
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure elucidation of the syringafactin lipopeptides provides insight in the evolution of nonribosomal peptide synthetases†
Sebastian
Götze‡
 a,
Johannes
Arp‡
a,
Gerald
Lackner‡
b,
Shuaibing
Zhang
a,
Hajo
Kries
c,
Martin
Klapper
a,
María
García-Altares
d,
Karsten
Willing
e,
Markus
Günther
a and
Pierre
Stallforth
*a
a,
Johannes
Arp‡
a,
Gerald
Lackner‡
b,
Shuaibing
Zhang
a,
Hajo
Kries
c,
Martin
Klapper
a,
María
García-Altares
d,
Karsten
Willing
e,
Markus
Günther
a and
Pierre
Stallforth
*a
aIndependent Junior Research Group Chemistry of Microbial Communication, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Beutenbergstrasse 11a, 07745 Jena, Germany. E-mail: pierre.stallforth@leibniz-hki.de
bIndependent Junior Research Group Synthetic Microbiology, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Beutenbergstrasse 11a, 07745 Jena, Germany
cIndependent Junior Research Group Biosynthetic Design of Natural Products, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Beutenbergstrasse 11a, 07745 Jena, Germany
dDepartment Biomolecular Chemistry, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Beutenbergstrasse 11a, 07745 Jena, Germany
eDepartment Bio Pilot Plant, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Beutenbergstrasse 11a, 07745 Jena, Germany
First published on 4th December 2019
Abstract
Modular biosynthetic machineries such as polyketide synthases (PKSs) or nonribosomal peptide synthetases (NRPSs) give rise to a vast structural diversity of bioactive metabolites indispensable in the treatment of cancer or infectious diseases. Here, we provide evidence for different evolutionary processes leading to the diversification of modular NRPSs and thus, their respective products. Discovery of a novel lipo-octapeptide family from Pseudomonas, the virginiafactins, and detailed structure elucidation of closely related peptides, the cichofactins and syringafactins, allowed retracing recombinational diversification of the respective NRPS genes. Bioinformatics analyses allowed us to spot an evolutionary snapshot of these processes, where recombination occurred both within the same and between different biosynthetic gene clusters. Our systems feature a recent diversification process, which may represent a typical paradigm to variations in modular biosynthetic machineries.
Introduction
Bacterial natural products or secondary metabolites are low molecular weight compounds of extraordinary structural complexity and diversity. Their structures have been shaped by evolutionary selection pressures to interact for instance with proteins at high affinity and specificity. Thus, they are indispensable leads in pharmaceutical research; and, as a matter of fact, most commercially available antibiotics are natural products or semisynthetic derivatives thereof.1 Simple building blocks are assembled to form the core structures of many bacterial natural products, which can be further modified by specific enzymes.2–4 This assembly process is often carried out by modular biosynthetic enzymes, whereby each module selects and incorporates a specific monomer into the nascent natural product. Polyketide synthases (PKSs)5 and nonribosomal peptide synthetases (NRPSs)6 are the paradigmatic examples of biosynthetic assembly lines. In the case of NRPSs, each module incorporates one amino acid into the growing peptide chain, which finally leads to the biosynthesis of a nonribosomal peptide (NRP). Within a module, the adenylation (A) domain selects an amino acid (governed by its amino acid specificity), activates it via adenylation and loads it on a thiolation (T) domain, which among other roles acts as flexible linker arm. Condensation (C) domains catalyze amide bond formation between the downstream amino acyl group and activated acyl chains (Cstarter), an amino acyl group, or a peptidyl group attached to the upstream T domain. C Domains are specific for the configuration of the substrates that they link and thus, they can be phylogenetically grouped according to functional categories. Eventually, the peptide chain is released, typically via a thioesterase (TE) domain, from the last module. Due to the modular nature of NRPSs, recombination is believed to be a major driving force in the evolution and diversification of these biosynthetic genes.7 While we have some understanding on how natural recombination may occur, we lack examples or snapshots that allow retracing these evolutionary events. Recombination is an inherently rare event and both negative and positive selection can easily remove recombination products that are non-functional or non-beneficial, respectively.8It is thus a matter of careful genome analyses and often serendipity to identify good candidates in which recombination events are obvious. These snapshots of evolutionary events are however crucial in order to understand the mechanisms of molecular diversification.
Over time, genetic drift, neutral mutations, and other recombination events will lead to rapidly amassing sequence dissimilarities within genetic elements that were once transferred from a donor to a recipient strain. Eventually, this leads to functional co-evolution, making recombination events hard to discern. Furthermore, it could be shown that in modular polyketide synthases, these evolutionary events can be accelerated by inducing recombination between or deletions of modules leading to the formation of new biosynthetic products.9 Thus, the ideal system to investigate molecular evolution should be simple, with recombination events of the recent past. The latter result in a high sequence similarity between the transferred genetic elements, which allows for phylogenetic characterization.
Bacteria of the genus Pseudomonas are rich in biosynthetic gene clusters (BGC), which is manifest in a highly diverse and large secondary metabolome.10 In particular their NRP diversity is staggering. NRPs and especially lipidated NRPs fulfill multiple ecological functions in different pseudomonads and thus may provide an evolutionary advantage to their producers. For example, lipidated NRPs are crucial for swarming, biofilm formation, accessibility of nutrients, and defense against competitors as well as predators just to name a few.11
Here, we describe a system in which we can clearly retrace how recombination has led to evolutionary diversification of bacterial BGCs. The Pseudomonas-derived octapeptides of the syringafactin family provide clear evidence for diversification via intra and inter cluster recombination. Serendipitously, we identified a strain, Pseudomonas sp. QS1027 in which both inter and intra cluster recombination could be observed and both donor and recipient BGCs are still present.
The information about the evolution of biosynthetic genes can be directly applied by molecular engineers to design more efficient biomimetic approaches for biosynthetic alterations.12,13 Recently, a promising strategy for engineering NRPSs for the production of artificial peptide variants was described by the group of Bode, where particularly efficient exchange units were identified.14 These observations raise the question, which exchange units natural evolution harnesses in the diversification of NRPS clusters.
Results
Isolation and structure elucidation of virginiafactins and identification of the corresponding BGC
Pseudomonas sp. QS1027, when cultured under various conditions, produces the nonribosomal peptide jessenipeptin15 and the polyketide mupirocin15 as well as a group of four previously uncharacterized compounds with similar composition according to high-resolution mass-spectrometry (HRMS) measurements (pseudomolecular ion peaks at m/z 1110.7755, 1096.7599, 1082.7422, and 1066.7135). Analysis of their 1D and 2D nuclear magnetic resonance (NMR) spectra in combination with tandem mass-spectrometric (MS-MS) measurements revealed these four compounds to be lipo-octapeptides bearing similarity to members of the cichofactins16 and to the syringafactin17 family (Fig. 1).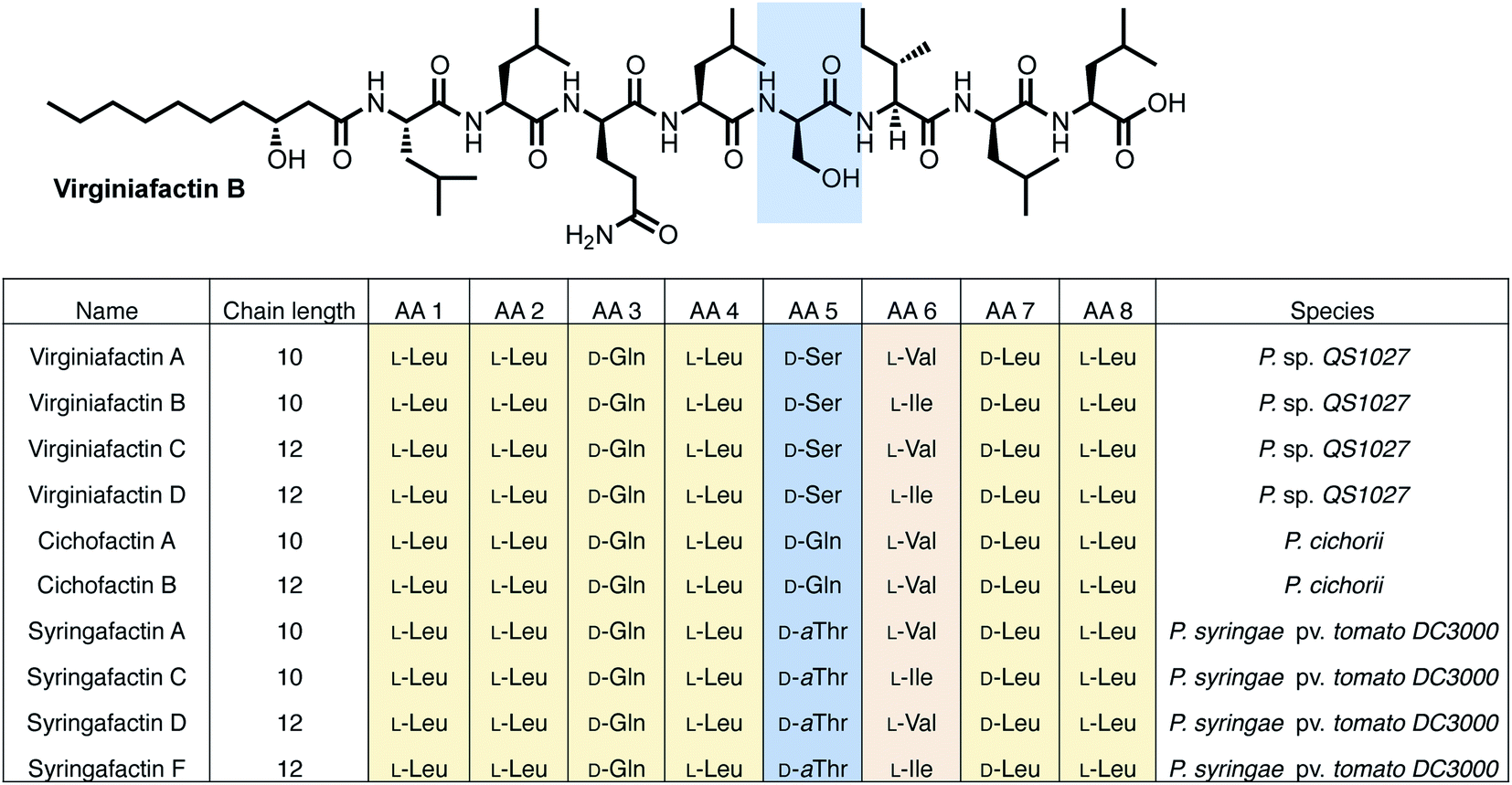 | ||
| Fig. 1 Structures of the lipo-octapeptides virginiafactins, cichofactins, and syringafactins. A representative structure of virginiafactin B is given on the top. Amino acids in positions 1, 2, 3, 4, 7, and 8 are identical. Amino acids in position 6 are either L-Val or L-Ile. Amino acids in position 5 are either D-Ser, D-Gln, or D-aThr, depending on their family affiliation. | ||
These compounds are lipo-octapeptides also found in other Pseudomonas species. Since Pseudomonas sp. QS1027 was isolated in Virginia, US, we named the new compounds virginiafactin A, B, C, and D. Bioinformatics analysis of the genome sequence (GenBank accession no. PHSU01000000) of the producing organism using antiSMASH18 uncovered a NRPS consisting of two adjacent core NRPS open reading frames vifA and vifB containing three and five modules respectively, which would lead to the production of a lipo-octapeptide (Fig. 2, the up- and downstream regions of the vif BGC can be found in the SI). We generated a mutant, Δvif, with an impaired vif BGC, which did not produce any virginiafactins (see Fig. S1† for metabolic profile of Δvif). This allowed us to link the vif BGC to the production of the virginiafactins. Interestingly, unlike the Pseudomonas mutants, which were unable to produce cichofactins and syringafactins through a similar genetic manipulation of their corresponding BGCs, the Δvif mutant was still able to swarm. Therefore, it is not clear if the virginiafactins are not surface-active or if the producer strain biosynthesizes additional biosurfactants, which compensate the lack of lipopeptides.
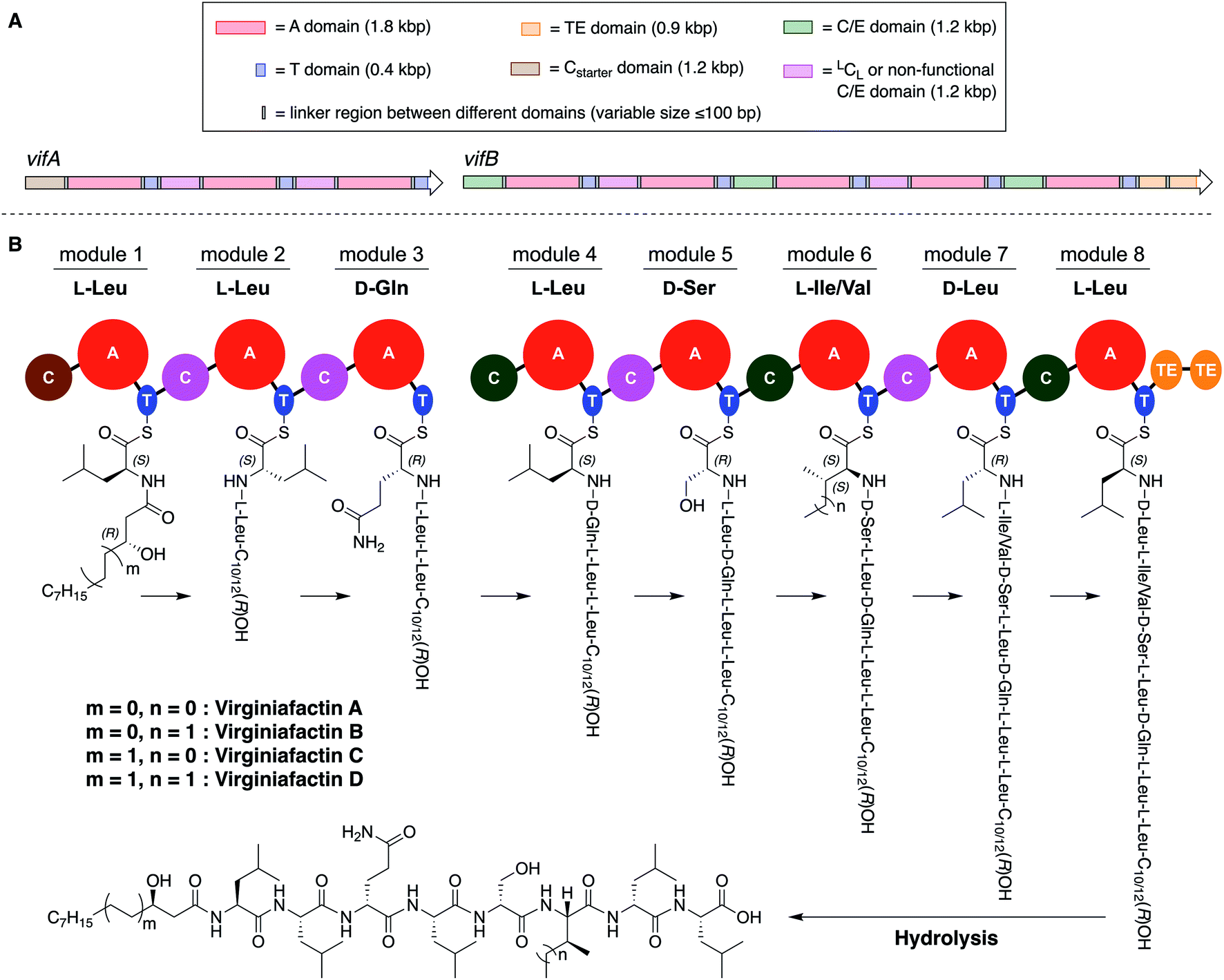 | ||
| Fig. 2 (A) Multidomain organization of the BGC encoding the virginiafactins. (B) Proposed biosynthesis of the virginiafactins. Different colors indicate the family of the C domain (brown = Cstarter; pink = LCL or non-functional C/E domain; green = C/E domain). Abbreviation: bp = base pair. | ||
NMR and MS-MS studies unveiled the planar structure of virginiafactin B to be 3-hydroxydecanoyl–Leu–Leu–Gln–Leu–Ser–Ile–Leu–Leu. So far, all lipopeptides isolated from Pseudomonas sp. containing 3-hydroxy fatty acids display an R-configuration – suggesting the same configuration for the virginiafactins.
Virginiafactin A, C, and D differed in the length of the fatty acid alkyl chain as well as in the amino acid in position 6, which can be valine or isoleucine (Fig. 1). Marfey's analysis revealed the presence of three D-configured and five L-configured amino acids: 1× D-Gln, 1× D-Ser, 1× D-Leu, 4× L-Leu, as well as 1× L-Ile, or L-Val. Bioinformatics analysis (using antiSMASH) of the condensation (C) domains of the vif BGC suggested, however, the sequence D-Leu–D-Leu–D-Gln–L-Leu–D-Ser–L-Ile–D-Leu–L-Leu, which was not in accordance with the results from Marfey's analysis. It is known that some bioinformatically predicted dual C domains, which epimerize L-configured amino acids to D-amino acids may be non-functional.19
We thus synthesized all permitted permutations of (3R)-hydroxydecanoyl–X-Leu–X-Leu–D-Gln–L-Leu–D-Ser–L-Ile–X-Leu–L-Leu (with X = D or L), constrained by the results of Marfey's analysis (i.e. 1× D-, 2× L-Leucin, ESI, compounds S1, S2, S4†). We synthesized the enantiopure fatty acid esters 1 and 2 (Fig. 3A),20 which were converted into the activated esters 3 and 4. These were condensed with synthetic peptide 5, obtained by solid-phase peptide synthesis (Fig. 3B). We then compared the retention times, NMR-, optical rotation- and MS-data of all synthetic lipo-octapeptides to that of virginiafactin B and D (Fig. 3C). Thus, we could confirm the structure of virginiafactin B and D (Fig. 3). We also synthesized virginiafactins A and C, all of which matched the isolated counterparts (ESI Fig. S18, S20 and S21†) and are unknown according to the database NORINE (https://bioinfo.lifl.fr/norine/).
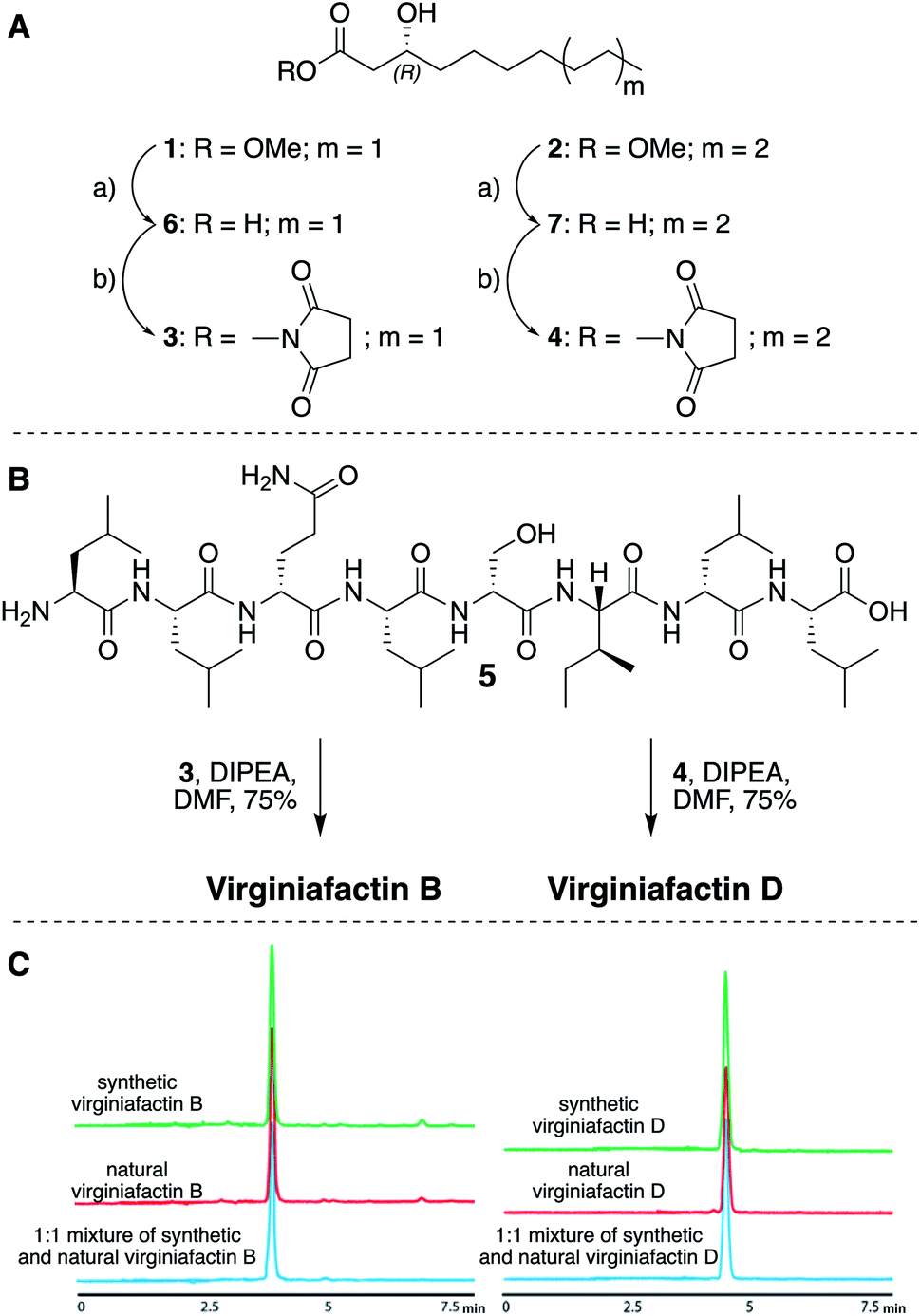 | ||
Fig. 3 (A) Synthesis of enantiopure fatty acid esters 3 and 4. Reagents and conditions: (a) LiOH, THF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rt, 90% for 6 and 94% for 7; THF, TSTU, DIPEA, rt, 46% for 3 and 28% for 4. (B) Acylation reaction to obtain virginiafactin B and D standards. (C) HPLC profiles of synthetic and natural virginiafactin B and D. Abbreviations: DIPEA = N,N-diisopropylethylamine; DMF = dimethylformamide; THF = tetrahydrofuran; TSTU = N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uroniumtetrafluoroborate. :1), rt, 90% for 6 and 94% for 7; THF, TSTU, DIPEA, rt, 46% for 3 and 28% for 4. (B) Acylation reaction to obtain virginiafactin B and D standards. (C) HPLC profiles of synthetic and natural virginiafactin B and D. Abbreviations: DIPEA = N,N-diisopropylethylamine; DMF = dimethylformamide; THF = tetrahydrofuran; TSTU = N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uroniumtetrafluoroborate. | ||
Structure elucidation of lipo-octapeptides of the cichofactin, and syringafactin families
The cichofactin, syringafactin, and the virginiafactin families differ in the nature of the amino acid in position 5 (AA 5, Fig. 1). An NCBI similarity search based on vifA and vifB using BLAST identified the core NRPSs associated with the production of cichofactins (cifA, cifB) and syringafactins (syfA, syfB) in P. cichorii (both P. cichorii JBC1 and SF1-54 produce the same compounds, for further analyses we used JBC1) and P. syringae DC3000 pv. tomato, respectively, as closely related based on their gene sequences. However, only the planar structures of the syringafactins and cichofactins were previously published.16,17 In order to determine their absolute configurations, we cultured the producing organisms and isolated representative lipo-octapeptides and compared them with synthetic lipo-octapeptides. Based on the high sequence similarity of all three biosynthetic gene clusters (vif, cif, and syf), we presumed that all shared amino acids (i.e. all except AA 5) between the three families would be identical in their configuration. Thus, we synthesized lipo-octapeptides with both the D- and L-configurations of AA 5: (3R)-hydroxydecanoyl–L-Leu–L-Leu–D-Gln–L-Leu–D/L-Xxx–L-Val–D-Leu–L-Leu (with Xxx = [allo]Thr or Gln). Comparison of HPLC profiles of isolated lipo-octapeptides and synthetic 5-D and 5-L lipo-octapeptides showed that indeed the configurations of the amino acids in position 5 were identical (D) for all three families of nonribosomal peptides (Fig. 1 and ESI Fig. S21–S23†).Phylogenetic analysis of A and C domains
The nucleotide sequences of gene clusters vif, syf, and cif are very similar and they differ mainly in certain regions of the coding sequence for module 5. This is no surprise since the corresponding natural products differ in AA5. Our hypothesis was that these three biosynthetic gene clusters share a common ancestor and subsequent diversification processes led to the three different BGCs. To test this hypothesis and to identify possible diversification processes, we conducted phylogenetic analyses of coding regions of the A and C domains using a Maximum Likelihood (ML) estimation. We expected the domains to form clades according to their substrate specificity (A domains) or their functional categories (C domains, Fig. 4A/B).21 We also expected corresponding domains at the same position of related BGCs (e.g. A8 of vif, cif, and syf) to descend from an ancestral domain that had the same position within the assembly line. Indeed, this expectation was fulfilled by the majority of the A and C domains (ESI Fig. S24 and S25†). In particular, the coding regions for both the A and C domains in modules 1, 2, 3, 6, and 8 clearly showed these relationships. This led to the conclusion that the clusters vif, cif, and syf indeed share a common ancestor with a highly similar scaffold of modules.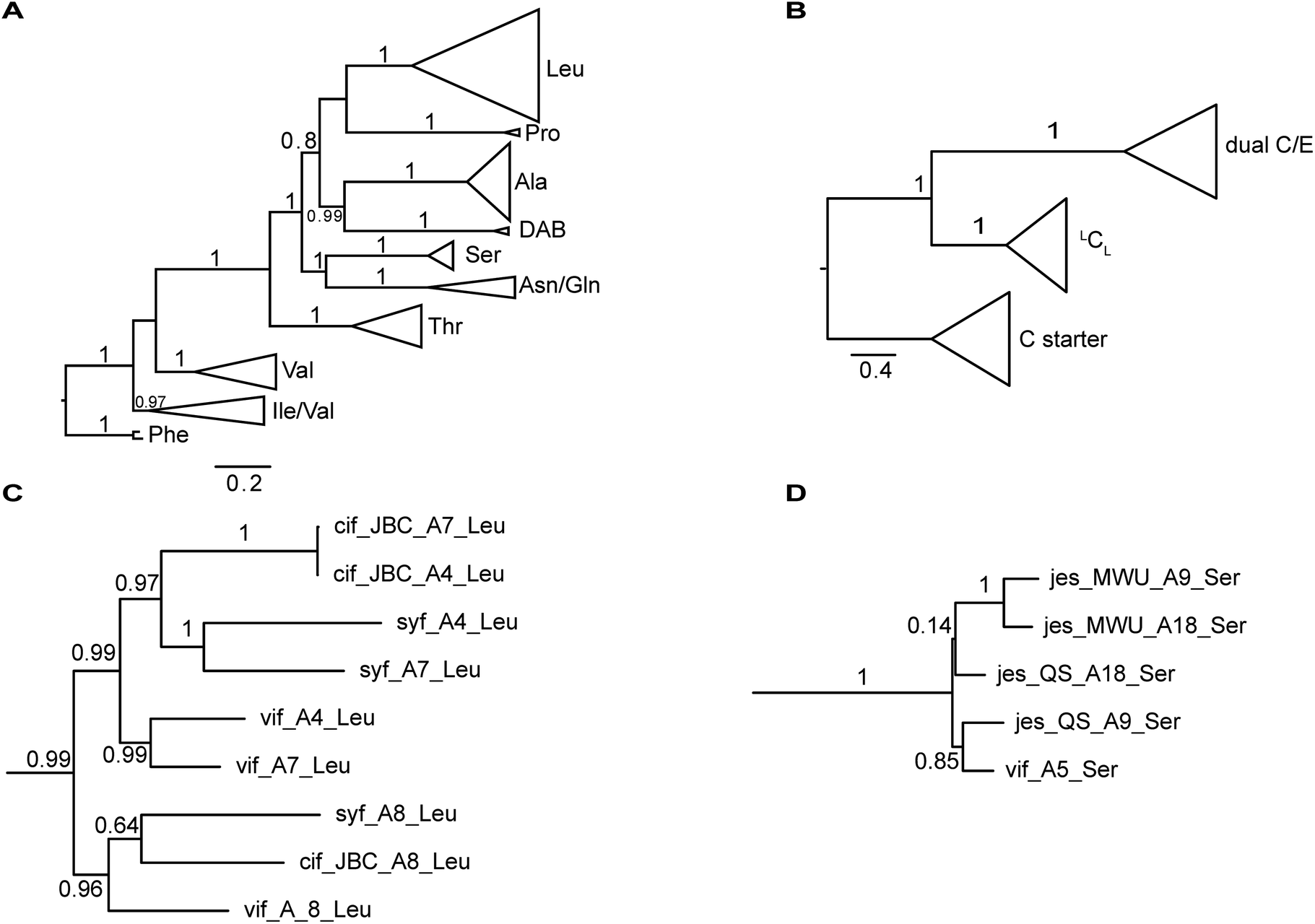 | ||
| Fig. 4 Phylogenetic analyses of A and C domains of the BGCs vif, cif, syf, jesQS, and jesMWU. (A) ML tree of sequences encoding A domains. A domains cluster according to their substrate specificities. The branch to phenylalanine-activating domains (jes-A19) was used to root the tree. (B) ML tree of sequences encoding C domains. C domains mainly cluster according to their functional categories (C starter, LCL and dual E/C domains) (C) Subtree of selected Leu-activating A domains. As expected, A domains in position A8 of the lipo-octapeptide BGC (cif, vif, syf) share a common ancestor suggesting a stable positioning within the assembly line. In contrast to this, A4 and A7 of each strain are more closely related to each other than to their common ancestor. This finding suggests up to three independent exchange events of A domains between position A4 and A7 in the lipo-octapeptide clusters. (D) Subtree of A domains of Ser-activating domains of JesQS and JesMWU. | ||
There are, however, a few remarkable exceptions. For instance, the coding regions of A4Leu and A7Leu form clades according to their BGC type (vif/cif/syf) rather than according to their position (4 or 7) within the assembly line (Fig. 4C). This tree topology clearly supports the hypothesis that one of these domains has duplicated and replaced the other one in each of the three BGCs. In particular the coding sequences cif-A4Leu and cif-A7Leu display 99% nucleotide identity with only a single nucleotide difference between the two A domains, while the average pairwise sequence identity of all A domains in cif is only 64%. Syf-A4Leu and syf-A7Leu as well as vif-A4Leu and vif-A7Leu also showed high sequence identity. This is in stark contrast to the corresponding C domain phylogeny, in which both C4 and C7 cluster according to their position rather than their similarity to each other (ESI Fig. S25†). Therefore, the A4 and A7 regions of vif, cif, and syf seem to be hot spots for recombination. It is conceivable that similar yet independent recombination events might have happened in evolution. The high sequence similarity would also be in agreement with dynamic exchanges between the A4 and A7 regions within a BGC.
Diversification of module 5
Mirroring their different substrate specificities, the DNA sequences of the A domains in modules 5 of cif, vif, and syf showed only little resemblance (55–61% pairwise identity) suggesting that these positions were acquired via recombination rather than by mutational diversification. Domains A5 of the Cif and Syf and Vif assembly lines activate Gln, Thr and Ser, respectively, and their coding sequences group with A domains displaying the same substrate predilections. Regarding the origin of the A5 domain in the vif BGC, we wondered if it could stem from another BGC within the same organism, Pseudomonas sp. QS1027. The NRPS JesQS, producing the natural product jessenipeptin (Fig. 5) actually contains two A domains with specificity for serine that might have been transferred to the vif cluster.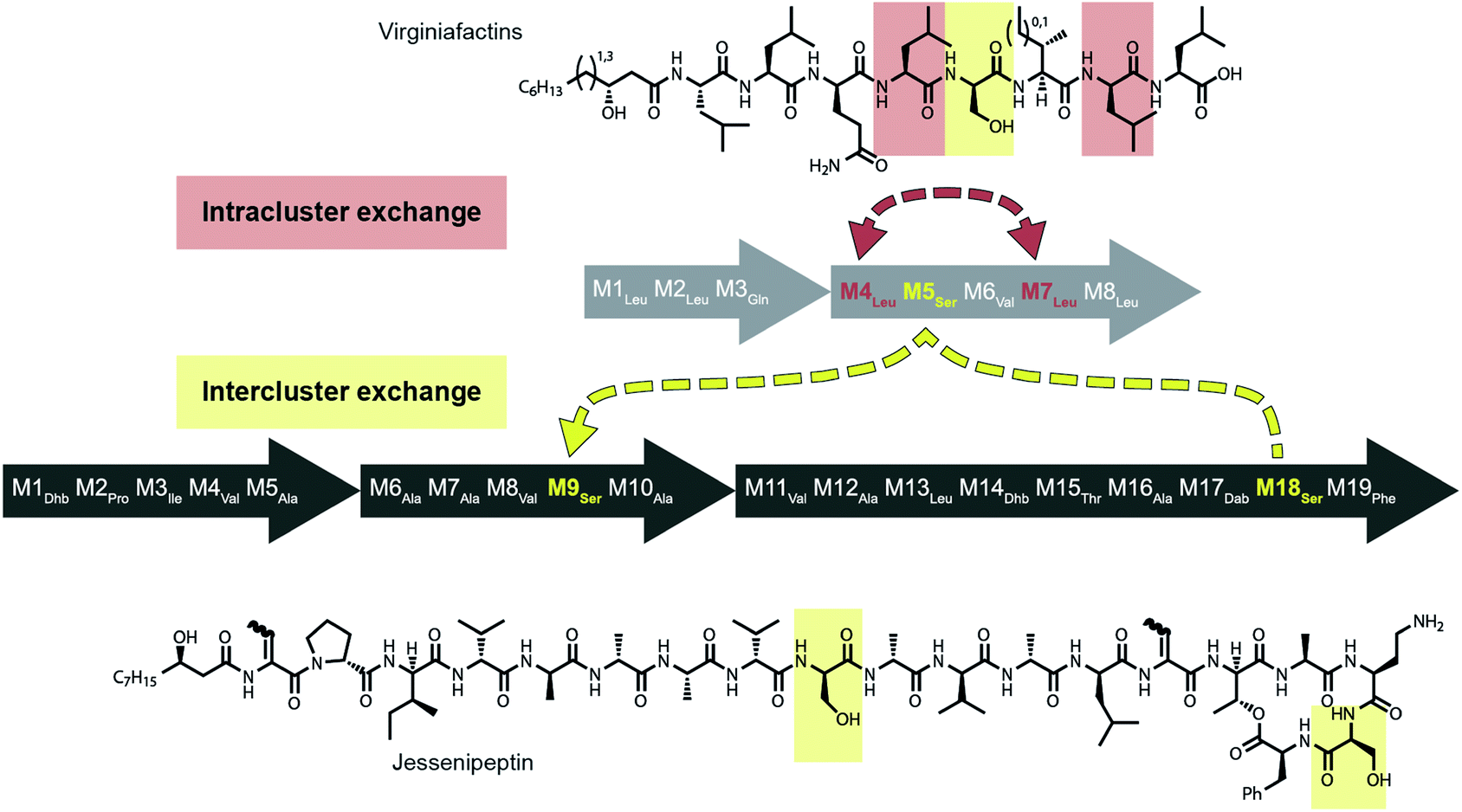 | ||
| Fig. 5 Recombination model leading to the diversification of the jes and vif BGC via intra (red) and inter (yellow) gene cluster recombination. | ||
Strikingly, sequence analysis indeed suggests transfer of genetic material between vif and jesQS. Whereas Pseudomonas sp. QS1027 contains an additional five BGCs that are classified as NRP BGCs (two of them are likely involved in pyoverdine production) none of their modules are predicted to incorporate serines, nor do they show high sequence similarity to the coding region of module 5 of vif.
The combined phylogenetic tree of the coding regions for the A domains of the lipo-octapeptide vif BGC and cyclic lipopeptide jesQS BGC clearly indicated that jesQS-A9Ser, jesQS-A18Ser, and vif-A5Ser are closely related (Fig. 4D and ESI Fig. S24†). Even on a nucleotide level, vif-A5Ser and jesQS-A18Ser display 93.7% identity while vif-A5Ser and jesQS-A9Ser display 93.5% identity. It is therefore highly likely, that at least two genetic transfer events affecting A domains took place between the serine-activating modules.
Intriguingly, the coding regions of condensation domain vif-C5 also deviate from the canonical phylogeny forming a clade together with jesQS-C18 but not C9 (ESI Fig. S25†). This is a clear indication that transfer of the entire C–A didomain has taken place between an ancestral jes module 18 (M18) and vif-M5. The topology of the ML tree contains a clade, albeit with slightly decreased support (0.85), where vif-A5Ser and jesQS-A9Ser share a common ancestor. This accounts for the reduced identity between jesQS-C9 and jesQS-C18 (92.8%). Taken together, the most parsimonious scenario consistent with the data would be a transfer of the C–A didomain from jes-M18 to vif-M5, followed by an A domain transfer from vif-M5 to jes-M9 (Fig. 5).
In order to shed more light on the directionality of the transfer vif-ASer ↔ jesQS-ASer, we searched for another jes-like BGC to be added to our phylogenetic analysis as an outgroup that might not have undergone domain transfers. A search in the NCBI database for BGC related to the jessenipeptin BGC using BLAST led us to the draft genome of Pseudomonas sp. MWU13-2860. The cluster was distributed over two contigs (NCBI accession numbers PPYB02000007 and PPYB02000026), but comparison of the contig ends allowed merging the contigs to yield a complete BGC that aligned very well with the jessenipeptin BGC (93% identity over a length of 64 kb). The overall domain structure was identical and antiSMASH predicted the same nonribosomally synthetized peptide backbone. Since the BGC was found in Pseudomonas sp. MWU13-2860, we designated this cluster jesMWU. As expected from the high similarity between the jesMWU and jesQS clusters, most of the jes domains cluster according to their positions within the assembly line. Surprisingly, however, both jesMWU-A9Ser and jesMWU-A18Ser regions formed their own clade descending from an ancestral jes-A18Ser domain in the subtree (Fig. 4D). Thus, they do not help to disentangle the directionality of domain exchanges. Rather, this finding is an indication for yet another exchange event that took place within the jesMWU BGC after the triplication in the ancestor of the Pseudomonas strain.
Molecular phylogeny of the lipopeptide-producing pseudomonas strains
In order to discriminate horizontal gene transfer from vertical transmission of gene clusters, we generated a species phylogeny of all the different strains used within this study (Fig. 6). To this end, we performed multilocus sequence typing (MLST) based on 16S rRNA genes and five housekeeping genes. Pseudomonas sp. QS1027 and Pseudomonas sp. MWU13-2860, which harbor the jesQS and jesMWU cluster, respectively, are closely related, thus a jessenipeptin BGC was most likely present in the genome of a common ancestor and was vertically transmitted. Both strains group with the Pseudomonas species P. jessenii, P. chlororaphis, and P. protegens.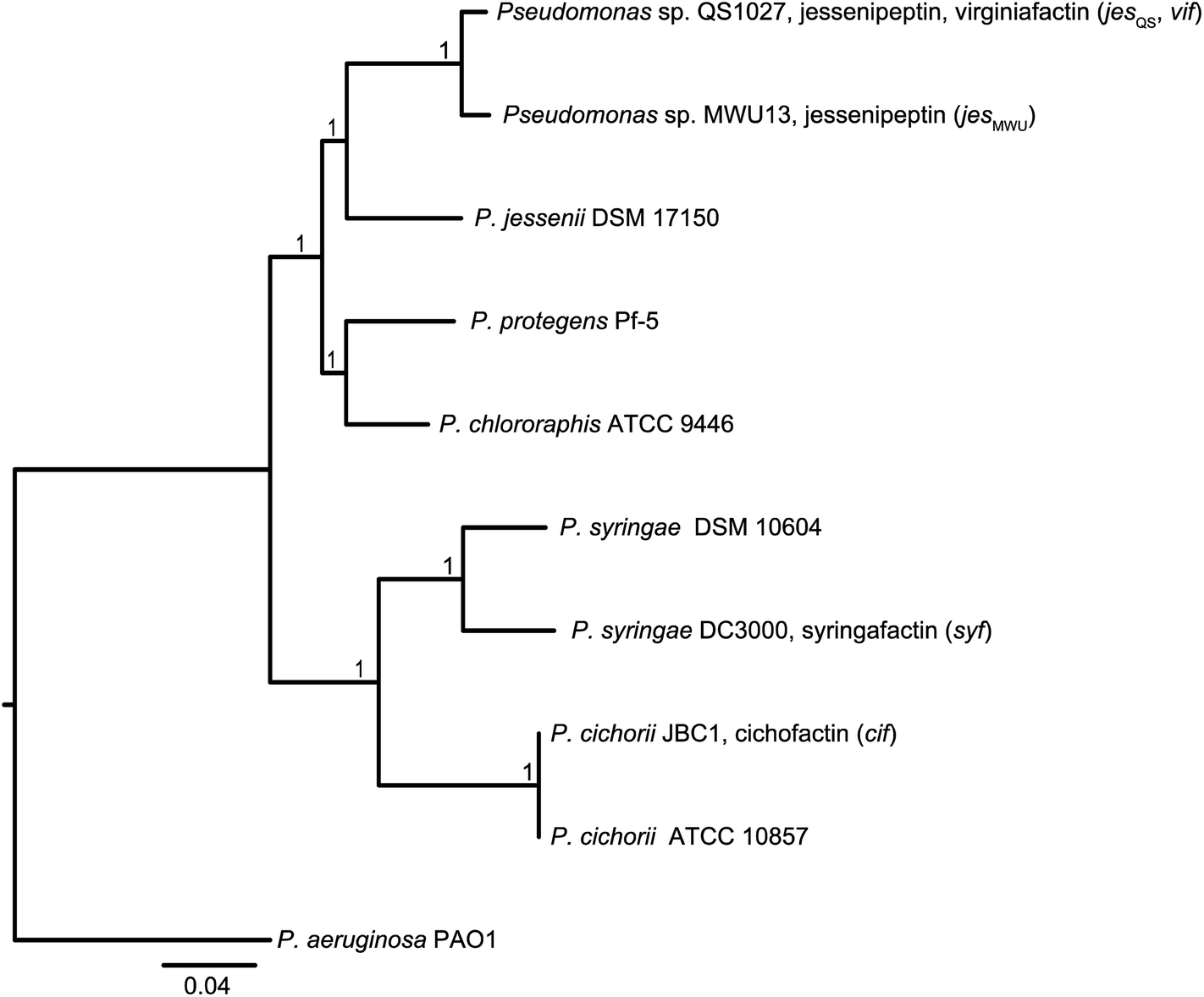 | ||
| Fig. 6 Top: Molecular phylogeny of the lipopeptide-producing Pseudomonas species based on multilocus sequence typing (MLST) of 16S rRNA genes and five housekeeping genes (gyrB, nuoCD, IleS, ppsA, rpoB). The tree was inferred by Maximum Likelihood estimation using a concatenated alignment as input. The P. aeruginosa PAO1 sequence was used as an outgroup to root the tree. The scale bar indicates substitutions per site. | ||
The producers of the cichofactins and the syringafactins, however, group together and are related to Pseudomonas syringae. While vertical transmission of the lipo-octapeptide BGCs in P. cichorii and P. syringae DC3000 may be possible, vertical transmission from these two species and Pseudomonas sp. QS1027 is unlikely due to the long phylogenetic distance. Here, a horizontal transmission appears much more plausible. Notably, no virginiafactin-like gene clusters were found in Pseudomonas sp. MWU13-2860. We cannot, however, discriminate between a recent loss of the vif cluster in Pseudomonas MWU13-2860 or a gain of the cluster by Pseudomonas sp. QS1027.
Identification of break points of recombination events
To better visualize putative regions of genetic exchange between modules, we also compared the Ser-activating modules using BLAST and created a Circos diagram (Fig. 7A). The Circos diagram also suggests that neighboring T domains might be part of the “exchange units”. To explain the high A domain sequence similarities among the three modules, at least two segment transfers between e.g. jesQS-A18Ser and jesQS-A9Ser or vif-A5Ser and jesQS-A9Ser must have taken place. As mentioned above, the topology of the ML tree suggests a scenario, where vif-A5Ser and jesQS-A9Ser share a common ancestor. The decreased support value of this clade (0.85), however, is indicative of certain conflicting phylogenetic signals within the subtree. A phylogenetic network helped visualizing the conflicting phylogenetic signals as reticulate structure in the center of the network (Fig. 7B), but consistently supported a slightly closer relationship between vif-A5 and jesQS-A9. Notably, a Phi test22 for recombination was positive within the subtree dataset so that one can assume further exchange of sequence segments between A domains.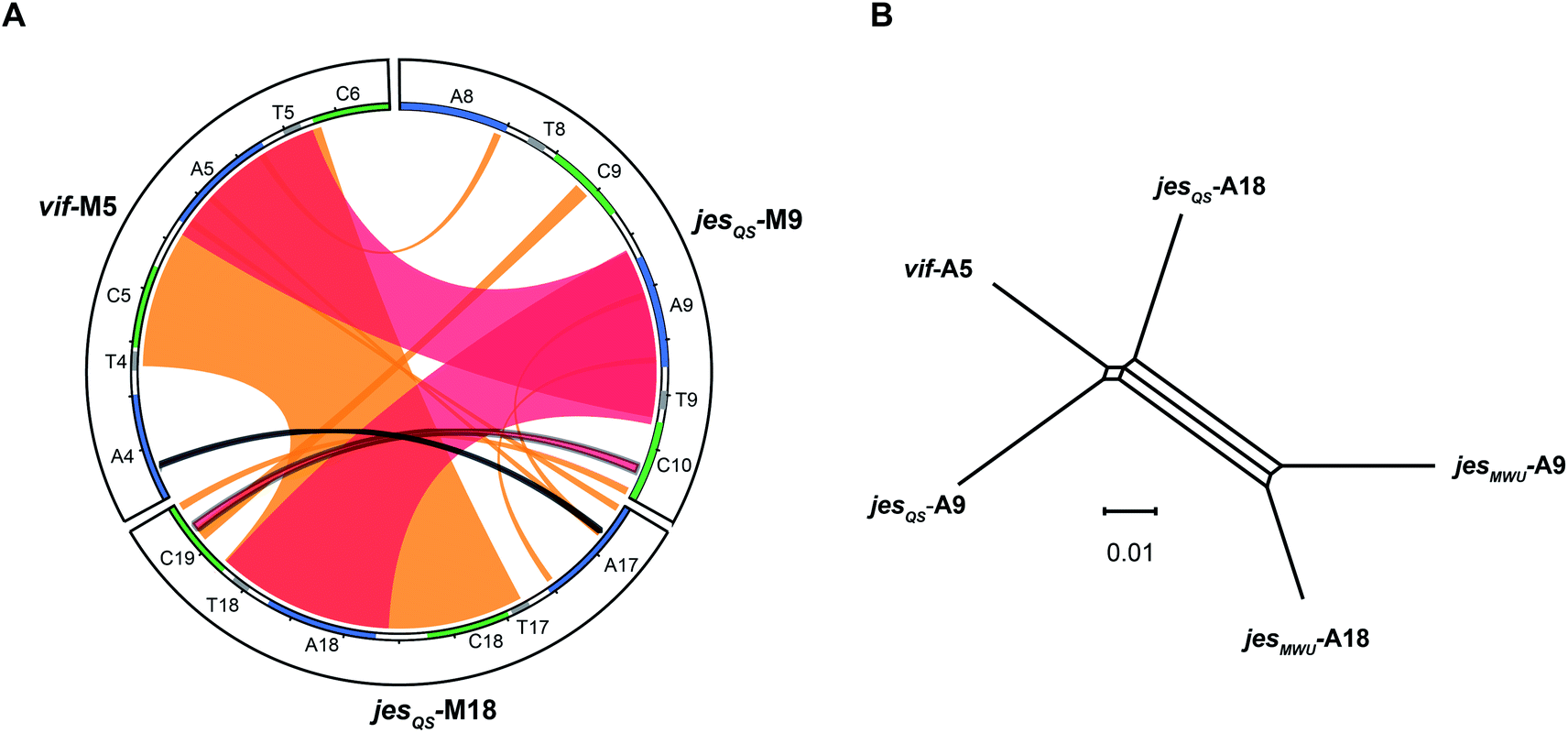 | ||
| Fig. 7 (A) Circos diagram comparing vif module 5 (vif-M5), jesQS-M9 and jesQS-M18. Module sequences with domain annotations are represented by circle segments as indicated. Regions of extended nucleotide identity are indicated as colored ribbons (red: >90% identity, orange: 80–90% identity according to BLAST). The triplicate A domain, more precisely the A–T didomain, is highly identical in all three modules as shown by broad red ribbons. An entire C–A didomain (or even a T–C–A–T multi-domain) was transferred between jesQS-M18 and vif-M5 as shown by broad orange ribbon. Thin ribbons represent conserved motifs. (B) Phylogenetic network analysis of repeated Ser-activating A domains. The central split mirrors phylogenetic separation of the “triplicate” jesQS domains from the “duplicate” jesMWU domains. The slightly closer relationship of jesQS-A9 and vif-A5 compared to jesQS-A18 is detectable as well. However, the reticulate structure in the middle of the network points towards conflicting phylogenetic signals suggesting further recombination events. The scale bar indicates uncorrected pairwise distances. | ||
While previous analyses revealed high sequence similarities as well as recombination of certain A and C domain coding regions, we were interested in identifying the exact boundaries of recombination events. To this end, we performed a search for repeated DNA sequences within the vif, jesQS, cif, and syf gene clusters. This would allow us to identify sequences flanking the A and C domain coding regions which are similar in different modules and which would have escaped phylogenetic analyses.
We performed a series of similarity searches with a lower percent-identity cut-off at 90%. A search for repeated sequences in the jesQS BGC led to the identification of a repeated sequence of 1364 bp and 94% sequence identity that include the A domains: jesQS-A6Ala and jesQS-A12Ala (Fig. 8). This segment duplication was not directly visible in the A domain tree, since the repeat region does not coincide with the domain boundaries.
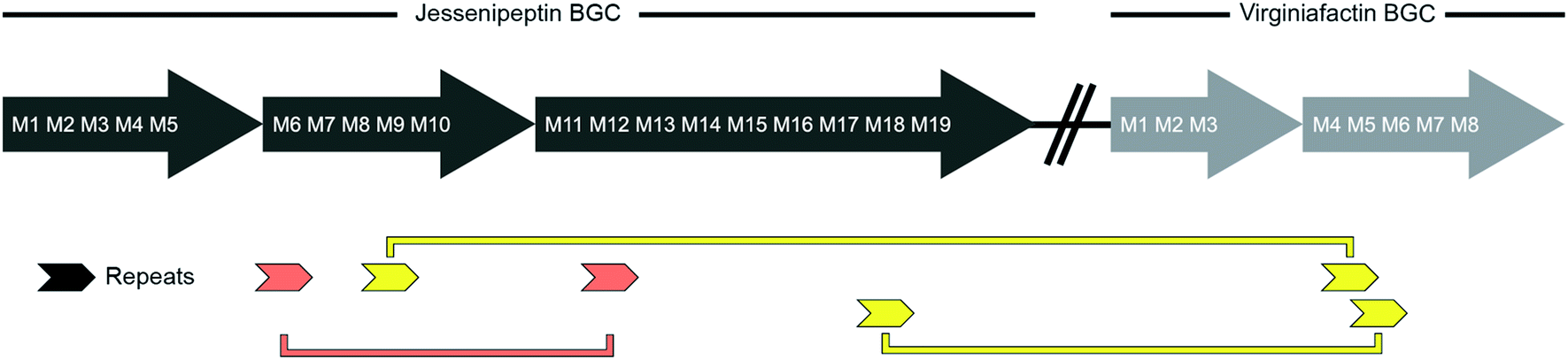 | ||
| Fig. 8 Schematic representation of repetitive sequences in the jessenipeptin (jesQS) and virginiafactin (vif) BGC. red: jesQS-A6Ala and jesQS-A12Ala, yellow: jesQS-M9Ser and vifQS-M5Ser; jesQS-M18Ser and vifQS-M5Ser (repetitive sequences were found using Repeat Finder of Geneious® Version 9.1.5). | ||
In accordance with the phylogenetic analysis, joint analysis of the vif and jesQS BGCs revealed three DNA stretches of high similarity (Fig. 8). The repeated sequence contains 1306 bp and displays a shared sequence identity of 92% between the three fragments. The repeats contain the A domain coding regions for jesQS-A9Ser, jesQS-A18Ser, and vif-A5Ser. A pairwise analysis of the three repeated sequences allowed us to identify a similar stretch of 1976 bp and a sequence identity of 91% which includes coding regions for jesQS-A18Ser, and vif-A5Ser as well as the corresponding downstream T and part of the C domain. Furthermore, long repeats (>1.2 kbp) with very high sequence identities (>99%) were found in the cif BGC. The 1251 bp repeats span the entire cif-A4Leu and cif-A7Leu domain coding regions and display a sequence identity of 99.4%. Due to the exceptionally high sequence identity, we amplified the respective regions by PCR and re-sequenced them using Sanger sequencing to preclude any errors in the whole genome sequencing (c.f. ESI†). The repeat regions could further be extended to include the downstream T domains when the sequence identity cut-off was set to 95%.
We then aligned the sequence fragments with high sequence identity to visualize the boundaries (Fig. 9). Sequence alignment of jes modules 6 and 12 showed the regions with high sequence identity that span a region beyond the A domain (ESI Fig. S26†). Remarkably, in the case of cif modules 4 and 7, a very sharp decrease in sequence identity allowed us to predict the putative recombination boundaries within a few nucleotides (Fig. 9A). Mapping of these exchanged fragments on a homology structure model showed that the exchange unit started C-terminally of the C–A linker in the first helices of the A domain (Fig. 9B) and ended in the middle of the C-terminal subdomain of the A domain.
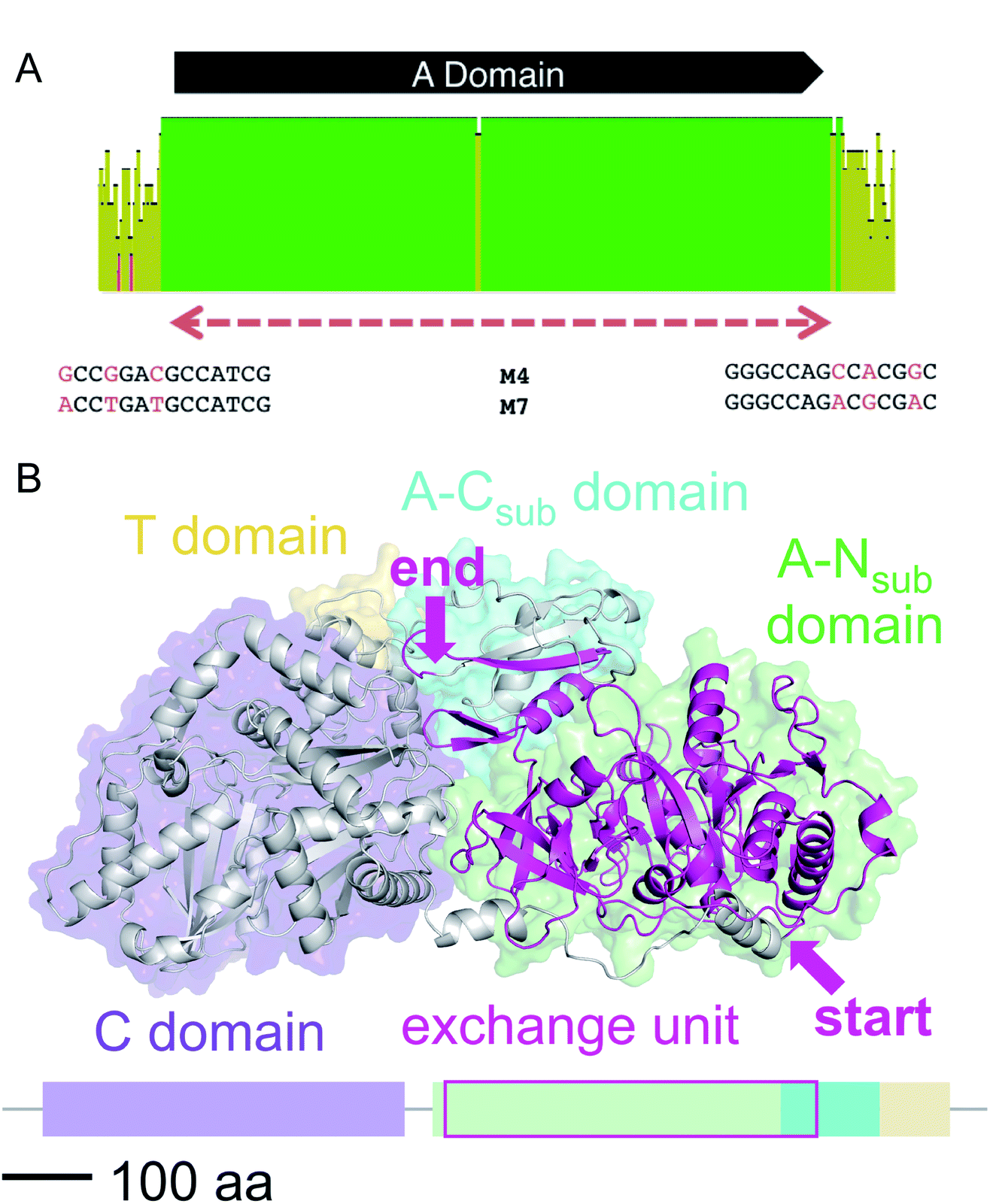 | ||
| Fig. 9 (A) Sequence alignment of cif modules 4 and 7 (see ESI Fig. S28† for nucleotide sequences). Shown are the mean pairwise identities (using Geneious® Version 9.1.5) green: 100%, green-brown: 30–99%, red: <30%. Sliding window size: 10 nt. The red, dashed bar indicates the repeated sequence with identity cut-off = 99%. (B) Homology model of Cif module 7 built with Swiss-Model23 based on the crystal structure of SrfA-C24 with the exchange unit highlighted in pink (see ESI Fig. S29† for protein sequence alignment). | ||
Discussion
Due to their modular nature, nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) are interesting and well-suited candidates to study the molecular mechanisms and evolution of natural product diversification. For PKSs it is evident that they share a common ancestor with fatty acid synthases (FAS).25–27 It is presumed that duplication events of a single ancestor module caused the enlargement and diversification of PKSs.28 Exchange between DNA fragments of PKSs may further lead to diversification of these biosynthetic machineries. Thus, it could be shown that for certain PKSs horizontal recruitment and assembly of particular domains effectively resulted in mosaic gene structures.29 The generation of several contracted and elongated rapalogs evidences the relevance of homologous recombination events in the synthetic biology of PKSs.9Computational analyses have shown that rates of evolutionary events that lead to diversification in BGCs are typically higher than corresponding rates in other clusters or genes e.g. from primary metabolism.30 This same study also suggests, based on bioinformatics approaches, different modes of diversification. For instance, module duplication and concerted evolution may lead to highly repetitive NRPS and domains can be swapped between related BGCs.30 Interestingly, two BGCs can also be combined and modified to yield a novel BGC, as was shown for the serratiochelins.31
Regarding NRPSs and NRPS/PKS hybrids, a great deal of attention has been attributed to analyzing the evolutionary history of the cyanobacterial biosynthetic machineries.32–36 In particular, the biosynthesis of microcystin served as benchmark for these investigations. Additional insight in the toolbox of evolutionary processes could be gained from Streptomyces-derived NRPSs. Piel and co-workers, for instance, found that small regions of the hormaomycin BGC, which code for substrate recognition sites, were exchanged within the same BGC.37,38 This may have resulted in a shift in the A domain specificity without the need of an entire module or A domain to be exchanged. Module exchange, however, and multiple recombination events are believed to have led to a plethora of nonribosomal peptides. Yet, a real snapshot of these evolutionary events, clarifying the processes that result in this large number of secondary metabolites has been missing to date.
Pseudomonas-derived nonribosomal peptides, in particular lipopeptides, are particularly diverse in their structures and consequently in their biological functions.10,39 Their evolutionary nature, however, remained obscure.
In this study, we could show that these biosynthetic machineries are ideally suited to retrace some of the underlying principles of diversification. We identified a novel lipo-octapeptide family, the virginiafactins, which shows strong resemblance to the syringafactins and the cichofactins. A full structure elucidation of all three families enabled us to show that these families only differ in one amino acid in position 5 – all other amino acids are identical in structure and configuration. This feature is mirrored in the underlying gene sequences coding for the respective NRPS (vif, cif, and syf). All modules except for module 5 show high sequence similarity suggesting a recent common ancestral BGC.
Additionally, we could show that in the case of virginiafactin, part of module 5 was most likely acquired via a recombinational event from another BGC, namely the jessenipeptin BGC (jesQS). BGCs are amongst the most mobile genes that are readily exchanged between different bacteria via horizontal gene transfer. The accumulation of multiple BGCs can be explained amongst other factors by synergistic effects between the corresponding secondary metabolites or for contingency reasons.40
We believe that certain strains may acquire different NRPS genes and exchange sections of modules and then one or the other BGC may be lost, e.g. if the corresponding metabolite leads to negative selection in a new environment (Fig. 10).
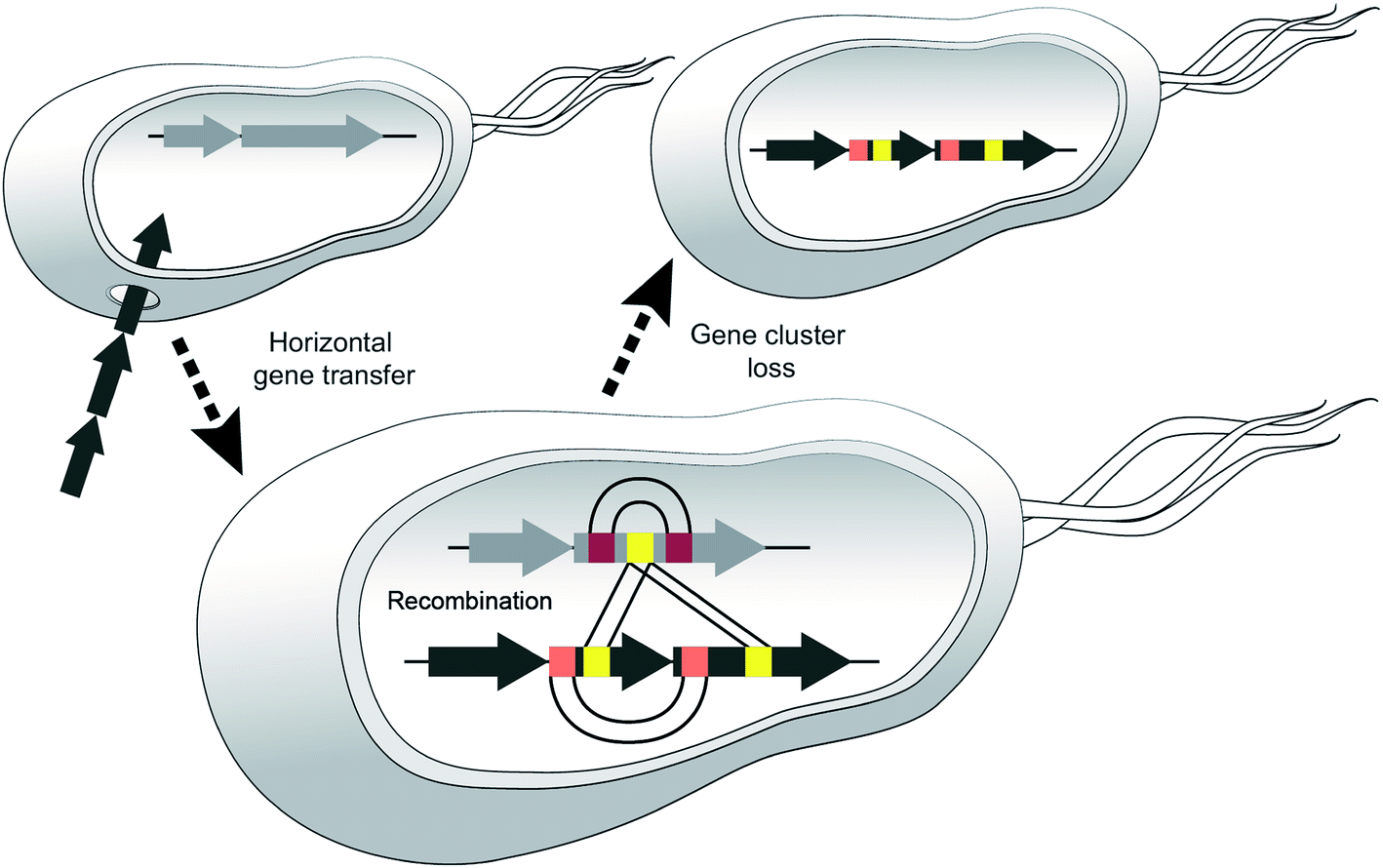 | ||
| Fig. 10 Model highlighting three events that diversify NRPSs. Horizontal gene transfer (HGT) disseminates BGCs between donor and recipient strains, diversification via recombination occurs between different NRPS clusters or within one cluster (duplication of modules/domains). Adaptation to new habitats may result in gene cluster loss. | ||
In support of this hypothesis, we could not identify any NRPS in the producing strains of the cichofactins or syringafactins, from which parts of their module 5 might have been acquired. In the virginiafactins, amino acid 5 is D-serine and most likely, the respective CA didomain was introduced from module 18 of the jesQS BGC. A second recombination event appears to have resulted in a transposition of vif-A5Ser to jesQS-A9Ser. Thus, at least two recombination events occurred between the two distinct BGCs. Similar events presumably would have led to the introduction of different A5 domains in the syringafactins and cichofactins, yet the NRPS from which parts of the module were obtained was lost or syf or cif were transferred to another strain via HGT. When looking for sequence stretches with high sequence similarity in the different BGCs, we noticed that the coding sequences of A4Leu and A7Leu within each BGC were more similar to each other than to their homologs from the other clusters. This suggests that in each of the three BGCs an exchange between the A4Leu and A7Leu could have occurred. Although it may seem unlikely that three separate recombination events would have occurred at similar positions, our results support that A4Leu and A7Leu are hotspots for recombination. Recombination within one strain was additionally seen for jesQS-A6Ala and jesQS-A12Ala, where bioinformatics analyses also suggest an exchange. Overall, these observations suggest dynamic processes by which NRPS parts interchange in an unexpectedly rapid fashion, like mobile genetic elements.
The boundaries for exchanges seem to vary: entire domains, parts of didomains, or only small fragments within a module can be exchanged. Natural recombination events can inspire and inform BGC engineering in the laboratory38,41 and for this purpose, more data describing these events are necessary. Since recombination in bacterial genomes is typically a rare event,8 which is greatly facilitated by sequence similarity, hot spots for recombination are likely to occur in particular in highly conserved regions. Consequently, an intra-species recombination event will drastically increase the likelihood for another recombination in the same place, perhaps explaining, for instance, repeated domain exchanges within the jessenipeptin-like (jesMWU) cluster. It is conceivable that these regions may also be ‘functional’ hot spots i.e. changes in these regions may result in functionally different natural products. A detailed structure–activity relationship would be required to shed light on this aspect.
These recombination events and the resulting mosaic nature of NRPSs with partial homology obfuscate phylogenetic analyses and may often lead to paradoxical results – thus phylogenetic analyses of modular BGC always have to be treated with care. In our case, a snapshot in evolution where recipient and donor BGCs were still in the same strain attests a recombination event beyond reasonable doubt. Hopefully, with larger numbers of genomes of prolific natural product producers like Pseudomonas, we will find more of these snapshots to further validate our hypotheses.
Conclusion
We propose a model for the evolutionary diversification processes that led to the different Pseudomonas-derived lipo-octapeptides virginiafactin, cichofactin, and syringafactin. The foundation was laid by a comprehensive structure elucidation of all compounds demonstrating that these lipo-octapeptide families differ mainly in the nature of a single amino acid. The high sequence identity of the respective biosynthetic gene clusters allowed us to perform detailed phylogenetic analyses and it enabled us to spot recombination events.Thus, we identified a snapshot of evolution in flagrante, where both intra and inter gene cluster exchanges caused natural product diversification. Currently, there is an impressive number of Pseudomonas genome sequences available, many of which display plenty NRPS biosynthetic genes. With reliable structural information of the respective nonribosomal peptides we could acquire a more global picture of recombinational events contributing to the vast diversity of NRPs. This calls for medium-to high-throughput techniques regarding the structure determination of NRPs, in particular assigning the absolute configuration of each amino acid, which is currently a bottleneck. Furthermore, application of big data in silico approaches to identify exchanges between or within biosynthetic genes would allow reconstituting the ancient mosaic of natural product diversification.
Author contributions
PS, GL, JA, SG, HK designed the research. SG, JA, SZ, MG-A performed experiments. KW performed large-scale fermentations. PS, SG, GL, JA, HK, MK, MG-A analyzed the data. PS, SG, GL, JA, HK, MK, MG wrote the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Stefanie Ranf, Monica Höfte, Harald Gross for sharing the bacterial strains P. cichorii JBC1, P. cichorii SF1-54, P. syringae pv. tomato DC3000 with us. We are grateful for financial support from the Leibniz Association. This work was supported by Deutsche Forschungsgemeinschaft Grant STA1431/2-1 and the collaborative research cluster ChemBioSys SFB1127. An Aventis Foundation PhD fellowship (to M. K.) is acknowledged. H. K. acknowledges financial support from the Daimler and Benz Foundation. G. L. acknowledges financial support from the Carl Zeiss Foundation. Matthias Steinacker and Klaus Menzel are thanked for their help in the fermentation of Pseudomonas sp. QS1027.References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS.
- C. T. Walsh and Y. Tang, Natural Product Biosynthesis: Chemical Logic and Enzymatic Machinery, Royal Society of Chemistry, 2017 Search PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS.
- C. T. Walsh and M. A. Fischbach, J. Am. Chem. Soc., 2010, 132, 2469–2493 CrossRef CAS.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- M. A. Fischbach, C. T. Walsh and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4601–4608 CrossRef CAS PubMed.
- J. P. Gogarten, W. F. Doolittle and J. G. Lawrence, Mol. Biol. Evol., 2002, 19, 2226–2238 CrossRef CAS PubMed.
- A. Wlodek, S. G. Kendrew, N. J. Coates, A. Hold, J. Pogwizd, S. Rudder, L. S. Sheehan, S. J. Higginbotham, A. E. Stanley-Smith and T. Warneck, et al. , Nat. Commun., 2017, 8, 2016 CrossRef.
- H. Gross and J. E. Loper, Nat. Prod. Rep., 2009, 26, 1408–1446 RSC.
- S. Götze and P. Stallforth, Nat. Prod. Rep., 2019 10.1039/C9NP00022D.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 11, 1210–1228 RSC.
- H. Kries, J. Pept. Sci., 2016, 22, 564–570 CrossRef CAS PubMed.
- (a) K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2017, 10, 275–281 CrossRef PubMed; (b) K. A. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y.-N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef PubMed.
- J. Arp, S. Götze, R. Mukherji, D. J. Mattern, M. Garcia-Altares, M. Klapper, D. A. Brock, A. A. Brakhage, J. E. Strassmann, D. C. Queller, B. Bardl, K. Willing, G. Peschel and P. Stallforth, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3758–3763 CrossRef CAS.
- E. Pauwelyn, C.-J. Huang, M. Ongena, V. Léclère, P. Jacques, P. Bleyaert, H. Budzikiewicz, M. Schäfer and M. Höfte, Mol. Plant-Microbe Interact., 2013, 26, 585–598 CrossRef CAS PubMed.
- A. D. Berti, N. J. Greve, Q. H. Christensen and M. G. Thomas, J. Bacteriol., 2007, 189, 6312–6323 CrossRef CAS PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Müller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef CAS PubMed.
- K. Scherlach, G. Lackner, K. Graupner, S. Pidot, T. Bretschneider and C. Hertweck, ChemBioChem, 2013, 14, 2439–2443 CrossRef CAS PubMed.
- M. De Vleeschouwer, D. Sinnaeve, J. Van den Begin, T. Coenye, J. C. Martins and A. Madder, Chem.–Eur. J., 2014, 20, 7766–7775 CrossRef CAS.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 78 CrossRef.
- T. C. Bruen, Genetics, 2005, 172, 2665–2681 CrossRef.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS.
- A. Tanovic, S. A. Samel, L.-O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS.
- C. P. Ridley, H. Y. Lee and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4595–4600 CrossRef CAS PubMed.
- S. Kroken, N. L. Glass, J. W. Taylor, O. C. Yoder and B. G. Turgeon.
- H. Jenke-Kodama, A. Sandmann, R. Müller and E. Dittmann, Mol. Biol. Evol., 2005, 22, 2027–2039 CrossRef CAS.
- H. Jenke-Kodama, T. Börner and E. Dittmann, PLoS Comput. Biol., 2006, 2, e132 CrossRef.
- T. Nguyen, K. Ishida, H. Jenke-Kodama, E. Dittmann, C. Gurgui, T. Hochmuth, S. Taudien, M. Platzer, C. Hertweck and J. Piel, Nat. Biotechnol., 2008, 26, 225–233 CrossRef CAS.
- M. H. Medema, P. Cimermancic, A. Sali, E. Takano and M. A. Fischbach, PLoS Comput. Biol., 2014, 10, e1004016 CrossRef PubMed.
- M. R. Seyedsayamdost, S. Cleto, G. Carr, H. Vlamakis, M. J. Vieira, R. Kolter and J. Clardy, J. Am. Chem. Soc., 2012, 134, 13550–13553 CrossRef CAS.
- K. Ishida, M. Welker, G. Christiansen, S. Cadel-Six, C. Bouchier, E. Dittmann, C. Hertweck and N. Tandeau de Marsac, Appl. Environ. Microbiol., 2009, 75, 2017–2026 CrossRef CAS.
- G. Christiansen, J. Fastner, M. Erhard, T. Borner and E. Dittmann, J. Bacteriol., 2003, 185, 564–572 CrossRef CAS PubMed.
- D. P. Fewer, L. Rouhiainen, J. Jokela, M. Wahlsten, K. Laakso, H. Wang and K. Sivonen, BMC Evol. Biol., 2007, 7, 183 CrossRef PubMed.
- S. Meyer, J.-C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Süssmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef CAS.
- A. Tooming-Klunderud, B. Mikalsen, T. Kristensen and K. S. Jakobsen, Microbiology, 2008, 154, 1886–1899 CrossRef CAS.
- I. Höfer, M. Crüsemann, M. Radzom, B. Geers, D. Flachshaar, X. Cai, A. Zeeck and J. Piel, Chem. Biol., 2011, 18, 381–391 CrossRef.
- M. Crüsemann, C. Kohlhaas and J. Piel, Chem. Sci., 2013, 4, 1041–1045 RSC.
- N. Geudens and J. C. Martins, Front. Microbiol., 2018, 9, 1867 CrossRef.
- G. L. Challis and D. A. Hopwood, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14555–14561 CrossRef CAS.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, ACS Synth. Biol., 2018, 7, 399–404 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03633d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |