transition state TS3 or TS3′. Finally, transient product PC or PC′ was obtained through site-selective carbenoid functionalization at the ortho-position of either the sulfonamide or thiazole moiety. Alternatively, the active catalyst RhCp*(OAc)2 might directly react with diazo compound 2a to form a metal–carbene complex via a similar nitrogen extrusion process in TScarbene.
The profiles of the potential energy surface proposed above are shown in Fig. 3. It can be noted that the energies of the direct metal–carbene formation transition state TScarbene (31.2 and 34.2 kcal mol−1 in toluene and DCE, respectively) were higher than the barriers depicted in Fig. 3, which reconfirmed the fact that the active Rh(III) catalysts would preferentially react with aryl substrates (details in the ESI†). As depicted in Fig. 3, the free energy barrier in the metal–carbene formation was higher than those of the C–H activation process and C–C formation step. The calculated results demonstrated that the C–H activation step was not rate-determining, which was consistent with experimental KIE results. In addition, stoichiometric C–H rhodation experiments were conducted (Scheme 5). As shown in Scheme 5A, 14% and 0% D, respectively, were incorporated at the ortho-positions of the sulfonamide and thiazole groups at a lower reaction concentration (0.005 M) in toluene, which was supported by the energy barrier of the ortho-sulfonamide C–H activation in toluene (TS1 is 8.6 kcal mol−1 lower than TS1′). Furthermore, the competitive C–H activation results shown in Scheme 5B confirmed the calculated narrow free energy gap between TS1 and TS1′ (2.1 kcal mol−1) in DCE. The concentration factor was considered in Scheme 3, which implied that the increased concentration of reaction complexes and more polarized solvent would favour thiazole-directed C–H activation. The experimental observations were in consistence with calculated C–H activation barriers, especially in the pathway of the more polarized solution (ε = 25.0) wherein a narrower free energy gap between TS1 and TS1′ was observed (1.2 kcal mol−1). More importantly, a stable thiazole-directed intermediate IM′-OAc was generated, which was responsible for more deuteration at the thiazole side in Scheme 3B.
 | ||
| Fig. 3 Profile of the potential energy surface (PES) of site-selective carbenoid functionalization in aryl sulfonamide 1a catalyzed by the RhCp*(OAc)2 complex. The energy of reactant RC is referred to as the total energy of dissociative reactants (1a, 2a, RhCp*(OAc)2). | ||
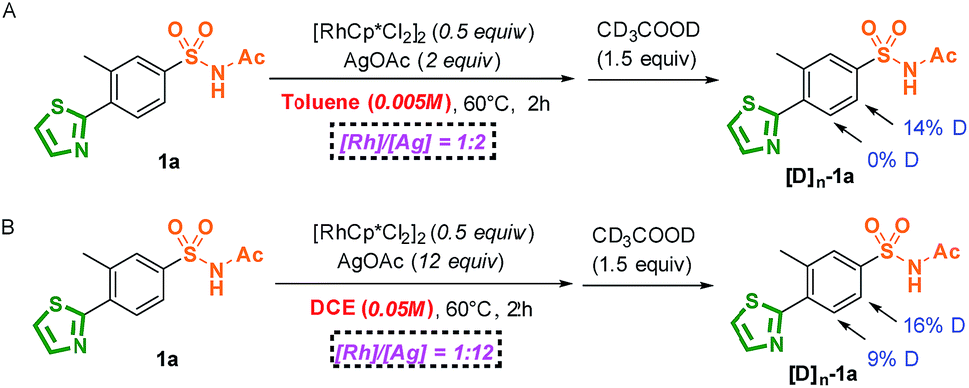 | ||
| Scheme 5 Stoichiometric C–H rhodation experiment. | ||
The reaction selectivities were further rationalized. As depicted in Fig. 3, the sulfonamide-directed pathway was more energetically favourable than the thiazole-directed pathway in toluene (red pathway). The calculated results were in good agreement with the experimental observations that ortho-sulfonamide C–H carbenoid functionalization product 3 was the major product in toluene (Table 1, entries 7, 9 and 10). Furthermore, close relative free energy barriers of C–H activation transition states TS1 (18.8 kcal mol−1) and TS1′ (20.9 kcal mol−1), and of metal–carbene formation transition states TS2 (25.1 kcal mol−1) and TS2′ (25.7 kcal mol−1) in DCE were observed (black pathway). The calculated competitive reaction pathways in DCE were in good agreement with experimental findings that both ortho-sulfonamide product 3 and ortho-thiazole product 3′ were obtained in DCE at a lower AgOAc concentration (Table 1, entry 6). The use of a more polarized solvent and/or high concentration of reaction complexes was also investigated, which together contributed to the increasing polarity of the reaction environment. As described above, the metal–carbene formation process was the rate-determining step, and therefore, the energy alterations of TS2 and TS2′ under various polarization conditions were investigated. As depicted in Fig. 4A, the thiazole-directed TS2′ was more polarized than the sulfonamide-directed TS2, which would rationalize the free energy changes in Fig. 4B wherein the thiazole-directed TS2′ was better stabilized in high polarity surroundings. Therefore, PES profiles in a more polarized solution are also provided in Fig. 3 (blue pathway). As demonstrated in Fig. 3, the energies of the thiazole-directed reaction species were decreased and the free energy of the rate-determining TS2′ was lower than that of TS2. Meanwhile, as mentioned above, the introduction of excess OAc− ligand would result in a more stable thiazole-directed intermediate IM2′-OAc. Thereby, the decreased thiazole-directed energy barriers and the important OAc− ligand participation demonstrated in Fig. 3 (blue), as well as the better stabilization of TS2′ in more polarized surroundings shown in Fig. 4B, would reasonably explain the reaction selectivities under the conditions of more polarized solvent and/or high concentration of reaction complexes demonstrated in Table 1.
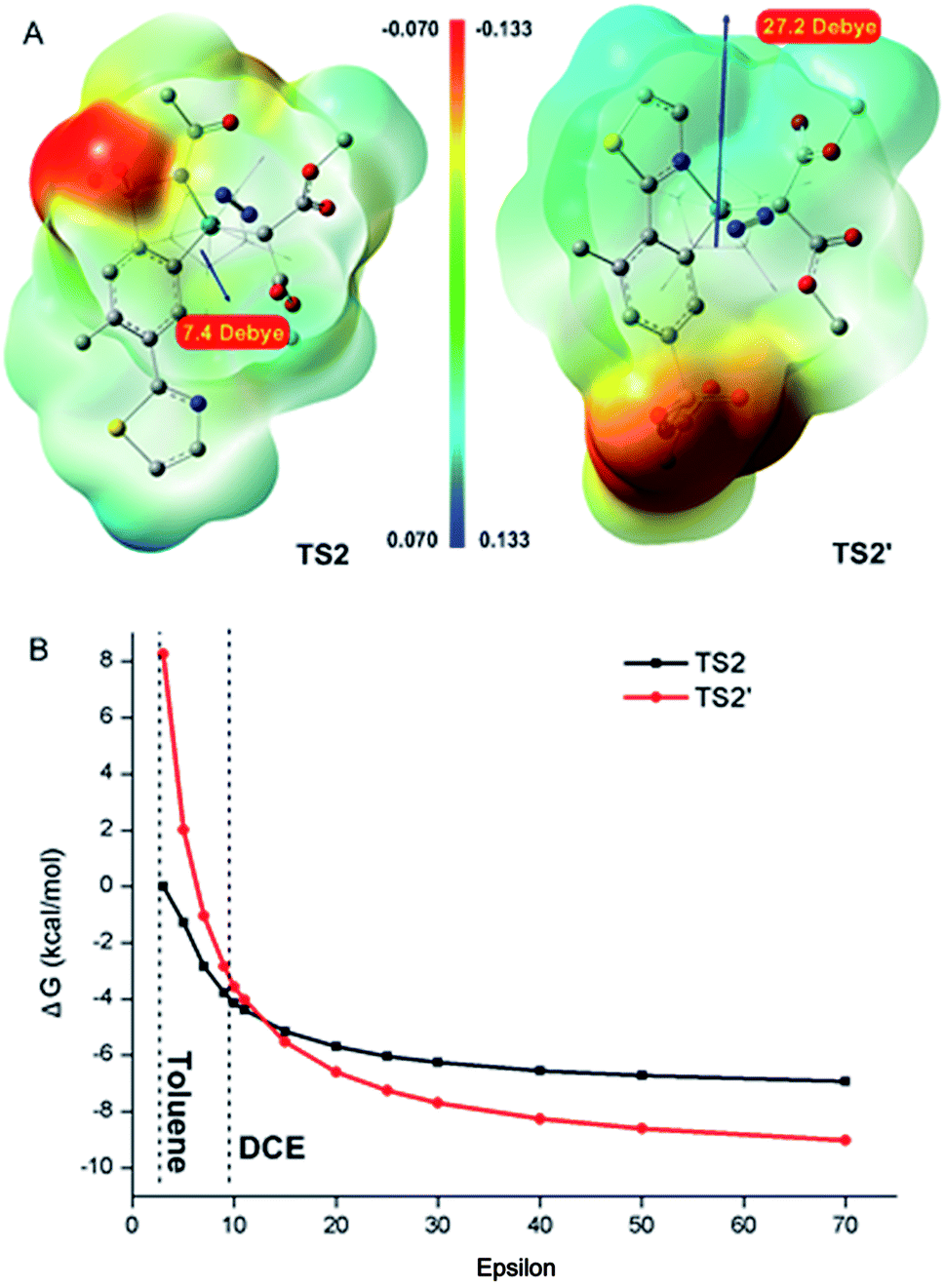 | ||
| Fig. 4 (A) Electrostatic potential (ESP) and dipole moment of TS2 and TS2′ in DCE. (B) Relative energy changes of TS2 and TS2′ with increasing solvent dielectric constant. The energy of TS2 in toluene (ε = 2.4) was set to zero. | ||
Conclusions
In summary, we have developed a Rh(III)-catalyzed arene C–H carbenoid functionalization with good yields and excellent site-selectivity switching for aryl sulfonamides containing strongly coordinating N-heterocycles. The polarity of the reaction environment was an important factor influencing the site-selectivity. Notably, the combination of a less-polar solvent, such as toluene, and a lower additive concentration favored C–H functionalization at the ortho-position relative to the sulfonamide group with excellent site-selectivity. Furthermore, strongly coordinating N-heterocycles, including pyridine, pyrrole, thiazole, pyrimidine, and pyrazine, were tolerated. This switchable site-selectivity carbenoid functionalization methodology is suitable for the late-stage modification of N-heterocycle-derived sulfonamide drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (21772235), the CAMS Innovation Fund for Medical Sciences (2017-I2M-3-011), the Drug Innovation Major Project (2018ZX09711-001-005), the Non-profit Central Research Institute Fund of the Chinese Academy of Medical Sciences (2018PT35003, 2019-RC-HL-008), and China Postdoctoral Science Foundation (2017M620496). We are grateful to Prof. Yun-Dong Wu (Peking University Shenzhen Graduate School) for a valuable discussion on the computational study.Notes and references
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- N. Kuhl, S. Schoöder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed.
- J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS PubMed.
- L. Ackermann, Top. Organomet. Chem., 2007, 24, 35 CrossRef CAS.
- I. Omae, Coord. Chem. Rev., 2004, 248, 995 CrossRef CAS.
- M. Wasa, B. T. Worrell and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 1275 CrossRef CAS PubMed.
- H. A. Malik, B. L. H. Taylor, J. R. Kerrigan, J. E. Grob, K. N. Houk, J. Du Bois, L. G. Hamann and A. W. Patterson, Chem. Sci., 2014, 5, 2352 RSC.
- Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7734 CrossRef CAS PubMed.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed.
- S. Duez, A. K. Steib, S. M. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 7686 CrossRef CAS PubMed.
- L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS PubMed.
- J.-P. Leclerc and K. Fagnou, Angew. Chem., Int. Ed., 2006, 45, 7781 CrossRef CAS PubMed.
- K. S. Kanyiva, Y. Nakao and R. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872 CrossRef CAS PubMed.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed.
- M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashral, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007 CrossRef CAS PubMed.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256 CrossRef PubMed.
- N. A. Meanwell, Chem. Res. Toxicol., 2011, 24, 1420 Search PubMed.
- Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Nature, 2014, 515, 389 CrossRef CAS PubMed.
- M. Shang, M.-M. Wang, T. G. Saint-Denis, M.-H. Li, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 5317 CrossRef CAS PubMed.
- H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386 CrossRef CAS PubMed.
- L. C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS PubMed.
- D. Lapointe, T. Markiewicz, C. J. Whipp, A. Toderian and K. Fagnou, J. Org. Chem., 2011, 76, 749 CrossRef CAS PubMed.
- J. M. Alderson, A. M. Phelps, R. J. Scamp, N. S. Dolan and J. M. Schomaker, J. Am. Chem. Soc., 2014, 136, 16720 CrossRef CAS PubMed.
- Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127 CrossRef CAS PubMed.
- R. D. Dolewski, P. J. Fricke and A. McNally, J. Am. Chem. Soc., 2018, 140, 8020 CrossRef CAS PubMed.
- H. J. Kim, J. Kim, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2011, 133, 16385 Search PubMed.
- J. Zhang, S.-C. Sha, A. Bellomo, N. Trongsiriwat, F. Gao, N. C. Tomson and P. J. Walsh, J. Am. Chem. Soc., 2016, 138, 4260 CrossRef CAS PubMed.
- V. K. Tiwari, N. Kamal and M. Kapur, Org. Lett., 2017, 19, 262 CrossRef CAS PubMed.
- S. Lee, S. Mah and S. Hong, Org. Lett., 2015, 17, 3864 CrossRef CAS PubMed.
- R. Boyaala, R. Touzani, T. Roisnel, V. Dorcet, E. Caytan, D. Jacquemin, J. Boixel, V. Guerchais, H. Doucet and J.-F. Soulé, ACS Catal., 2019, 9, 1320 CrossRef CAS.
- W. J. Kerr, M. Reid and T. Tuttle, ACS Catal., 2015, 5, 402 CrossRef CAS.
- S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Niardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- A. Ammazzalorso, B. De Filippis, L. Giampietro and R. Amoroso, Chem. Biol. Drug Des., 2017, 90, 1094 CrossRef CAS PubMed.
- F. Carta, C. T. Supuran and A. Scozzafava, Future Med. Chem., 2014, 6, 1149 CrossRef CAS PubMed.
- P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Angew. Chem., Int. Ed., 2017, 56, 4100 CrossRef CAS PubMed.
- M. Pal, M. Madan, S. Padakanti, V. R. Pattabiraman, S. Kalleda, A. Vanguri, R. Mullangi, N. V. S. Rao Mamidi, S. R. Casturi, A. Malde, B. Gopalakrishnan and K. R. Yeleswarapu, J. Med. Chem., 2003, 46, 3975 CrossRef CAS PubMed.
- F. Delbecq, G. Cordonnier, N. Pommery, D. Barbry and J.-P. Hénichart, Bioorg. Med. Chem. Lett., 2004, 14, 1119 CrossRef CAS PubMed.
- N. C. Goodwin, G. Cianchetta, H. A. Burgoon, J. Healy, R. Mabon, E. D. Strobel, J. Allen, S. Wang, B. D. Hamman and D. B. Rawlins, ACS Med. Chem. Lett., 2015, 6, 53 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC.
- Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS.
- F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986 RSC.
- Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed.
- W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed.
- Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204 CrossRef CAS PubMed.
- T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed.
- F. Hu, Y. Xia, F. Ye, Z. Liu, C. Ma, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 1364 CrossRef CAS PubMed.
- B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896 CrossRef CAS PubMed.
- Y. Liang, K. Yu, B. Li, S. Xu, H. Song and B. Wang, Chem. Commun., 2014, 50, 6130 RSC.
- B. Zhou, Z. Chen, Y. Yang, W. Ai, H. Tang, Y. Wu, W. Zhu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 12121 CrossRef CAS PubMed.
- Y. Xia, Z. Liu, S. Feng, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 223 CrossRef CAS PubMed.
- Á. Gutiérrez-Bonet, F. Juliá-Hernández, B. de Luis and R. Martin, J. Am. Chem. Soc., 2016, 138, 6384 CrossRef PubMed.
- J. H. Kim, S. Greβies and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5577 CrossRef CAS PubMed.
- R. B. Dateer and S. Chang, Org. Lett., 2016, 18, 68 CrossRef CAS PubMed.
- Q. Zhou, S. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 16013 CrossRef CAS PubMed.
- Z. Long, Z. Wang, D. Zhou, D. Wan and J. You, Org. Lett., 2017, 19, 2777 CrossRef CAS PubMed.
- Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 15539 CrossRef CAS PubMed.
- B. Shen, B. Wan and X. Li, Angew. Chem., Int. Ed., 2018, 57, 15534 CrossRef CAS PubMed.
- Y. Dong, J. Chen and H. Xu, Chem. Commun., 2019, 55, 2027 RSC.
- H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef CAS PubMed.
- M. V. Pham, B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 10610 CrossRef CAS PubMed.
- W. Xie, J. Yang, B. Wang and B. Li, J. Org. Chem., 2014, 79, 8278 CrossRef CAS PubMed.
- Q. Ding, T. Liu, Q. Zheng, Y. Zhang, L. Long and Y. Peng, RSC Adv., 2014, 4, 51309 RSC.
- X. Li, Y. Dong, F. Qu and G. Liu, J. Org. Chem., 2015, 80, 790 CrossRef CAS PubMed.
- D. Kalsi and B. Sundararaju, Org. Lett., 2015, 17, 6118 CrossRef CAS PubMed.
- O. Planas, C. J. Whiteoak, A. Company and X. Ribas, Adv. Synth. Catal., 2015, 357, 4003 CrossRef CAS.
- W. Liu, D. Wang, Y. Zhao, F. Yi and J. Chen, Adv. Synth. Catal., 2016, 358, 1968 CrossRef CAS.
- R. Vanjari, T. Guntreddi and K. N. Singh, Chem.–Asian J., 2016, 11, 696 CrossRef CAS PubMed.
- T. Lan, L. Wang and Y. Rao, Org. Lett., 2017, 19, 972 CrossRef CAS PubMed.
- N. Thrimurtulu, R. Nallagonda and C. M. R. Volla, Chem. Commun., 2017, 53, 1872 RSC.
- T. T. Nguyen, L. Grigorjeva and O. Daugulis, Chem. Commun., 2017, 53, 5136 RSC.
- N. Barsu, D. Kalsi and B. Sundararaju, Catal. Sci. Technol., 2018, 8, 5963 RSC.
- Y. Ran, Y. Yang, H. You and J. You, ACS Catal., 2018, 8, 1796 CrossRef CAS.
- L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed.
- P. Zhao, F. Wang, K. Han and X. Li, Org. Lett., 2012, 14, 3400 CrossRef CAS PubMed.
- Y. Li, Z. Qi, H. Wang, X. Yang and X. Li, Angew. Chem., Int. Ed., 2016, 55, 11877 CrossRef CAS PubMed.
- J. Wang, M. Wang, K. Chen, S. Zha, C. Song and J. Zhu, Org. Lett., 2016, 18, 1178 CrossRef CAS PubMed.
- Y. Yang, X. Wang, Y. Li and B. Zhou, Angew. Chem., Int. Ed., 2015, 54, 15400 CrossRef CAS PubMed.
- H. Jiang, S. Gao, J. Xu, X. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2016, 358, 188 CrossRef CAS.
- Y. Li, F. Wang, S. Yu and X. Li, Adv. Synth. Catal., 2016, 358, 880 CrossRef CAS.
- T. Zhou, W. Guo and Y. Xia, Chem.–Eur. J., 2015, 21, 9209 CrossRef CAS PubMed.
- H. Xu, X. Zhang, Z. Ke and C. Zhao, RSC Adv., 2016, 6, 29045 RSC.
- C. Wang, Y. Zhou and X. Bao, J. Org. Chem., 2017, 82, 3751 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848 CrossRef CAS PubMed.
- T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed.
- S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1874184 and 1874186. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03691a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |