
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge-driven tripod somersault on DNA for ratiometric fluorescence imaging of small molecules in the nucleus†
Kang-Nan
Wang
 ,
Qian
Cao
*,
Liu-Yi
Liu
,
Zi-Jian
Zhao
,
Wenting
Liu
,
Dan-Jie
Zhou
,
Cai-Ping
Tan
,
Wei
Xia
,
Liang-Nian
Ji
and
Zong-Wan
Mao
*
,
Qian
Cao
*,
Liu-Yi
Liu
,
Zi-Jian
Zhao
,
Wenting
Liu
,
Dan-Jie
Zhou
,
Cai-Ping
Tan
,
Wei
Xia
,
Liang-Nian
Ji
and
Zong-Wan
Mao
*
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou, 510275, P. R. China. E-mail: caoqian3@mail.sysu.edu.cn; cesmzw@mail.sysu.edu.cn; Fax: +86 20 8411 2245
First published on 6th September 2019
Abstract
Although fluorescence tracing of small bioactive molecules in living cells has been extensively studied, it is still a challenging task to detect their variations in the nucleus mainly due to the impermeable nuclear membrane and nucleic acid interference. Herein, we take advantage of the nucleic acid enriched environment in the nucleus to establish a strategy, named “charge-driven tripod somersault on DNA”, for ratiometric fluorescence imaging of small bioactive molecules in the nucleus. Taking SO2 derivatives as a typical target analyte, a tripodal probe has been constructed by conjugating two DNA binding groups containing a SO2 derivative reaction site. Mechanism studies demonstrate that upon encountering and reacting with SO32−/HSO3−, a charge variation occurs at the responsive arm of the tripodal probe, triggering a tripod somersault on DNA, resulting in the conformational rearrangement of the DNA binding modes with DNA-modulated fluorescence change, which allows the second emission feature to emerge. In this strategy, probe–DNA binding is not influenced by RNA or non-specific protein association, thus making it ideal for tracing nucleus-localized analytes. The application of this strategy has realized both in vitro and in vivo ratiometric fluorescence imaging of the variations of endogenous SO2 derivatives in the nucleus for the first time, with high specificity and selectivity. Also, in theory, this strategy opens up a new avenue for the design of fluorescence probes for the nucleus-localized biological analytes.
Introduction
The nucleus is the most important organelle in eukaryotic cells and is generally recognized as the regulatory centre of cell activity, heredity, and metabolism.1,2 The occurrence and development of many fundamental diseases are usually accompanied by abnormal nuclear behaviour.3–5 It has been found that many endogenous bioactive molecules at an imbalanced level can interfere with genetic materials within the nucleus and cause nuclear dysfunction, although at physiological concentrations they also act as messengers featuring a cytoprotective effect and maintaining cellular homeostasis. For example, excess ClO− and H2O2 can cause the accumulation of ROS-mediated oxidative damage to the nuclear genetic materials, resulting in aging, Alzheimer's disease and cancer;6,7 nuclear metal ions such as Zn2+, Cr3+, and Al3+ can form a hinge with nucleic acids and hinder gene expression;8–10 inhalation of SO2 results in abnormal nuclear behaviour11 and DNA damage in multiple organs,12,13 contributing to the development and progression of ischemic stroke in the brain. Therefore, directly and specifically tracking the flux of these small bioactive molecules in the nucleus offers a potentially reliable indicator for the progression of disease states.As is known to all, fluorescence combined with confocal microscopy is an indispensable tool for visualizing biological events with high sensitivity and high-contrast spatiotemporal resolution.14,15 Compared with the conventional fluorescence “turn-on” strategy,16 self-referencing ratiometric fluorescence offers greatly improved sensitivity and reliability, relying on the intensity changes of two or more emission features, thus eliminating a variety of interference factors, e.g. fluorophore concentrations, instrumental parameters, cellular auto-fluorescence, etc.17–21 Currently, ratiometric fluorescence sensing is mainly achieved by the following strategies: (1) internal charge transfer (ICT) for e.g. H2S and H2O2 sensing in living cells;22,23 (2) Förster resonance energy transfer (FRET) for quantifying e.g. phosphodiesterase activity;24 (3) excited state intramolecular proton transfer (ESIPT) for probing biologically relevant cations and anions (Zn2+, F−, CH3CO2−, H2PO4−, etc.);25,26 (4) through-bond energy transfer (TBET) for measuring e.g. in vivo intracellular pH values;27,28 (5) monomer–excimer based ratiometric sensing driven by analyte-induced changes in the separation of excimers, etc.29,30 However, utilizing these strategies to construct probes requires complex design and synthesis as well as the accurate matching of energy levels, which makes the difficulty and workload relatively great. Moreover, although the ratiometric fluorescence sensing of these bioactive molecules in the whole cell, cytoplasm, and mitochondria has been widely explored,19,31,32 it is still a challenging task to trace their flux in the nucleus. Indeed, the rare examples only include nuclear-localized probes NucPE1, SNAP-PG and a highly sensitive probe NP1 responding to the changes of nuclear H2O2 fluxes,33–35 a photo-stable pH probe NaOCy and a ratiometric fluorescence pH probe hoeFL for selectively staining nuclei in living cells;36,37 a sulfur-containing fluorogenic probe JG45 for selectively detecting nuclear methylmercury (MeHg2+).38 The difficulty in visualizing the flux of bioactive molecules in the nucleus is mainly due to the impermeable nuclear membrane and the interference of the complex nucleic acid environment with the fluorescence of probes.39,40
On the other hand, numerous DNA and RNA binding agents have been reported, exhibiting nucleic acid induced fluorescence turn-on properties for selectively staining the DNA-rich nucleus and RNA-rich nucleolus.41–45 This provides an idea that conjugating two DNA binding groups with different DNA affinities in one probe may exhibit two different emission features based on the DNA binding modes and the DNA-modulated fluorescence. Simultaneously, utilizing DNA binding agents to construct fluorescent probes can also resolve the nuclear-targeting problem.46 With these criteria in mind, we establish a ratiometric fluorescence imaging strategy, named “charge-driven tripod somersault on DNA” by taking advantage of the nucleic acid enriched environment in the nucleus and the conjugation of two DNA binding groups. Herein, SO2 derivatives (sulfite SO32− and bisulfite HSO3−) are taken as a typical target analyte to clarify the mechanism of action of this novel strategy. Generally, sulphite and bisulphite in neutral fluid and plasma exist in equilibrium (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, M/M), and the concentration of total serum sulphite is 0–9.85 μM in healthy donors.47,48 Measurements of nuclear SO2 derivatives are undoubtedly valuable, because exposure to HSO3− can cause abnormal nuclear behavior including increased incidence rates of chromosomal aberrations, sister chromatid exchanges and micronuclei. SO2 derivatives can also induce DNA damage in multiple organs (the brain, lungs, liver, spleen, kidneys, intestines, and testicles) in vivo in a dose-dependent manner.11–13 However, detecting analytes in the nucleus is always very challenging due to the impermeable nuclear membrane and the interference of nucleic acids with the fluorescent probes.49–51 So far, reports on the variations of SO2 derivatives in the nucleus are still lacking although the cellular and mitochondrial SO2 derivatives have been widely explored.52–57
:1, M/M), and the concentration of total serum sulphite is 0–9.85 μM in healthy donors.47,48 Measurements of nuclear SO2 derivatives are undoubtedly valuable, because exposure to HSO3− can cause abnormal nuclear behavior including increased incidence rates of chromosomal aberrations, sister chromatid exchanges and micronuclei. SO2 derivatives can also induce DNA damage in multiple organs (the brain, lungs, liver, spleen, kidneys, intestines, and testicles) in vivo in a dose-dependent manner.11–13 However, detecting analytes in the nucleus is always very challenging due to the impermeable nuclear membrane and the interference of nucleic acids with the fluorescent probes.49–51 So far, reports on the variations of SO2 derivatives in the nucleus are still lacking although the cellular and mitochondrial SO2 derivatives have been widely explored.52–57
As shown in Scheme 1, a tripodal probe named [PAST]+ ((E)-2-(4-(bis(4-(pyridin-4-yl)phenyl)amino)styryl)-3-methylbenzo[d]thiazol-3-ium iodide) has been designed by conjugating a dipyridyltriphenylamine fluorophore and a benzothiazole salt, both of which are DNA binding agents, through an ethylene group, which is the reaction site with SO2 derivatives. Before encountering the target analyte, the tripod binds to DNA via the positively charged benzothiazole moiety and one uncharged arm of dipyridyltriphenylamine, thus giving the first emission feature; after reacting with the negatively charged target analyte, the newly formed tripod [PAT]− ((Z)-1-(4-(bis(4-(pyridin-4-yl)phenyl)amino)phenyl)-2-(3-methylbenzo[d]thiazol-2(3H)-ylidene)ethanesulfonate) gradually dissociates from the DNA system due to the Coulomb repulsion between the negatively charged benzothiazole–SO2 group and DNA backbone, and then becomes free to rotate in the solvent and ultimately seize back the DNA system via the two inherent neutral arms of dipyridyltriphenylamine whereas the benzothiazole–SO2 group stretches out. Such a change in the DNA binding mode results in the disappearance of the first emission feature (red emission) and the formation of the second emission feature (yellow emission), thus giving the ratiometric fluorescence spectra. Accordingly, we name this strategy the “charge-driven tripod somersault on DNA”. By using this, both in vitro and in vivo ratiometric fluorescence imaging of the variations of endogenous SO2 derivatives in the nucleus have been realized for the first time.
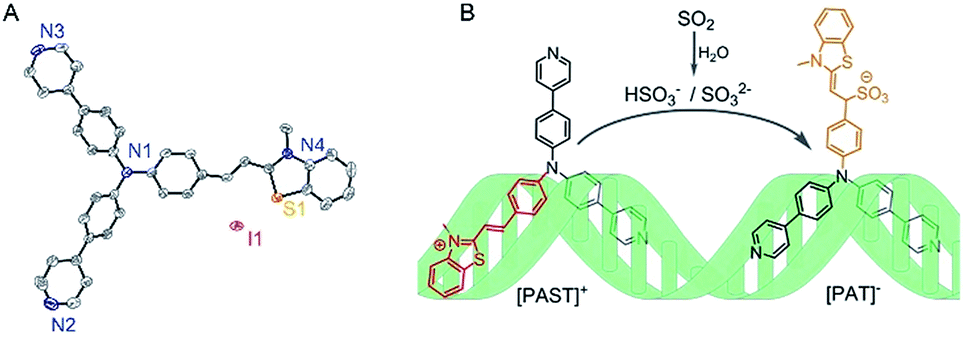 | ||
| Scheme 1 (A) X-Ray crystal structure of [PAST]+ (CCDC number: 1893794†), in which the hydrogen atoms are omitted for clarity and the counter ion is I−; (B) schematic illustration of the strategy named “charge-driven tripod somersault on DNA” for ratiometric fluorescence imaging of small bioactive molecules in the nucleus: the tripodal probe [PAST]+ is constructed by connecting two DNA binding moieties with an ethylene group; once the DNA–[PAST]+ reacts with the negatively charged target analyte, the newly formed tripod [PAT]− gradually dissociates from the DNA system, freely rotates, and ultimately seizes back the DNA with different binding modes, thus resulting in the disappearance of the first emission feature from DNA–[PAST]+ and the emergence of the second emission feature from DNA–[PAT]−. | ||
Results and discussion
Synthesis and characterization of [PAST]+ and [PAT]−
The probe [PAST]+ was synthesized and isolated (Scheme S1, ESI†) and fully characterized by HRMS, 1H NMR, 13C NMR, elemental analysis and X-ray crystallography (Scheme 1A, Fig. S1–S3 and Tables S1–S3, ESI†). 1H NMR investigation (DMSO-d6/D2O, 4:1) shows that upon HSO3− addition, the resonance signals corresponding to the alkene protons of [PAST]+ disappear, (Fig. S4, ESI†), indicating the nucleophilic addition of HSO3− to the double bond of the benzothiazole ethylene salt, resulting in the formation of [PAT]−, as briefly depicted in Scheme 1. Pure [PAT]− was also isolated for structural characterization by HRMS, 1H NMR, 13C NMR, and elemental analysis (Scheme S1 and Fig. S5–S7, ESI†). Furthermore, the reaction between [PAST]+ and HSO3− to produce [PAT]− was tracked by HPLC, and the HPLC retention time compared with those of independently synthesized [PAST]+ (4.21 min) and [PAT]− (33.89 min) further demonstrated the purity of both compounds (Fig. S8, ESI†). The photophysical properties of [PAST]+ and [PAT]− including the quantum yields, extinction coefficients, and fluorescence lifetimes were recorded in various solvents (Fig. S9 and Table S4, ESI†).
Spectral response of [PAST]+ toward SO2 derivatives in PBS or DNA enrichment buffer
Before spectral response studies, the interference of [PAST]+ self-aggregation was first excluded. Crystal data show that π–π stacking forces (distance: 4.163 Å and 4.384 Å) and intermolecular hydrogen bonding interactions (bond length: 2.501 Å and bond angle 157.991°) between adjacent [PAST]+ molecules contribute to the self-aggregation of [PAST]+ (Fig. S10, ESI†). The concentration-dependent absorption/fluorescence spectra show that the linear relationship is only observed in the concentration range of 0.1–120 μM (Fig. S11, ESI†). This indicates that [PAST]+ only undergoes self-aggregation at high concentrations (>120 μM), thus excluding the influence of self-aggregation on the reliability of the fluorescence response in the following imaging experiments using 20 μM [PAST]+.The response of [PAST]+ to HSO3− was initially investigated in PBS (pH = 7.4). With increasing concentrations of HSO3− (0–10 μM), the absorption of [PAST]+ at 515 nm gradually decreases (Fig. 1A) and the colorimetric changes (A515/A345) show a good linear relationship (Fig. S12A, ESI†). Upon 515 nm excitation, [PAST]+ exhibits a near-infrared emission centered at 735 nm, which is gradually quenched upon HSO3− titration (Fig. 1B). The quick fluorescence response (<1 min) of [PAST]+ to SO2 derivatives demonstrates its potential for real-time detection (Fig. S12B, ESI†). And the limit of detection (LOD) value of [PAST]+ for SO2 derivatives is determined to be 25.4 nM in a PBS environment (Fig. S13A, ESI†). The mechanism of the absorbance/emission change of [PAST]+ towards SO2 derivatives is explained by the HOMO and LUMO distributions of [PAST]+ and [PAT]− (Fig. S14, ESI†). For [PAST]+, the HOMO resides on the whole conjugated structure while the LUMO primarily resides on the benzothiazole ligand. However, the 1,4-addition of HSO3− to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond breaks the conjugated structure and generates [PAT]−, in which the HOMO resides on the benzothiazole ligand while the LUMO is primarily delocalized on the dipyridyltriphenylamine fluorophore. Accordingly the energy gaps between the HOMO and LUMO are greatly influenced and calculated to be 2.26 eV and 2.56 eV for [PAST]+ and [PAT]−, respectively. The corresponding calculated spectra are consistent with the observed excitation spectra of [PAST]+ and [PAT]− in PBS (Fig. S15, ESI†). The Job-plot further demonstrates this reaction with a 1:1 ratio (Fig. S16, ESI†), similar to the previously reported nucleophilic addition reaction.58,59
C bond breaks the conjugated structure and generates [PAT]−, in which the HOMO resides on the benzothiazole ligand while the LUMO is primarily delocalized on the dipyridyltriphenylamine fluorophore. Accordingly the energy gaps between the HOMO and LUMO are greatly influenced and calculated to be 2.26 eV and 2.56 eV for [PAST]+ and [PAT]−, respectively. The corresponding calculated spectra are consistent with the observed excitation spectra of [PAST]+ and [PAT]− in PBS (Fig. S15, ESI†). The Job-plot further demonstrates this reaction with a 1:1 ratio (Fig. S16, ESI†), similar to the previously reported nucleophilic addition reaction.58,59
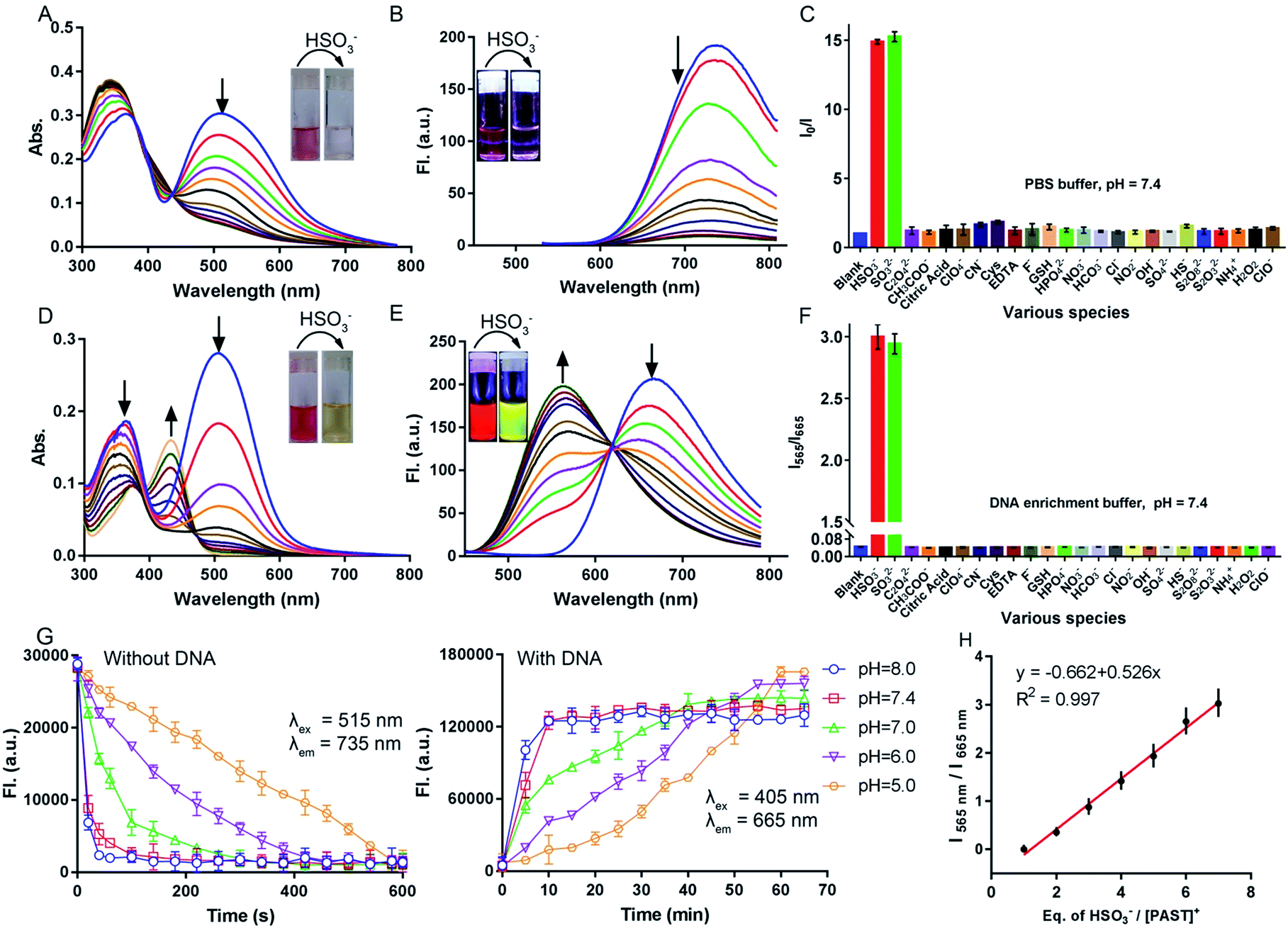 | ||
| Fig. 1 UV-Vis absorption and emission spectra of [PAST]+ (10 μM) upon HSO3− titration in (A and B) PBS solution and (D and E) DNA enrichment buffer solution (dsDNA 6.0 μM), respectively, (the HSO3− concentration for (A and B) is 0–10 μM and for (D and E) is 0–100 μM). Inset: photograph showing the corresponding absorption and fluorescence (under a 365 nm UV lamp) response seen by the naked eye; (C and F) changes in the fluorescence intensity of [PAST]+ (10.0 μM) after incubation with various biologically relevant analytes (2 mM, 30 min) in PBS (I/I0, λex = 515 nm) and DNA enrichment buffer solution (I565 nm/I665 nm, λex = 405 nm); (G) fluorescence response rate of [PAST]+ (10.0 μM) to HSO3− in aqueous solution with or without dsDNA (6.0 μM) at various pH values (5.0–8.0). (H) Linear relationship between the changes of the I565 nm/I665 nm emission ratio (λex = 405 nm) and equivalents of HSO3−/[PAST]+, (the [PAST]+ concentration: 10 μM, the data were collected from (E)). | ||
Subsequently the response of [PAST]+ to HSO3− was investigated in DNA enrichment buffer, which is PBS containing 6.0 μM duplex DNA (dsDNA, 5′-C1T2T3T4T5G6C7A8A9A10A11G12-3′) (Fig. S17A, ESI†). Fluorescence titration data indicate the binding constant Ka = 5.1 × 106 mol−1 (Fig. S17B, ESI†). The DNA/[PAST]+ stoichiometry has been determined from the Job-plot to be 1:2 (Fig. S17C, ESI†), meaning that 2 molecules of [PAST]+ bind with 1 molecule of 12 mer DNA. Upon HSO3− titration, the absorption at 345 nm and 515 nm gradually decreases and a new absorption peak emerges at 425 nm (Fig. 1D). Under 515 nm excitation, [PAST]+ exhibits intense red emission centered at 665 nm, which also gradually decreases upon HSO3− titration similar to that in PBS without DNA (Fig. S18, ESI†). However, under 405 nm excitation it is appealing to observe a new band appearing and gradually blue-shifting (ca. 115 nm) to 565 nm accompanied by reduced emission at 665 nm, the iso-emission point of which is at 625 nm (Fig. 1E). Moreover, the LOD value of [PAST]+ for SO2 derivatives in the DNA enrichment environment is determined to be 320 nM (Fig. S13B, ESI†). There is a good linear relationship (R2 = 0.997) between the emission intensity ratio I565/I665 of [PAST]+ and HSO3− concentrations in the range of 10–100 μM (Fig. 1H). When we changed the concentrations of [PAST]+ (5 μM, 10 μM, and 20 μM), the linear relationship between the HSO3−/[PAST]+ equivalent and the fluorescence ratio overlaps (Fig. S19†). Accordingly, this calibration curve can be used for calculating SO2 derivatives in solution experiments. Similar ratiometric fluorescence response (λex = 405 nm) of [PAST]+ toward HSO3− is also observed in calf thymus DNA (ctDNA, 40 μg mL−1) enrichment buffer (Fig. S20, ESI†), and the response is completed in 10 min (Fig. S21, ESI†). The new peaks produced by [PAST]+ reacting with HSO3− in enrichment buffer coincide with the new peaks produced by [PAT]− binding with DNA (Fig. S22, ESI†). By contrast, the ratiometric phenomenon (λex = 405 nm) was not observed in PBS, 40% PEG200 solution (mimicking the crowded environment in nuclei),60 RNA (56 μg mL−1) enrichment buffer, and HSA (56 μg mL−1) enrichment buffer, thus excluding the interference of nuclear viscosity, RNA binding and non-specific protein–probe association, respectively (Fig. S23 and S24, ESI†). These results demonstrate that [PAST]+ can be utilized for ratiometric sensing of SO2 derivatives in a DNA enriched environment, for example the cellular nucleus.
Selectivity of [PAST]+ toward SO2 derivatives and pH-dependency
The fluorescence response of [PAST]+ (10 μM) to various biologically relevant analytes was investigated, including various reactive sulfur species (S2O82−, SO42−, S2O32−, and HS−), reactive oxygen and nitrogen species (H2O2, ClO−, ClO4−, NO2−, NO3−, NH4+, and CN−), bio-relevant species (Cys, GSH, EDTA, and citric acid) and several common anions (F−, Cl−, HCO3−, HPO42−, OH−, C2O42−, and CH3COO−), at 200-times higher concentrations (2.0 mM) than that of HSO3−/SO32− (10 μM). As shown in Fig. 1C and F, none of these analytes could change the fluorescence of [PAST]+ either in the absence of DNA (I0/I) or in the presence of DNA (I565/I665), indicating the good selectivity of [PAST]+ toward SO2 derivatives. The selectivity was further confirmed by competition experiments, in which [PAST]+ (10 μM) was mixed with various biologically relevant analytes (2.0 mM) with or without DNA (40 μg mL−1), and then SO2 derivatives were added and the fluorescence response was monitored. It is found that fluorescence changes of [PAST]+ in the absence of DNA (I0/I) or the presence of DNA (I565/I665) are only observed when SO2 derivatives are present, indicating that all the tested biologically relevant analytes will not affect the fluorescence response even at concentrations as high as 2.0 mM (Fig. S25, ESI†).Considering that SO32− and HSO3− have a chance to reach pH-equilibrium in aqueous solution and the intracellular pH values vary in the range of 5.0–8.0, we also investigated the pH effect on the fluorescence responsiveness of [PAST]+ toward SO2 derivatives. In the absence of DNA, the red emission from [PAST]+ is always significantly quenched at pH = 5.0 and pH = 8.0 upon NaHSO3 addition, indicating the occurrence of the addition reaction and the formation of [PAT]− (Fig. S26, ESI†). However, the addition reaction rate is pH-dependent and is much slower in acidic solution than that in neutral and alkaline solution. For example, the addition reaction is completed in 10 min at pH 5.0, 1 min at pH 7.4 and 1.5 min at pH 8.0 (Fig. 1G). In DNA enrichment buffer, similar ratiometric fluorescence response of [PAST]+ to NaHSO3 titration is observed at pH = 5.0 and pH = 8.0, indicating its potential for use in a cellular environment with varied pH values (Fig. S26, ESI†). Also the ratiometric fluorescence response is much slower in acidic solution (e.g. 60 min at pH 5.0) than in neutral (10 min at pH 7.4) and alkaline solution (8 min at pH 8.0), as shown in Fig. 1G. All these pH-dependency experiments demonstrate that although the addition reaction rates in the absence and presence of DNA are both pH-dependent (slower in acidic solution), the ratiometric fluorescent response of [PAST]+ selectively toward SO2 derivatives always works in the cellular pH range.
Ratiometric fluorescence mechanism in a DNA enriched environment
To further understand the binding mode of the probe to DNA, we also performed NMR (1D and 2D) and circular dichroism (CD) spectroscopy of DNA in the absence and presence of [PAST]+ and [PAT]−, respectively. As shown in Fig. 2A, adding [PAST]+ in a DNA solution at pH 7.4 led to notable line broadening of DNA signals, indicative of a medium or fast exchange rate on the NMR time scale. In addition, small but interesting chemical shift changes were observed in the H1/H3 proton signals of the T4, T5, and G24. [PAST]+ titration was performed only in the concentration range of 0.0–1.0 equivalents because a higher concentration of [PAST]+ leads to DNA precipitation. In contrast, significant chemical shift changes in the H1/H3 proton signals of the T2, T3, T4, T5, and G6 as well as line broadening were observed for [PAT]− titration into DNA under the same conditions. Corresponding 2D H–H NOESY spectra were also collected. Consistent with the 1D NMR spectra, severe line broadening was observed for [PAST]+ binding (Fig. S27, ESI†). Signal shifts and some new NOE signals were observed after [PAT]− binding (Fig. S28, ESI†). Although the line broadening in the NMR spectra prevents the detailed structural analysis of DNA–[PAST]+ and DNA–[PAT]− complexes, the different chemical shift changes demonstrated that [PAST]+ and [PAT]− binding led to different conformational changes of DNA, indicative of distinct binding modes of these two molecules to DNA.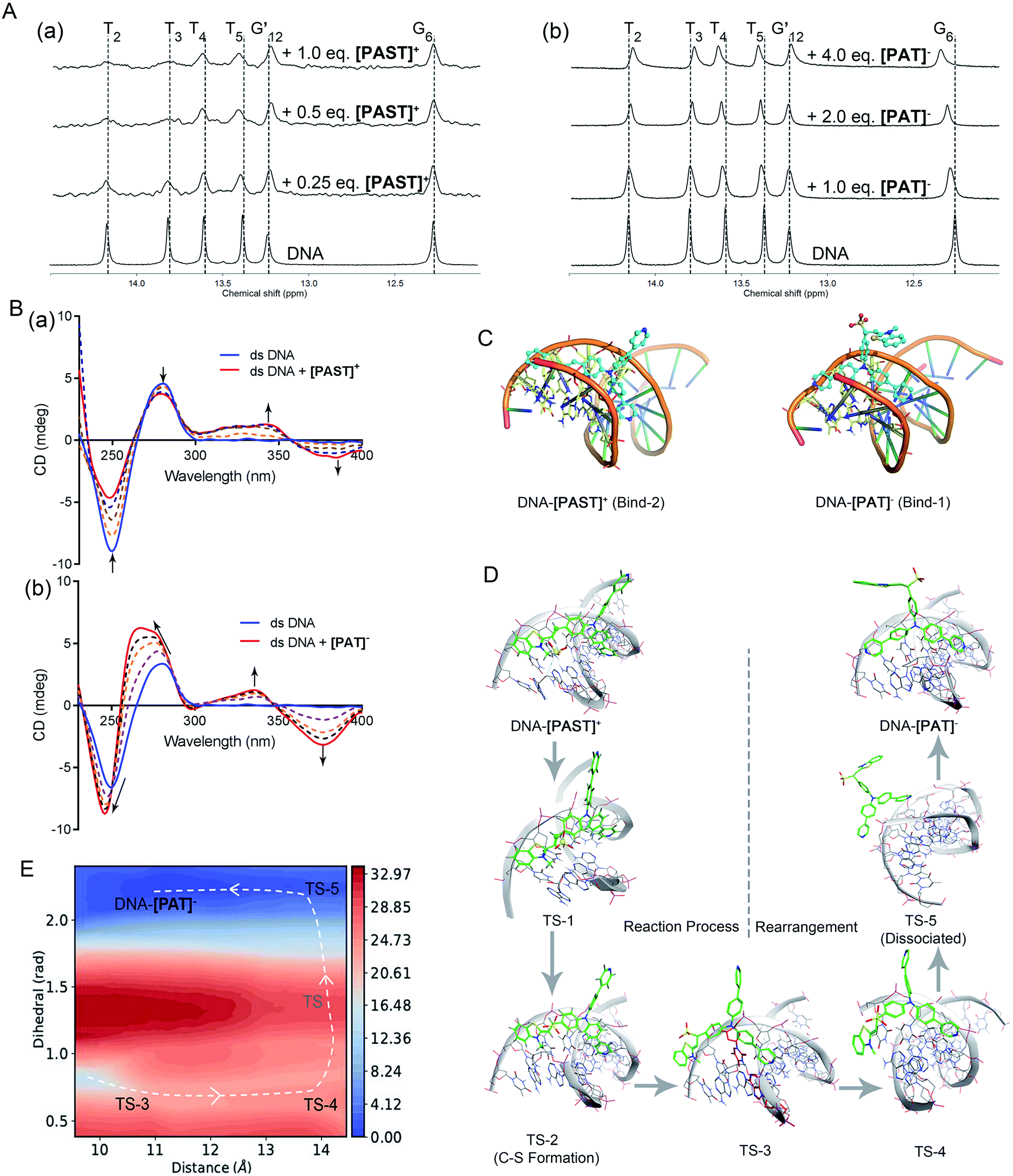 | ||
| Fig. 2 (A) 1H NMR spectra of dsDNA in buffer solution (D2O:H2O = 1:9, 5 °C) in the presence of [PAST]+ and [PAT]−, respectively; (B) CD spectra of dsDNA in PBS buffer solution in the presence of increasing concentrations of [PAST]+ and [PAT]−, respectively; (C) the most stable binding modes of the DNA–[PAST]+ complex and DNA–[PAT]− complex. See Fig. S29 in ESI† for other binding modes with binding energies. The nucleotides T2, T3, T4, T5, A21, A22 and A23 were colored in blue/yellow and highlighted in the stick model; (D) QM/MM simulations of the nucleophilic addition reaction process and the subsequent conformational rearrangement; (E) the two-dimensional free energy map of the conformational rearrangement process with two defined reaction coordinates, which are the distance (Å) between the core N atom in the tripod and the centroid of the nucleotides A21 and T4 in the DNA system and the dihedral angle (rad) between two 4-phenylpyridine rings, respectively. The energy is in units of kcal mol−1 and the interval between two consecutive contours is 1.0 kcal mol−1. | ||
In CD spectra (Fig. 2B), dsDNA is characterized by a positive band at ∼275 nm and a negative band at ∼245 nm, indicative of base stacking and the right-handed helix structure, respectively. With increasing concentrations of [PAST]+, both peak signals gradually weaken, indicating that DNA–[PAST]+ interaction can change the helical nature of DNA and cause DNA unwinding, so that it may be an external binding manner rather than classical intercalation. The newly emerged positive band at ∼340 nm and negative band at ∼380 nm indicate a minor groove binding which is consistent with the molecular docking results. By contrast, with increasing concentrations of [PAT]−, both negative (∼245 nm) and positive (∼275 nm) peaks of DNA are greatly enhanced and blue-shifted, indicating that DNA–[PAT]− interaction can enhance the stacking between bases and stabilize the right-handed superhelix structure. Meanwhile, the newly emerged positive and negative peak signals are also observed at ∼340 nm and ∼380 nm indicative of the occurrence of minor groove binding. The different changes in the CD spectra of DNA induced by [PAST]+ and [PAT]− indicate that both compounds can alter the helical nature of DNA and affect the base accumulation of DNA but in a different manner, which is indicative of distinct bonding modes of these two molecules to DNA (Fig. 2B).
In order to authenticate the ratiometric sensing mechanism, [PAST]+ and [PAT]− were respectively docked by Glide docking to a 12-mer dsDNA (5′-C1T2T3T4T5G6C7A8A9A10A11G12-3′), which had exactly the same base sequence as that used in the experiments, and the docked poses were ranked by XP scoring. MM/MD simulations give four highest scored binding modes with various binding free energies as shown in Fig. S29, (ESI†), and among them two binding modes DNA–[PAST]+ Bind-2 and DNA–[PAT]− Bind-1 are shown in Fig. 2C as typical examples. Based on the energy decomposition, hydrophobic interactions and stacking between DNA nucleotides and the aromatic rings of tripods allow the formation of DNA–[PAST]+ and DNA–[PAT]− complexes, and the nucleotides T2, T3, T4, T5, A21, A22 and A23 always exhibit a stronger interaction force than other nucleotides. Thus [PAST]+ and [PAT]− share the same binding position (minor groove) in the DNA system. Such favorable binding also likely arises from a nice shape complementarity between the DNA groove and the tripodal structure.
Although [PAST]+ and [PAT]− share the same DNA skeleton, the calculated binding energy indicate the stronger DNA binding of [PAST]+ than of [PAT]−. More interestingly, these two tripods have distinct DNA binding modes. For the DNA–[PAST]+ complex, the benzothiazole group in [PAST]+ has a better DNA binding affinity than the 4-phenylpyridine since the positive charge in benzothiazole has a strong Coulomb attraction force with the phosphate backbones of DNA. Thus [PAST]+ prefers to bind with DNA via the benzothiazole moiety and one 4-phenylpyridine arm to form a stable DNA–[PAST]+ complex (Bind-1, -2, and -3 in Fig. 2C and S29A, ESI†). As for the DNA–[PAT]− complex, the negative charge of the sulfite ion results in a Coulomb repulsion force between the benzothiazole–SO2 group and DNA backbone, resulting in significantly reduced binding energies which make the DNA–[PAT]− partially (Bind-1 and Bind-2 in Fig. S29B, ESI†) or completely dissociate (Bind-3 and -4 in Fig. S29G, ESI†). The most stable DNA–[PAT]− complex (Bind-1 in Fig. 2C and S29B, ESI†) shows that [PAT]− uses both of the 4-phenylpyridine groups to bind with DNA while the benzothiazole–SO2 moiety stretches out. The distinct binding modes of DNA–[PAST]+ and DNA–[PAT]− indicate a conformational rearrangement of the DNA-tripod complex after the sulfite ion addition reaction.
Furthermore, QM/MM simulations were performed to investigate the detailed process of the nucleophilic addition reaction and the DNA binding mode rearrangement (Fig. 2D). Herein, although [PAST]+ Bind-1 has a lower binding free energy, the nucleophilic carbon orienting inside the DNA system impedes the attack of the sulfite ion, making Bind-1 less suitable for the sulfite ion addition reaction. Thus [PAST]+ Bind-2 obtained in the MM/MD simulations is chosen as the initial structure for the QM/MM simulations. The relative potential energy surface in Fig. S30 (ESI†) shows that the energy barrier of sulfite ion addition reaction is ca. 9 kcal mol−1, which is feasible in biological systems, and in the view of thermodynamics this is an exothermal reaction releasing ca. 16 kcal mol−1 energy. The transition state structures obtained in QM/MM simulations show that at the beginning of the nucleophilic addition reaction, the oxygens in the sulfite ion first flip over (TS-1) and then the carbon sulfur bond is formed (TS-2) to generate [PAT]−. Due to the Coulomb repulsion between the negatively charged benzothiazole–SO2 group and DNA backbone, this transition state has a very short life time (<100 ns) and the DNA–[PAT]− binding mode rearrangement is expected to occur right after the addition reaction. TS-2 gradually dissociates with the less stable DNA–[PAT]− Bind-2 as a partially dissociated state (TS-3). Then after complete dissociation (TS-5), the ligands of [PAT]− become free to rotate in the solvent, and once the negatively charged benzothiazole–SO2 group rotates away from the DNA system, the free energy decreases a lot. The barrier of this rotation is about 7 kcal mol−1 and it releases ca. 15 kcal mol−1. After the rotation [PAT]− seizes back DNA while releasing another 1 kcal mol−1, generating the most stable DNA–[PAT]− complex (Bind-1). Such a feasible and exothermal binding mode rearrangement to produce DNA–[PAT]− Bind-1 is indicated by the two-dimensional free energy map shown in Fig. 2E. To further evaluate whether the negatively charged responsive arm is required for the charge-driven tripod somersault on DNA, we synthesized a control compound, TPPA, possessing a symmetric structure with three phenyl-pyridyl groups but without a responsive group (Fig. S29F, ESI†). This compound only exhibits one DNA binding mode via two phenyl-pyridyl arms with a binding free energy of −16.7 kcal mol−1, and its DNA binding capability can be confirmed by the significantly (40-fold) enhanced fluorescence in the DNA titration experiment. This provides additional evidence confirming that [PAT]− binding with DNA via two neutral phenyl-pyridyl arms is very reasonable. We also performed MM/MD simulations of neutral [HPAT]0 and protonated [H2PAT]+ and [H3PAT]2+ compounds docked to the 12-mer dsDNA (Fig. S29C–E, ESI†). It is found that the most stable DNA binding mode of [HPAT]0 is similar to that of [PAT]−, in which the two phenyl-pyridyl arms bind with DNA at minor grooves, and their binding free energies (−11.8 and −11.1 kcal mol−1) are also similar, indicating that an electron-neutral responsive arm is sufficient for the change of the DNA binding mode. The positively charged [H2PAT]+ and [H3PAT]2+ also exhibit similar DNA binding modes to neutral [HPAT]0 and [PAT]− but with stronger binding free energies (−20.4 and −21.5 kcal mol−1), indicating that DNA–[PAT]− might be more stable in an acidic microenvironment, which was consistent with the pH-dependency investigations in Fig. S29, ESI.†
Ratiometric fluorescence imaging of endogenous SO2 derivatives using [PAST]+ in the nucleus
Subsequently, we used this strategy for in vitro and in vivo ratiometric fluorescence imaging of the variations of endogenous SO2 derivatives in the nucleus of living cells. At the cellular level, [PAST]+ (20 μM) can effectively enter living A549 cells in 20 min (Fig. S31, ESI†), the intensive red emission of which exhibits a high degree of co-localization with the nucleus-specific stain Hoechst 33342 (Pearson's correlation coefficient is 0.96, Fig. 3A), but non-colocalization with LysoTracker or MitoTracker (Fig. S32, ESI†), indicating the effective and specific accumulation of [PAST]+ in the nucleus. Similar nuclear-specific accumulation is also observed in HeLa and PC3 cells (Fig. S33, ESI†). In the cytotoxicity experiment, A549 cells exposed to as high as 100 μM [PAST]+ for 6 h and 24 h still show cellular viability greater than 90% and 60%, respectively (Fig. S34, ESI†). When the imaging concentration (20 μM) is performed, [PAST]+ induced alternations of intracellular ROS levels and mitochondria membrane potential after 6 h treatment are similar to those of control cells (Fig. S35 and S36, ESI†); when the treatment time is increased (12–24 h), [PAST]+ treated cells only exhibit 1.1-fold higher intracellular ROS levels. Considering the interaction between [PAST]+ and DNA, we also investigated the influence of [PAST]+ on the A549 cell cycles by propidium iodide (PI) staining using flow cytometry. As shown in Fig. S37 (ESI†), [PAST]+ (10 and 20 μM) treated cells show similar cell cycles after 6–24 h incubation compared with the vehicle-treated cells, and exhibit only slight increases in the proportion of cells in the S-phase after 18–24 h incubation. These results indicate that although [PAST]+ has some long-term cytotoxicity, it will not induce alternations of the intracellular microenvironment or adverse effects at least during the imaging process.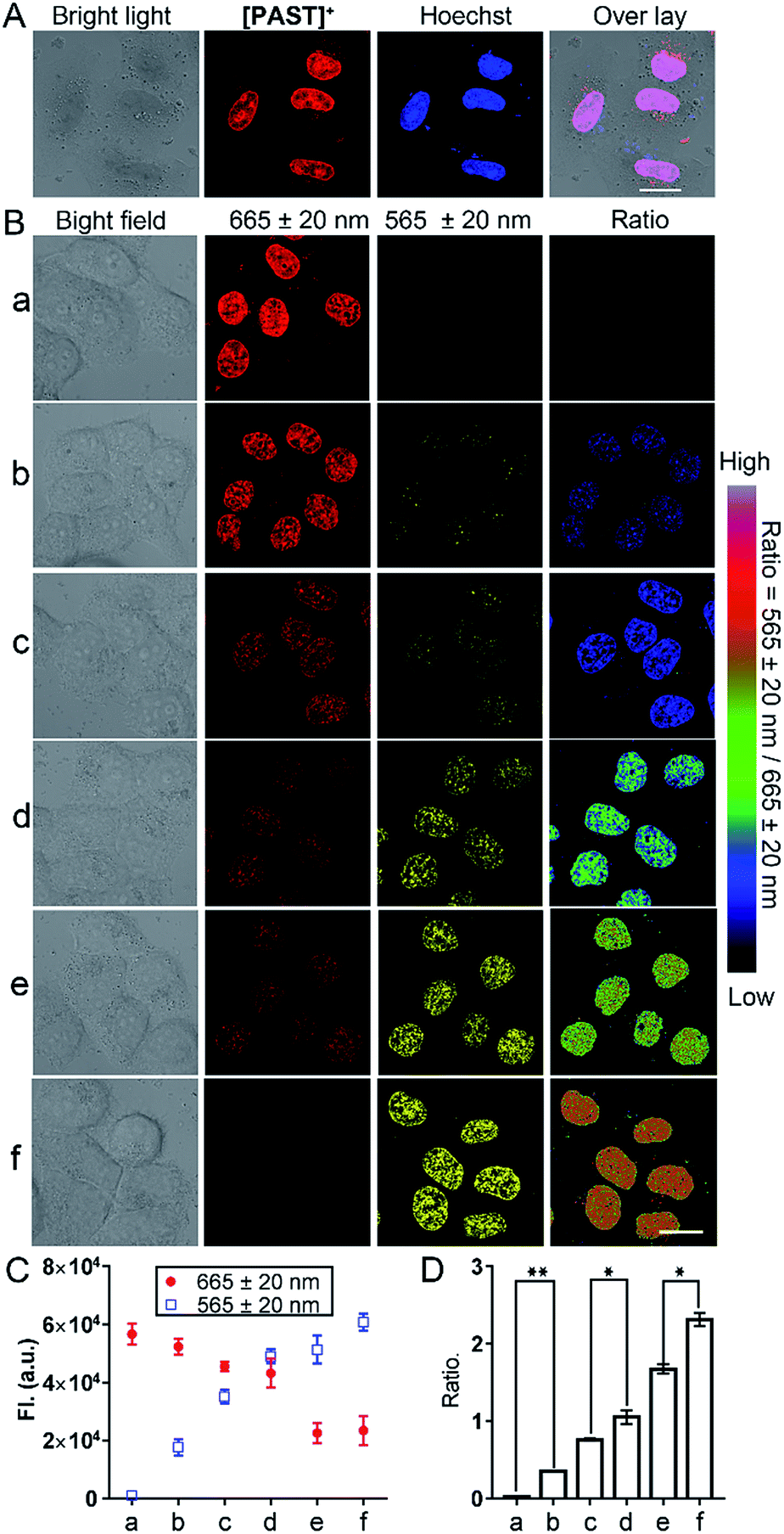 | ||
| Fig. 3 (A) Colocalization between [PAST]+ and the nucleus-specific stain Hoechst (Hoechst 33342) in living cells (probe: λex/λem = 543 nm/665 ± 20 nm; Hoechst: λex/λem = 405 nm/460 ± 20 nm). (B) Ratiometric fluorescence imaging of the variations of endogenous SO2 derivatives in the nucleus of living cells. A549 cells were incubated with [PAST]+ (20 μM) for 20 min and then treated with thiosulphate/GSH at (a) 0 μM/0 μM (control), (b) 60 μM/30 μM, (c) 120 μM/60 μM, (d) 180 μM/90 μM, (e) 240 μM/120 μM, and (f) 300 μM/150 μM (concentrations) for another 30 min to stimulate endogenous SO2 derivatives; scale bar: 20 μm. (C and D) The mean values of the fluorescence photon number collected from the red and yellow channel, respectively, and the ratio of their photon numbers were calculated. Data were calculated from 3–5 fields of view and each experimental condition was performed in triplicate. Data are represented as mean ± SD of three independent experiments, **P < 0.05, *P < 0.02. | ||
Ratiometric fluorescence imaging of nuclear SO2 derivatives in A549 cells was performed in dual-emission mode (λex = 405 nm) by mediating the yellow channel image (band path of 565 ± 20 nm) with the corresponding red channel image (band path of 665 ± 20 nm). It has been reported that biological sulphite can be produced endogenously from thiosulphate and GSH via thiosulphate sulphur transferase (TST) and can then be converted to sulphate via sulphate oxidase in mammals.61,62 It is worth mentioning that separately added thiosulphate or GSH will not induce the generation of endogenous SO2 derivatives, and thus will not affect the [PAST]+ emission (Fig. S38, ESI†). Therefore A549 cells were stimulated with various concentrations of thiosulphate/GSH (2:1) for 30 min to induce different concentrations of endogenous SO2 derivatives.63,64 As shown in Fig. 3B, non-stimulated A549 cells stained with [PAST]+ (20 μM, 20 min) show intense fluorescence in the red channel and dim fluorescence in the yellow channel, giving a low Fyellow/Fred ratio implying a very low sulfite/bisulfite level. However, after stimulating cells with increasing concentrations of thiosulphate/GSH, a distinct increase in yellow fluorescence is observed accompanied by a dramatic decrease in the red fluorescence, thus leading to a gradual increase of the ratiometric signals (Fyellow/Fred). This indicates the gradually increased sulfite/bisulfite level in the nucleus. Each cellular imaging experiment was performed in triplicate under the same conditions with the same microscopic settings, and 3–5 fields of view were taken for each dish for calculating the mean values of fluorescence intensity with error bars and statistical significance (Fig. 3C and D). We have further investigated the effect of concentration of the probe on the ratiometric signals. A549 cells were incubated with different concentrations of [PAST]+ (10 μM, 20 μM and 30 μM) for 20 min, as shown in Fig. S39 (ESI†). The results show that the ratiometric signals changed obviously when the concentration of [PAST]+ changed from 10 μM to 20 μM, but when the concentration of [PAST]+ changed from 20 μM to 30 μM, the ratiometric signals almost did not change. In other words, [PAST]+ at 10 μM does not produce the same ratiometric signals in cells as it does at higher concentrations (20–30 μM). This may be due to the restrictions of cell uptake. The same results were also obtained with HeLa cells.
Once cells were pre-treated with a thiosulphate sulphur transferase inhibitor, TNBS (1.0 mM, 2 h), to prevent the generation of endogenous SO2 derivatives, the red emission from intracellular [PAST]+ did not show any changes with thiosulphate/GSH (500 μM/250 μM) stimulation (Fig. S38, ESI†). This further demonstrates the reliability of the ratiometric fluorescence imaging of endogenous SO2 derivatives. It is worth mentioning that the ratiometric signals only represent the variations of nuclear rather than cytoplasmic sulfite/bisulfite levels, which can be confirmed by parallel experiments with [PAT]− prepared in advance in solution (Fig. S40, ESI†). Living cells incubated with [PAT]− only show dim fluorescence; however, after PBS washing, culture medium refreshing, and cell permeation, the nuclei are lit up by yellow fluorescence. This demonstrates that even if [PAST]+ picks sulfite/bisulfite up on the way, the formed [PAT]− in the cytoplasm only shows dim fluorescence and cannot enter the nuclei to interfere with the ratiometric signals.
Furthermore, [PAST]+ can also visualize variations of sulfite/bisulfite levels on chromosomes with various structural variations of nuclear rather than cytoplasmic sulfite/bisulfite levels, which can be confirmed by parallel characteristics. This is exciting because SO2 directly relates to chromosomal aberrations and sister chromatid exchanges.11,51 As shown in the higher resolution micrographs of single cells, when asynchronous cells are imaged, [PAST]+ is always co-localized with chromosomes as cells progress through the cell cycle (Fig. 4), and ratiometric fluorescence images in living cells undergoing prophase, metaphase, anaphase and telophase can be obtained as an indication of local chromosomal SO2 derivatives (Fig. 4B). Mean values of every single sample taken from 3–5 fields of view were calculated and are shown as bar graphs with error bars and statistical significance in Fig. 4C. Considering the important role of DNA in this ratiometric sensing strategy, we also used nuclease-treated A549 cells, in which most nucleic acids were digested. As shown in Fig. S41 (ESI†), the red fluorescence from [PAST]+ in nuclease-treated cells is much weaker than that in control cells and mostly distributed in the cytoplasm. After incubating the nuclease-treated cells with 200 μM NaHSO3 for 30 min, a distinct decrease in the red channel fluorescence is observed without the ratiometric phenomenon. These results further confirm that DNA is an indispensable factor in this ratiometric fluorescence imaging strategy.
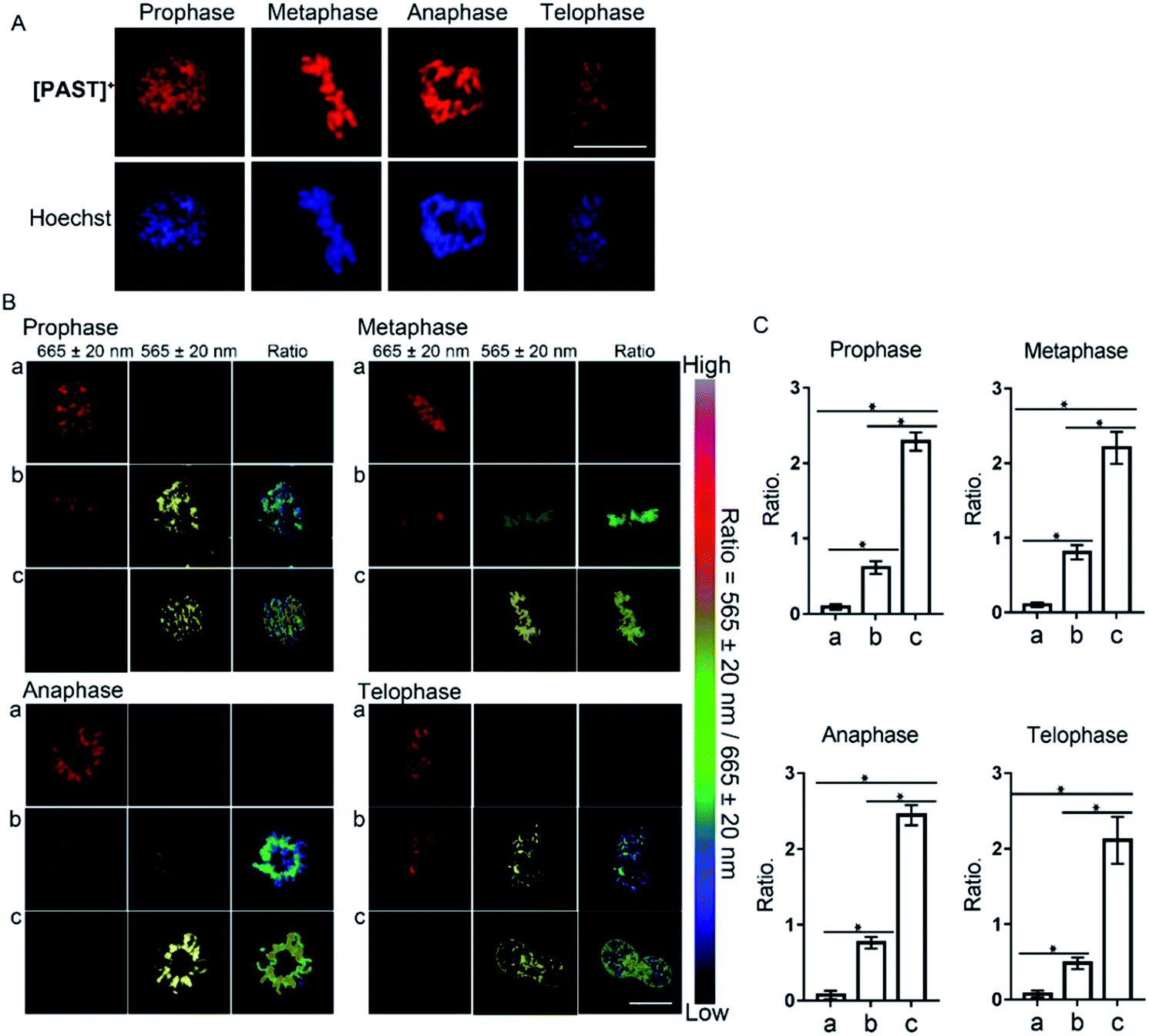 | ||
| Fig. 4 (A) Confocal microscope images of single asynchronous cells co-stained with [PAST]+ and the nucleus-specific stain Hoechst 33342. (B) Ratiometric fluorescence imaging of endogenous SO2 derivatives in living cells undergoing prophase, metaphase, anaphase and telophase, respectively. A549 cells during the split period were incubated with [PAST]+ (20 μM) for 20 min and then treated with thiosulphate/GSH at (a) 0 μM/0 μM, (b) 250 μM/125 μM, and (c) 500 μM/250 μM for another 30 min to stimulate endogenous SO2 derivatives; scale bar: 10 μm. (C) The mean values of the ratiometric fluorescence intensity obtained from different division phases. Data are represented as mean ± SD of three independent experiments, *P < 0.02. | ||
Ratiometric fluorescence imaging of endogenous SO2 derivatives using [PAST]+in vivo
Lastly, the feasibility of [PAST]+ for in vivo imaging of endogenous SO2 derivatives was also investigated. [PAST]+ (20 μM, 300 μL) was injected into the thigh roots (both left and right) of living nude mice; 30 min later, thiosulphate/GSH (1.0 mM/500 μM, 300 μL) was solely injected into the right thigh to stimulate endogenous SO2 derivatives while the left thigh was injected with 300 μL PBS as a control. As shown in Fig. 5A(a), under 530 nm excitation, the fluorescence intensity in the left thigh (control) does not change during the measurement whereas that in the right thigh is gradually quenched in 2 h. This is consistent with the reduced Fred channel observed in the cellular experiment (Fig. 3). Simultaneously, under 430 nm excitation (Fig. 5A(b)), the fluorescence intensity in the left thigh (control) is negligible whereas that in the right thigh root is gradually enhanced in 2 h. This is consistent with the gradually increased Fyellow channel observed in living cells (Fig. 3). Mean values of fluorescence signals were calculated for three mice per condition, shown as bar graphs with error bars and statistical significance (Fig. 5B). These data demonstrate that [PAST]+ can be used for measuring the variations of endogenous SO2 derivatives in vivo in a ratiometric fluorescence manner.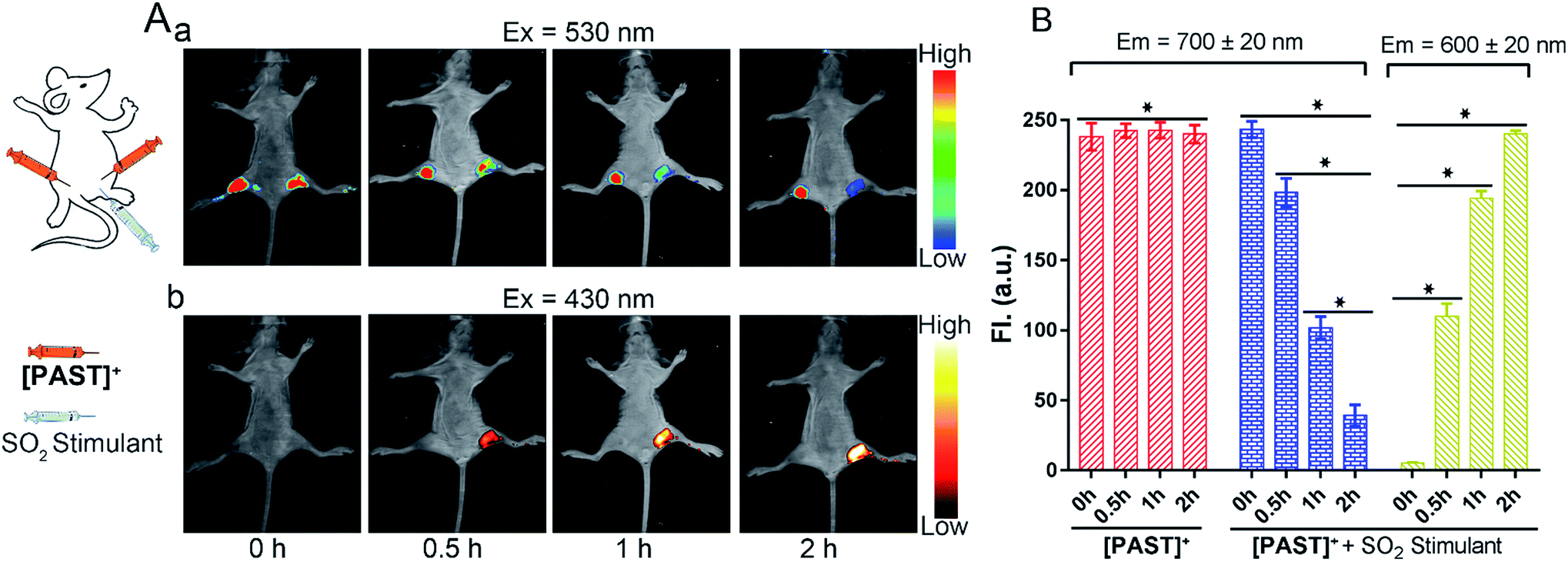 | ||
| Fig. 5 (A) In vivo time-dependent fluorescence images for visualizing endogenous SO2 derivatives. λem = 700 ± 20 nm (a); λem = 600 ± 20 nm (b). The probe [PAST]+ (con. 20 μM) was injected into the thigh roots (both left and right) of living nude mice; 30 min later, thiosulphate/GSH (1.0 mM/500 μM) was injected into the right thigh root only to stimulate endogenous SO2 derivatives while the left thigh was used as a control without stimulation. (B) The mean values of the fluorescence intensity calculated from the red channel of the probe (left thigh root, λem = 700 ± 20 nm) and the red channel (right thigh root, λem = 700 ± 20 nm) and yellow channel (right thigh root, λem = 600 ± 20 nm) of the probe + SO2 stimulant, respectively. Data are collected from three mice and represented as mean ± SD of three independent experiments, *P < 0.02. | ||
Conclusions
In summary, we take advantage of the DNA enriched environment in the nucleus to establish a ratiometric fluorescence imaging strategy, the so called “charge-driven tripod somersault on DNA”. Based on this, a nucleus-targeted tripod [PAST]+ has been synthesized by conjugating two DNA binding agents containing a reaction site for the target analyte. Once the DNA–[PAST]+ reacts with the negatively charged target analyte, the newly formed tripod [PAT]− binds DNA with different binding modes, thus resulting in the disappearance of the first emission feature from DNA–[PAST]+ and the emergence of the second emission feature from DNA–[PAT]−. By using this, we realized the in vitro and in vivo ratiometric fluorescence imaging of endogenous SO2 derivatives in the nucleus for the first time. In theory, as long as the analyte could react with the responsive group and change the charge of the responsive arm of the tripodal probe, the tripod somersault could occur to realize the ratiometric fluorescence imaging. Moreover, the change from positive-to-negative charge is not necessary and the analyte-induced formation of an electron-neutral responsive arm is sufficient for the change of the DNA binding mode. Therefore, we anticipate that this strategy can be further extended for ratiometric fluorescence imaging of a wide range of bioactive molecules in the nucleus. Although [PAST]+ has some long-term cytotoxicity, it will not induce alternations of the intracellular microenvironment or adverse effects at least during the imaging process. Further modification may help to improve the biocompatibility of the tripodal probe, e.g. methylation and tert-butylation of the phenyl-pyridine arms, which is also helpful for avoiding the pH dependency of the probe. Furthermore, since the charge-driven tripod somersault on DNA is feasible, this “somersault on DNA” strategy can also be extended to other structures, such as triangles, propellers, clovers, and multi-arm structures with inherent nucleic acid binding capacity. This provides an opportunity for opening up a new avenue for the design of nucleus-localised responsive probes for biological analytes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China [21837006, 21572282 and 21401217], the Ministry of Education of China (No. IRT-17R111), 973 Program [2014CB845604, 2015CB856301], Natural Science Foundation of Guangdong Province [2017A030313041], Fundamental Research Funds for the Central Universities, Special Funds for the Cultivation of Guangdong College Students' Scientific and Technological Innovation [pdjha0005]. We thank Dr Hulin Tai (SYSU) for assistance with 2D NMR and Prof. Duxia Cao (University of Jinan), Dr Xi-Juan Chao (SYSU), Dr Yu Guo (SYSU) and Teng-Fang Long (SYSU) for their guidance and assistance with the fluorescence experiments.Notes and references
- G. G. Gundersen and H. J. Worman, Nuclear positioning, Cell, 2013, 152, 1376–1389 CrossRef CAS PubMed.
- Z. Dou, et al., Cytoplasmic chromatin triggers inflammation in senescence and cancer, Nature, 2017, 550, 402–406 CrossRef CAS PubMed.
- F. Gasset-Rosa, et al., Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport, Neuron, 2017, 94, 48–57 CrossRef CAS PubMed.
- Q. Zhou, et al., Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease, Nat. Genet., 2016, 48, 67–73 CrossRef CAS PubMed.
- N. Tanaka, T. Aoyama, S. Kimura and F. J. Gonzalez, Targeting nuclear receptors for the treatment of fatty liver disease, Pharmacol. Ther., 2017, 179, 142–157 CrossRef CAS PubMed.
- S. Loft and H. E. Poulsen, Cancer risk and oxidative DNA damage in man, J. Mol. Med., 1996, 74, 297–312 CrossRef CAS PubMed.
- J. A. Imlay, Cellular defenses against superoxide and hydrogen peroxide, Annu. Rev. Biochem., 2008, 77, 755–776 CrossRef CAS PubMed.
- S. Nemmiche, Oxidative signaling response to cadmium exposure, Toxicol. Sci., 2017, 156, 4–10 CAS.
- P. Chabosseau, J. Woodier, R. Cheung and G. A. Rutter, Sensors for measuring subcellular zinc pools, Metallomics, 2018, 10, 229–239 RSC.
- G. Simic, et al., Monoaminergic neuropathology in Alzheimer's disease, Prog. Neurobiol., 2017, 151, 101–138 CrossRef CAS PubMed.
- Z. Meng and L. Zhang, Cytogenetic damage induced in human lymphocytes by sodium bisulfite, Mutat. Res., 1992, 298, 63–69 CAS.
- N. Sang, et al., SO2 inhalation contributes to the development and progression of ischemic stroke in the brain, Toxicol. Sci., 2010, 114, 226–236 CrossRef CAS PubMed.
- G. Qin, J. Wang and N. Sang, Sulfur dioxide inhibits expression of mitochondrial oxidative phosphorylation genes encoded by both nuclear DNA and mitochondrial DNA in rat lungs, Environ. Sci. Pollut. Res., 2016, 24, 2527–2534 CrossRef PubMed.
- D. W. Domaille, E. L. Que and C. J. Chang, Synthetic fluorescent sensors for studying the cell biology of metals, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- T. Ueno and T. Nagano, Fluorescent probes for sensing and imaging, Nat. Methods, 2011, 8, 642–645 CrossRef CAS PubMed.
- D. Wu, et al., Fluorescent chemosensors: the past, present and future, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- K. Kikuchi, Design, Synthesis and biological application of chemical probes for bio-imaging, Chem. Soc. Rev., 2010, 39, 2048–2053 RSC.
- K. Gu, et al., Real-time tracking and in vivo visualization of beta-galactosidase activity in colorectal tumor with a ratiometric near-infrared fluorescent probe, J. Am. Chem. Soc., 2016, 138, 5334–5340 CrossRef CAS.
- M. H. Lee, J. S. Kim and J. L. Sessler, Small molecule-based ratiometric fluorescence probes for cations, anions, and biomolecules, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- L. He, B. Dong, Y. Liu and W. Lin, Fluorescent chemosensors manipulated by dual/triple interplaying sensing mechanisms, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC.
- F. Qian, et al., Visible light excitable Zn2+ fluorescent sensor derived from an intramolecular charge transfer fluorophore and its in vitro and in vivo application, J. Am. Chem. Soc., 2009, 131, 1460–1468 CrossRef CAS PubMed.
- D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, An ICT-based approach to ratiometric fluorescence imaging of hydrogen peroxide produced in living cells, J. Am. Chem. Soc., 2008, 130, 4596–4597 CrossRef CAS.
- Y. Chen, et al., A ratiometric fluorescent probe for rapid detection of hydrogen sulfide in mitochondria, Angew. Chem., Int. Ed., 2013, 52, 1688–1691 CrossRef CAS PubMed.
- H. Takakusa, et al., Design and synthesis of an enzyme-cleavable sensor molecule for phosphodiesterase activity based on fluorescence resonance energy transfer, J. Am. Chem. Soc., 2002, 124, 1653–1657 CrossRef CAS PubMed.
- M. M. Henary, Y. Wu and C. J. Fahrni, Zinc(II)-selective ratiometric fluorescent sensors based on inhibition of excited-state intramolecular proton transfer, Chem.–Eur. J., 2004, 10, 3015–3025 CrossRef CAS PubMed.
- V. S. Padalkar and S. Seki, Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- W. Lin, L. Yuan, Z. Cao, Y. Feng and J. Song, Through-bond energy transfer cassettes with minimal spectral overlap between the donor emission and acceptor absorption: coumarin-rhodamine dyads with large pseudo-Stokes shifts and emission shifts, Angew. Chem., Int. Ed., 2010, 49, 375–379 CrossRef CAS PubMed.
- L. Zhou, et al., Molecular engineering of a TBET-based two-photon fluorescent probe for ratiometric imaging of living cells and tissues, J. Am. Chem. Soc., 2014, 136, 9838–9841 CrossRef CAS PubMed.
- J. A. Sclafani, M. T. Maranto, T. M. Sisk and S. A. Van Arman, An aqueous ratiometric fluorescence probe for Zn(II), Tetrahedron Lett., 1996, 37, 2193–2196 CrossRef CAS.
- Z. Xu, et al., Unique sandwich stacking of pyrene-adenine-pyrene for selective and ratiometric fluorescent sensing of ATP at physiological pH, J. Am. Chem. Soc., 2009, 131, 15528–15533 CrossRef CAS PubMed.
- J. B. Grimm, et al., A general method to improve fluorophores for live-cell and single-molecule microscopy, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- Z. Yang, et al., Macro-/micro-environment-sensitive chemosensing and biological imaging, Chem. Soc. Rev., 2014, 43, 4563–4601 RSC.
- Y. Wen, et al., A highly sensitive ratiometric fluorescent probe for the detection of cytoplasmic and nuclear hydrogen peroxide, Anal. Chem., 2014, 86, 9970–9976 CrossRef CAS PubMed.
- B. C. Dickinson, Y. Tang, Z. Chang and C. J. Chang, A nuclear-localized fluorescent hydrogen peroxide probe for monitoring sirtuin-mediated oxidative stress responses In Vivo, Chem. Biol., 2011, 18, 943–948 CrossRef CAS PubMed.
- D. Srikun, A. E. Albers, C. I. Nam, A. T. Iavarone and C. J. Chang, Organelle-targetable fluorescent probes for imaging hydrogen peroxide in living cells via SNAP-tag protein labeling, J. Am. Chem. Soc., 2010, 132, 4455–4465 CrossRef CAS PubMed.
- L. Zeng, et al., A novel and photostable pH probe for selectively staining nuclei in living cells, Analyst, 2013, 138, 7083 RSC.
- A. Nakamura and S. Tsukiji, Ratiometric fluorescence imaging of nuclear pH in living cells using Hoechst-tagged fluorescein, Bioorg. Med. Chem. Lett., 2017, 27, 3127–3130 CrossRef CAS PubMed.
- B. Diaz de Grenu, et al., Chemical speciation of MeHg (+) and Hg (2+) in aqueous solution and HEK cells nuclei by means of DNA interacting fluorogenic probes, Chem. Sci., 2015, 6, 3757–3764 RSC.
- H. Zhu, J. Fan, J. Du and X. Peng, Fluorescent Probes for Sensing and Imaging Within Specific Cellular Organelles, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- T. Morisaki, et al., Real-time quantification of single RNA translation dynamics in living cells, Science, 2016, 352, 1425–1429 CrossRef CAS PubMed.
- Y. V. Suseela, N. Narayanaswamy, S. Pratihar and T. Govindaraju, Far-red fluorescent probes for canonical and non-canonical nucleic acid structures: current progress and future implications, Chem. Soc. Rev., 2018, 47, 1098–1131 RSC.
- Y. Liu, J. Niu, W. Wang, Y. Ma and W. Lin, Simultaneous imaging of ribonucleic acid and hydrogen sulfide in living systems with distinct fluorescence signals using a single fluorescent probe, Adv. Sci., 2018, 5, 1700966 CrossRef PubMed.
- G. Lukinavicius, et al., SiR-Hoechst is a far-red DNA stain for live-cell nanoscopy, Nat. Commun., 2015, 6, 8497 CrossRef CAS PubMed.
- D. Li, et al., Nucleic acid-selective light-up fluorescent biosensors for ratiometric two-photon imaging of the viscosity of live cells and tissues, Chem. Sci., 2016, 7, 2257–2263 RSC.
- B. Dumat, et al., DNA switches on the two-photon efficiency of an ultrabright triphenylamine fluorescent probe specific of AT regions, J. Am. Chem. Soc., 2013, 135, 12697–12706 CrossRef CAS PubMed.
- J. L. Kolanowski, F. Liu and E. J. New, Fluorescent probes for the simultaneous detection of multiple analytes in biology, Chem. Soc. Rev., 2018, 47, 195–208 RSC.
- Z. Meng and L. Zhang, Cytogenetic damage induced in human lymphocytes by sodium bisulfite, Mutat. Res. Genet. Toxicol., 1992, 298, 63–69 CrossRef CAS.
- G. Qin, J. Wang and N. Sang, Sulfur dioxide inhibits expression of mitochondrial oxidative phosphorylation genes encoded by both nuclear DNA and mitochondrial DNA in rat lungs, Environ. Sci. Pollut. Res., 2017, 24, 2527–2534 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Oxidants, oxidative stress and the biology of ageing, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- N. Sang, Y. Yun, H. Li, L. Hou, M. Han and G. Li, SO2 inhalation contributes to the development and progression of ischemic stroke in the brain, Toxicol. Sci., 2010, 114, 226–236 CrossRef CAS PubMed.
- Z. Meng, G. Qin, B. Zhang and J. Bai, DNA damaging effects of sulfur dioxide derivatives in cells from various organs of mice, Mutagenesis, 2004, 19, 465–468 CrossRef CAS PubMed.
- Y. Yue, et al., Dual-Site Fluorescent Probe for Visualizing the Metabolism of Cys in Living Cells, J. Am. Chem. Soc., 2017, 139, 3181–3185 CrossRef CAS PubMed.
- V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chemical probes for molecular imaging and detection of hydrogen sulfide and reactive sulfur species in biological systems, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC.
- Y. Liu, et al., A mitochondria-targeted colorimetric and ratiometric fluorescent probe for biological SO2 derivatives in living cells, Chem. Commun., 2015, 51, 10236–10239 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Luminescent chemodosimeters for bioimaging, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- L. Tang, et al., A mitochondria-targetable fluorescent probe for ratiometric detection of SO2 derivatives and its application in live cell imaging, Sens. Actuators, B, 2017, 247, 421–427 CrossRef CAS.
- Y. Zhang, et al., Reversible Fluorescent Probe for Selective Detection and Cell Imaging of Oxidative Stress Indicator Bisulfite, Anal. Chem., 2016, 88, 4426–4431 CrossRef CAS PubMed.
- W. Xu, et al., A mitochondria-targeted ratiometric fluorescent probe to monitor endogenously generated sulfur dioxide derivatives in living cells, Biomaterials, 2015, 56, 1–9 CrossRef CAS PubMed.
- J. Xu, et al., A mitochondria-targeted ratiometric fluorescent probe for rapid, sensitive and specific detection of biological SO2 derivatives in living cells, Biosens. Bioelectron., 2016, 77, 725–732 CrossRef CAS PubMed.
- Y. Xue, et al., Human telomeric DNA forms parallel-stranded intramolecular G-quadruplex in K+ solution under molecular crowding condition, J. Am. Chem. Soc., 2007, 129, 11185–11191 CrossRef CAS PubMed.
- H. Mitsuhashi, et al., Oxidative stress-dependent conversion of hydrogen sulfide to sulfite by activated neutrophils, Shock, 2005, 24, 529–534 CrossRef CAS PubMed.
- M. H. Stipanuk, J. De la Rosa and L. L. Hirschberger, Catabolism of cyst(e)ine by rat renal cortical tubules, J. Nutr., 1990, 120, 450–458 CrossRef CAS PubMed.
- G. Li, et al., A dinuclear iridium(III) complex as a visual specific phosphorescent probe for endogenous sulphite and bisulphite in living cells, Chem. Sci., 2013, 4, 4426 RSC.
- D. P. Li, Z. Y. Wang, H. Su, J. Y. Miao and B. X. Zhao, Fluorescence detection of endogenous bisulfite in liver cancer cells using an effective ESIPT enhanced FRET platform, Chem. Commun., 2017, 53, 577–580 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization; DNA titration experiments, cytotoxicity of probes, and so on. CCDC 1893794. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03594j |
| This journal is © The Royal Society of Chemistry 2019 |