
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulphide as a leaving group: highly stereoselective bromination of alkyl phenyl sulphides†
Daniele
Canestrari
a,
Caterina
Cioffi
a,
Ilaria
Biancofiore
ab,
Stefano
Lancianesi
a,
Lorenza
Ghisu
a,
Manuel
Ruether
c,
John
O'Brien
c,
Mauro F. A.
Adamo
 *a and
Hasim
Ibrahim
*a
*a and
Hasim
Ibrahim
*a
aCentre for Synthesis and Chemical Biology (CSCB), Department of Chemistry, Royal College of Surgeons in Ireland, 123 St. Stephen's Green, Dublin 2, Ireland. E-mail: madamo@rcsi.ie; hasimibrahim@rcsi.ie
bIRBM Science Park S.p.A, Department of Medicinal Chemistry, Via Pontina, 30.600, 00071 Pomezia RM, Italy
cTrinity Biomedical Sciences Institute, School of Chemistry, The University of Dublin, Trinity College, Dublin 2, Ireland
First published on 16th August 2019
Abstract
A conceptionally novel nucleophilic substitution approach to synthetically important alkyl bromides is presented. Using molecular bromine (Br2), readily available secondary benzyl and tertiary alkyl phenyl sulphides are converted into the corresponding bromides under exceptionally mild, acid- and base-free reaction conditions. This simple transformation allows the isolation of elimination sensitive benzylic β-bromo carbonyl and nitrile compounds in mostly high yields and purities. Remarkably, protic functionalities such as acids and alcohols are tolerated. Enantioenriched benzylic β-sulphido esters, readily prepared by asymmetric sulpha-Michael addition, produce the corresponding inverted bromides with high stereoselectivities, approaching complete enantiospecificity at −40 °C. Significantly, the reported benzylic β-bromo esters can be stored without racemisation for prolonged periods at −20 °C. Their synthetic potential was demonstrated by the one-pot preparation of γ-azido alcohol (S)-5 in 90% ee. NMR studies revealed an initial formation of a sulphide bromine adduct, which in turn is in equilibrium with a postulated dibromosulphurane intermediate that undergoes C–Br bond formation.
Introduction
Alkyl bromides are versatile and extensively utilised synthetic intermediates, constitute precursors for a wide range of C–C1 and C–heteroatom2 bond forming reactions, and are motifs found in biologically active natural products.3 Given their importance, numerous methods have been developed for their preparation,4 with protocols relying on the nucleophilic substitution of alkyl alcohols with bromide ions remaining the most widely applied and studied.5,6 Among them, the use of phosphorus(V) or phosphorus(III) reagents is particularly widespread (Scheme 1a).7 Catalytic variants of the phosphorus(V) based Appel reaction have been developed recently,8 including a catalytic system for the direct deoxybromination of alcohols.8b Other innovative modes for catalytic activation of alkyl alcohols towards bromide ion substitution have recently emerged;6b,9 however, these protocols proceed through in situ substitution of catalytically formed alkyl chloride intermediates by added exogenous bromide ions (Finkelstein reaction).6b,9d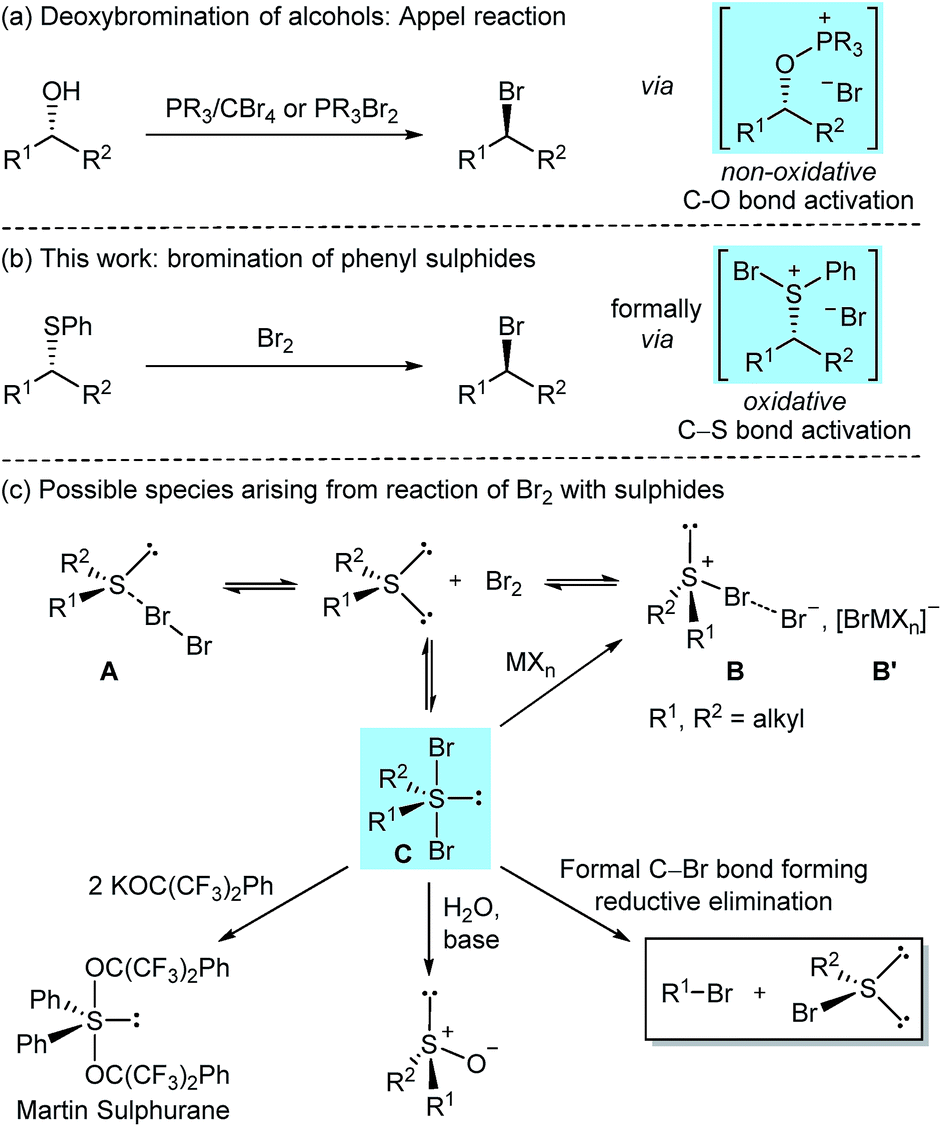 | ||
| Scheme 1 Nucleophilic bromination of alkyl phenyl sulphides in context. | ||
The above methods showcase the recent progress made in the deoxybromination of alkyl alcohols; however, challenges remain such as alkene side product formation from elimination sensitive bromides, limited scope and functional group tolerance, difficulties in removing by-products such as phosphine oxides, requirement of multiple reagents and bromide racemisation from optically active alkyl alcohols.6a,10
We have recently reported a novel nucleophilic chlorination approach to obtaining alkyl chlorides from readily available alkyl phenyl sulphides.11 This mild and rapid (dichloroiodo)benzene (PhICl2) promoted transformation proceeded formally via a SN2 chloride ion attack on a proposed, oxidatively generated chlorosulphonium chloride intermediate.
Considering the above-mentioned limitations in the preparation of alkyl bromides from alcohols, we posed the question whether sulphides could be used as precursors for alkyl bromides. Sulphides can be accessed by a number of methods, especially via thiol addition to widely available and inexpensive Michael acceptors. However, it was unclear if this was a viable approach, since Br2 and other Br+ equivalents are weaker oxidising agents than PhICl2 (vide infra).
Herein, we delineate how the concept of ‘sulphide as leaving group’ was exploited for the bromination of alkyl phenyl sulphides using Br2, an inexpensive laboratory commodity (Scheme 1b). This study culminated in the development of an exceptionally mild, chemoselective and highly stereoselective nucleophilic bromination reaction.
The reaction of diorganosulphides with Br2 has been known for over 100 years to oxidise the sulphur(II) centre;12 however, conflicting reports on the nature of the formed species have been disclosed. Depending on sulphide substitution, temperature, methods of generation, isolation and structural analysis (i.e. in the solid state or in solution),13 it has been suggested that the reaction could, via an equilibrium, generate tetrahedral sulphur(II) molecular complexes A (MCs), trigonal pyramidal sulphur(IV) bromosulphonium bromides B or trigonal bipyramidal (TB) sulphur(IV) dibromosulphurane adducts C (Scheme 1c).13,14 NMR spectroscopic studies in support of adducts A and/or C in solution have been reported.15
In the case of dialkyl substitution, the treatment of bromine adducts with Lewis acidic metal halides (MXn) was reported to form bromosulphonium [BrMXn] salts B′ by irreversible bromide ion complexation.13b,14c,16 These salts have found widespread application in organic synthesis as, for example, bromonium ion (Br+) equivalents and/or oxidants.16,17 Dibromosulphuranes C have been proposed as intermediates in several transformations. For instance, in his seminal work on organosulphuranes, Martin employed dibromosulphuranes as intermediates en route to alkoxysulphuranes,18 such as the versatile Martin sulphurane reagent.19 These reactions proceed via alkoxide displacement of the apical bromide ligands. In a related transformation, sulphuranes C were converted into the corresponding sulphoxides upon treatment with water and a base (Scheme 1c).20 However, that dibromosulphurane intermediates could reductively collapse to form concurrently alkyl bromides and organosulphenyl bromides has to the best of our knowledge, not been reported. Herein, we show that, with the right choice of the substituents on the sulphur, such a transformation is indeed feasible, and in doing so uncover that aryl sulphides can be considered a valuable, heretofore unexplored leaving group for highly stereoselective, non-neighbouring group assisted, nucleophilic bromination.
Results and discussion
As part of our studies on desulphurative chlorination with PhICl2, we examined the ability of other electrophilic chlorinating reagents, and specifically that of elemental chlorine, to promote chlorination of β-sulphido esters. In a preliminary experiment, we treated β-sulphido ester 1a with Cl2 gas and observed a rapid consumption of the starting material within 3 minutes. 1H NMR analysis of the crude material showed the formation of β-chloro ester 2a as the major product, accompanied by dehydrochlorination-derived methyl cinnamate (3a) in a ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 (Scheme 2). In spite of significant alkene side-product formation, this experiment clearly demonstrated that molecular chlorine could promote desulphurative chlorination. This reactivity formed the basis for our efforts to examine elemental bromine in the desulphurative bromination of β-sulphido esters.
:30 (Scheme 2). In spite of significant alkene side-product formation, this experiment clearly demonstrated that molecular chlorine could promote desulphurative chlorination. This reactivity formed the basis for our efforts to examine elemental bromine in the desulphurative bromination of β-sulphido esters.
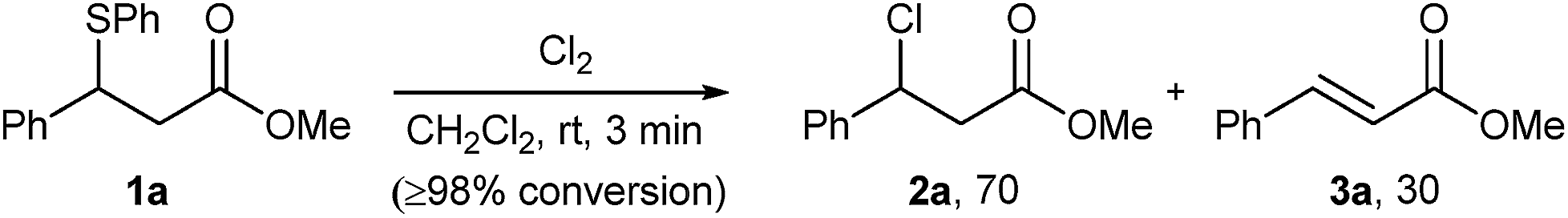 | ||
| Scheme 2 Reaction of β-sulphido ester 1a with Cl2. | ||
In an initial experiment, adding 1.0 equivalent of neat Br2 to our test substrate β-sulphido ester 1a in dry dichloromethane and monitoring the progress of the reaction by 1H NMR spectroscopy showed, within 15 minutes, complete conversion to β-bromo ester 4a, accompanied by small amounts of dehydrobromination-derived methyl cinnamate (3a) (Table 1, entry 1). This experiment clearly indicated that Br2 could not only promote bromination of sulphide 1a, but also, gratifyingly, with very little alkene side-product formation. Moreover, this surprising outcome was even more profound when considering the large difference in oxidising power between Cl2 and Br2.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Timec (min) | Ratiod4a:3a |
| a Conditions: 1a (0.5 mmol), Br2 (0.5 mmol) as 1.0 M solution in reaction solvent, and dry solvent (3.0 mL, 0.17 M); all reactions proceeded to complete conversion (≥98%). b Styrene (0.65 mmol, 1.3 equiv.) was added to quench the reaction. c Reaction progress was monitored by 1H NMR spectroscopy using a stock solution of styrene in CDCl3 (0.03 M). d Determined by 1H NMR spectroscopy of the crude material. e Neat Br2 (0.5 mmol) used. f Isolated yield after SiO2 chromatography. g Non-purified CH2Cl2. CM = complex mixture. | ||||
| 1e | CH2Cl2 | rt | 15 | 98:2 |
| 2 | CH2Cl2 | rt | 25 | 100:0 (95)f |
| 3 | CH2Cl2g | rt | 15 | 98:2 |
| 4 | Toluene | rt | 30 | 100:0 |
| 5 | DCE | rt | 30 | 98:2 |
| 6 | THF | rt | 90 | CM |
| 7 | MeCN | rt | 5 | 99:1 (72)f |
| 8 | CH2Cl2 | 0 | 60 | 100:0 |
| 9 | CH2Cl2 | −20 | 180 | 100:0 |
| 10 | CH2Cl2 | −40 | 900 | 100:0 |
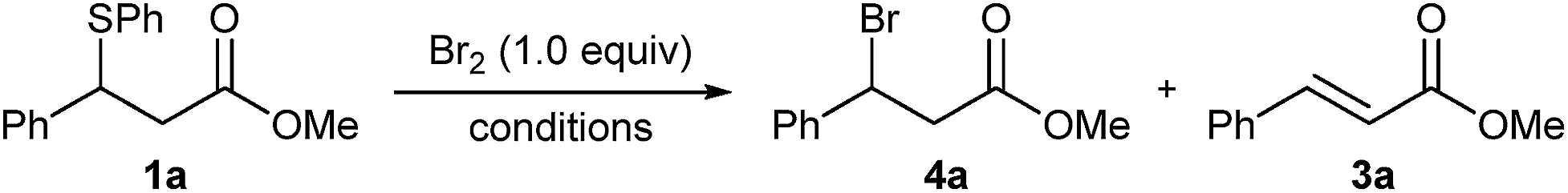
To simplify the procedure and ensure accurate addition of Br2, we repeated this reaction with a 1.0 M solution of Br2 in dry DCM, which gave complete conversion to the brominated product in 25 minutes, and significantly, without the formation of alkene 3a (entry 2). We were delighted to isolate β-bromo ester 4a in an excellent yield of 95% after SiO2 chromatography. Performing the same reaction in non-purified DCM gave complete conversion in a shorter reaction time of 15 min but with small amounts of alkene 3a (entry 3). The reaction proceeded equally well in toluene or dichloroethane (DCE), albeit with slightly longer reaction times (entries 4 and 5), whereas THF as solvent gave a complex mixture (entry 6). A marked increase in the reaction rate was observed in MeCN; however, workup and purification gave bromo ester 4a in a lower yield of 72% (entry 7).
Conducting the reaction in DCM at lower temperatures resulted in longer reaction times but gave clean conversions to bromide 4a, with the reaction at −40 °C requiring 15 hours for completion (entries 8–10). This is in stark contrast to desulphurative chlorination with PhICl2, which showed an increase in alkene side-product formation upon lowering the reaction temperature to 0 °C.11 Moreover, in contrast with the limited solubility of PhICl2 in chlorinated solvents at lower temperatures,21 reactions with Br2 remained homogeneous throughout. Crucially, in all of the above reactions only small amounts or no dehydrobrominated alkene 3a was observed,22 which is a testament to the exceptionally mild, base- and acid-free reaction conditions.
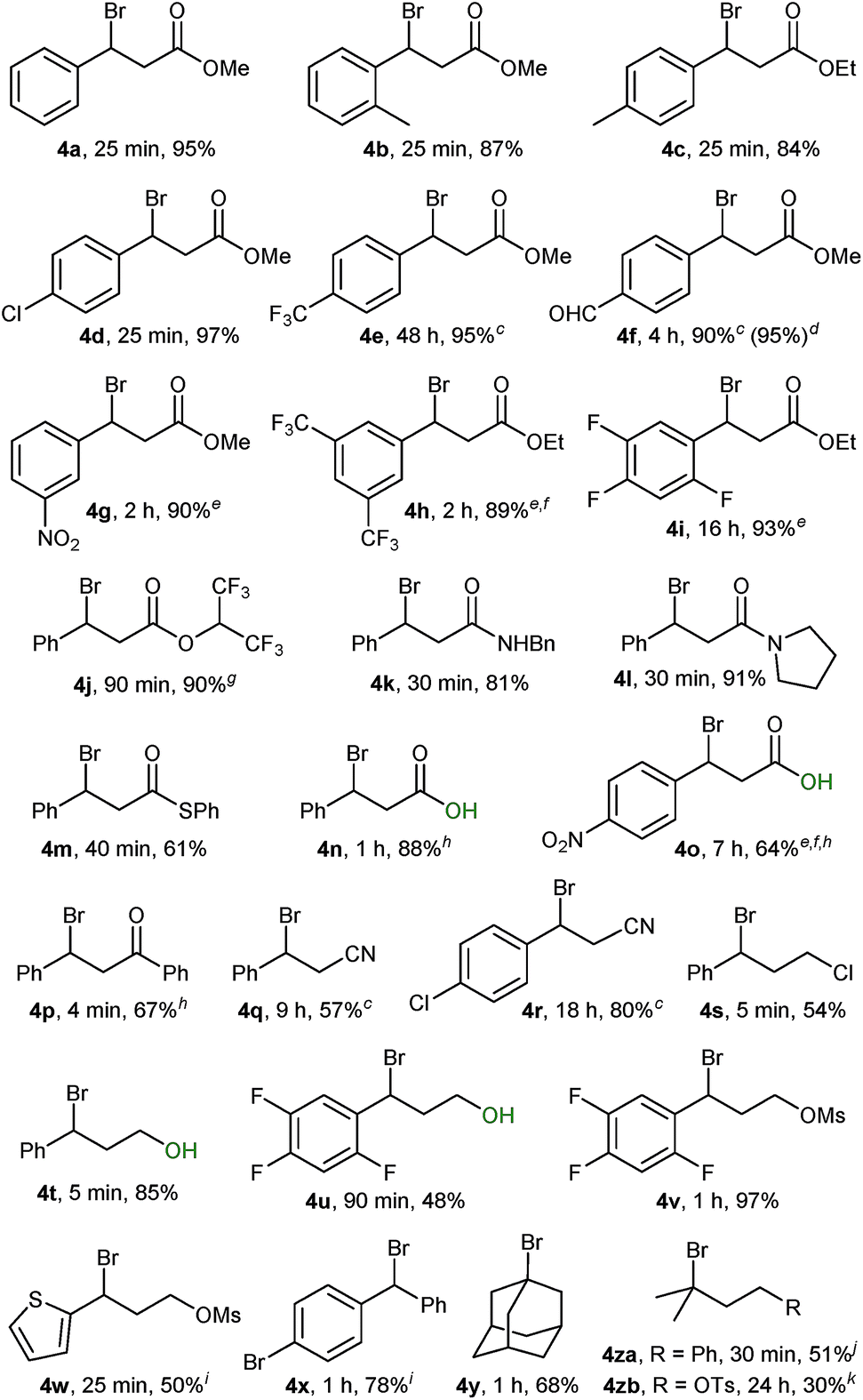
Using conditions from entry 2 in Table 1 we proceeded to examine the scope of our bromination with various β-sulphido carbonyl compounds, which generally gave good to excellent yields of the corresponding bromide products (Table 2, 4a–4p). Reaction times varied with substitution on the aryl ring, with aryl groups having deactivating substituents requiring longer reaction times (4 hours for 1f). Bromination of slow-reacting sulphides could be accelerated by running the reaction at double the concentration in DCM (1e), or in DCE as solvent at 50 °C (1g and 1i). Both heating at 50 °C and excess of Br2 were needed for the bromination of sulphides 1h and 1o.
|
|
|---|
| a Unless otherwise stated, reactions were performed on a 0.5 mmol scale at 0.17 M (3 mL) using 0.5 mmol of Br2 as 1.0 M solution in CH2Cl2; reaction progress was monitored by 1H NMR using a stock solution of styrene in CDCl3 (0.03 M). b Isolated yield after SiO2 chromatography. c Run at 0.33 M (1.5 mL). d 5.0 mmol scale. e Reaction performed in DCE at 50 °C. f Run with 1.0 mmol (2.0 equiv.) of Br2. g Contains 8% alkene. h Isolated yield after trituration or recrystallisation. i Yield of the corresponding trifluoroethyl ether derivative. j 1H NMR yield using CH2Br2 as the internal standard. k Isolated yield contains 26% 3,4-dibromide side-product. |
|
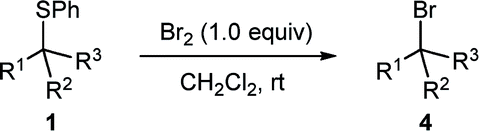
Remarkably, desulphurative bromination also occurred with substrates having protic functionalities such as β-sulphido acids 1n and 1o and sulphido alcohols 1t and 1u, a transformation that would be incompatible with the use of the aforementioned phosphorus(V) or phosphorus(III) based deoxybrominating reagents. The chemoselectivity for sulphido alcohols 1t and 1u is even more striking, when considering that the productive intermediate is formally a bromosulphonium ion analogous to the one generated in Swern oxidation. These brominations proceeded to complete conversion, with bromo alcohol 4t obtained in a high 85% yield. Bromo alcohol 4u was isolated in a moderate yield after purification; nevertheless, it could be generated in situ and taken further in a follow-up reaction. For instance, a one-pot bromination/mesylation sequence of sulphido alcohol 1u gave bromo mesylate 4v in 45% yield (97% yield from sulphide 1v).
Performing the bromination on easily accessible β-sulphido nitriles 1q and 1r gave rise to the corresponding β-bromonitriles in fair to good yields. Our method provides a direct and simple route to these novel and potentially versatile compounds,23 especially when considering that the only direct literature-known method relies on the mostly low-yielding halodehydration of the corresponding alcohol precursors.24
Scale-up of the above reactions proceeded without problems. For instance, a ten-fold scale-up, at 5 mmol, of the bromination of sulphide 1f gave bromide 4f in an excellent 95% yield, thus underlining the practicality of the herein reported bromination protocol.
Bromination of sulphides containing electron-rich heteroarenes such as sulphido thiophene 1w proceeded to complete conversion; however, attempts to purify the corresponding bromide 4w by chromatography resulted in decomposition. Ultimately, 2,2,2-trifluoroethanol solvolysis afforded the 2,2,2-trifluoroethoxy derivative 4wa in 50% yield (see the ESI†). Likewise, bromide 4x was sensitive towards purification by chromatography and was isolated as the 2,2,2-trifluoroethoxy derivative 4xa in 78% yield.
Initial experiments with adamantyl sulphide 1y indicated that tertiary alkyl sulphides were suitable substrates. Bromination of sulphide 1y proceeded with clean complete conversion as indicated by the 1H NMR spectrum of the crude material. However, the moderate yield of 68% for bromide 4y (as well as for 4s) was due to the chromatographic separation from (PhS)2, which possessed similar polarity. Bromination of tertiary dimethylalkyl-derived sulphides proceeded to complete conversion, but was accompanied by elimination side-products. Thus, sulphide 1za gave within 30 min the corresponding bromide in 51% 1H NMR yield together with minor elimination side-products, whereas bromo tosylate 4zb could be isolated in 30% yield as an inseparable 3:1 mixture together with its corresponding Br2 alkene addition side-product (see the ESI†).
Optically active bromides
There are relatively few methods to access optically active, racemisation-sensitive benzylic bromides. By far the most widely applied methods rely on nucleophilic SN2 bromination of optically active benzylic alcohols; however, these reactions require accurate monitoring of reaction conditions and usually yield partially racemised bromide products. Potentially very useful, but rare, asymmetric protocols for obtaining benzylic bromides are emerging. These include the recently reported Rh-catalysed asymmetric Kharasch addition to styrenes,25 and the Cu-catalysed formal asymmetric hydrobromination of styrenes.26 However, these methods require the use of precious metal-chiral phosphine catalysis and display limited substrate scope.Given the exceptionally mild reaction conditions to access elimination-sensitive bromides, we proceeded to examine the suitability of our method for the synthesis of highly versatile optically active benzylic β-bromo esters from enantiomerically enriched benzylic β-sulphido esters. Sulphide substrates were prepared according to Wang's asymmetric sulpha-Michael addition of thiophenol to hexafluoroisopropyl cinnamates.27 As the bromination of β-sulphido hexafluoroisopropyl esters 1j and 1ea gave, after purification by chromatography, bromides containing 8% and 25% alkene, respectively, we decided to investigate the corresponding β-sulphido methyl esters. Thus, acid-catalysed methanolysis was achieved without erosion of the enantiomeric purity for sulphides (S)-1j, (S)-1da and (S)-1ea (Scheme 3). However, methanolysis of sulphido ester (S)-1ba having 99% ee gave, under the studied conditions, partially racemised (S)-1b in 78% ee.
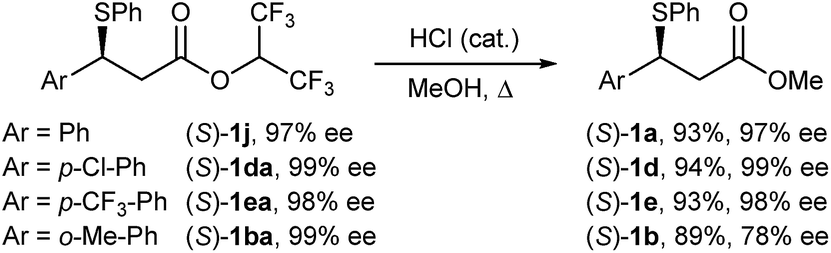 | ||
| Scheme 3 Synthesis of enantiomerically enriched β-sulphido methyl esters. | ||
Subjecting sulphide (S)-1a to the optimised bromination at room temperature (Table 1, entry 2) gave inverted bromide (R)-4a in 83% ee and 86% es (Table 3, entry 1), which was subsequently confirmed to have an R absolute configuration (vide infra). We were delighted to find that lowering the reaction temperature had a significant effect on the enantioselectivity of the reaction (Table 3, entries 2–4), with the bromination ran at −40 °C affording, after 15 hours, bromide (R)-4a in an excellent 93% ee and 96% es. Similarly, bromination of sulphide (S)-1d at −40 °C gave bromide (R)-4d in 93% ee and 94% es (entry 5).28 The slow reacting sulphide (S)-1e gave at room temperature bromide (R)-4e in high 86% ee and 88% es (entry 6). Lowering the reaction temperature to 0 °C gave bromide (R)-4e with a slightly improved enantiomeric excess of 87%, but at the expense of a significant drop in the reaction rate (entry 7). Finally, running sulphide (S)-1b having 78% ee at −40 °C gave bromide (R)-4b in 66% ee, which still corresponded to 85% es, showing that the reaction tolerated sterically encumbered ortho-substituted aryl groups (entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Ent. | (S)-1 | Temp. (°C) | Timec | (R)-4 yieldd (%) | eee (%) | es (%) |
| a Conditions: (S)-1 (0.25 mmol), Br2 (0.25 mmol) as 1.0 M solution in CH2Cl2, and dry CH2Cl2 (1.5 mL, 0.17 M). b Styrene (0.3 mmol, 1.3 equiv.) was added to quench the reaction. c Reaction progress was monitored by 1H NMR spectroscopy using a stock solution of styrene in CDCl3 (0.03 M). d Isolated yield after SiO2 chromatography. e ee values were determined by HPLC analysis on the chiral stationary phase. | ||||||
| 1 | (S)-1a | rt | 25 min | 82 | 83 | 86 |
| 2 | (S)-1a | 0 | 1 h | 94 | 87 | 90 |
| 3 | (S)-1a | −20 | 3 h | 87 | 92 | 95 |
| 4 | (S)-1a | −40 | 15 h | 89 | 93 | 96 |
| 5 | (S)-1d | −40 | 24 h | 97 | 93 | 94 |
| 6 | (S)-1e | rt | 48 h | 95 | 86 | 88 |
| 7 | (S)-1e | 0 | 48 h | 36 | 87 | 89 |
| 8 | (S)-1b | −40 | 15 h | 99 | 66 | 85 |
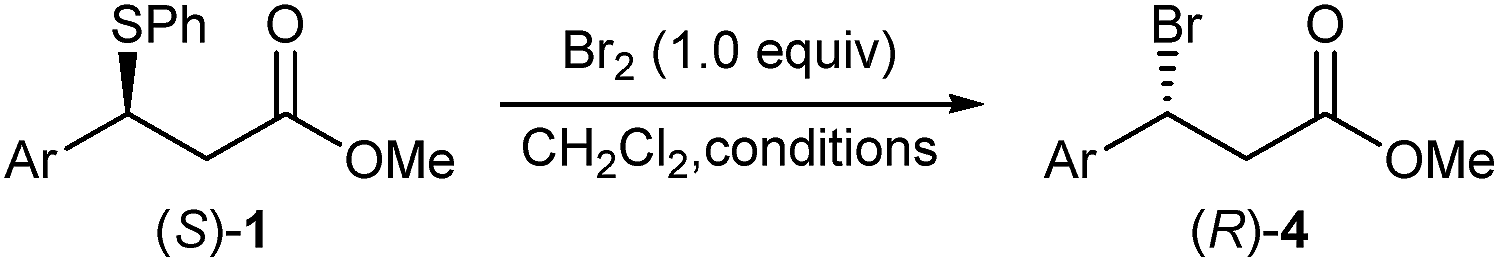
Optically active benzylic bromides have been reported to be prone to racemisation.1c,e,10 For instance, a sample of (R)-1-(bromoethyl)benzene having 88% ee kept at 0 °C was shown to fully racemise within 8 hours.1c We therefore monitored the configurational stability of a sample of β-bromo ester (R)-4a having 93% ee over a three-month period at −20 °C and room temperature, and uncovered that when stored at −20 °C, no racemisation was detectable. In contrast, the sample stored at room temperature showed partial racemisation with a drop in enantiomeric excess to 78% after the same period (see the ESI† for further information). This is a very significant finding as it shows that optically active benzylic β-bromo esters can be conveniently prepared using our method and stored for prolonged periods at −20 °C (for at least three months) without measurable racemisation.
In order to demonstrate the versatility of our enantiomerically enriched β-bromo esters and to corroborate that the desulphurative bromination occurred with the proposed inversion of configuration, we converted bromo ester (R)-4a in a reduction/azidation sequence into azido alcohol (S)-5 (Scheme 4). After examining several reducing reagents, we discovered that DIBAL readily reduced the ester functionality in the presence of a benzylic bromide to afford bromo alcohol (R)-4t in an excellent 93% yield, but with a partially eroded optical purity of 83% ee (Scheme 4). We believe that this is due to the propensity of optically active bromo alcohol (R)-4t towards racemisation.29 Subsequent treatment of the crude product with sodium azide in DMF gave azido alcohol (S)-5 in 86% yield and 83% ee.
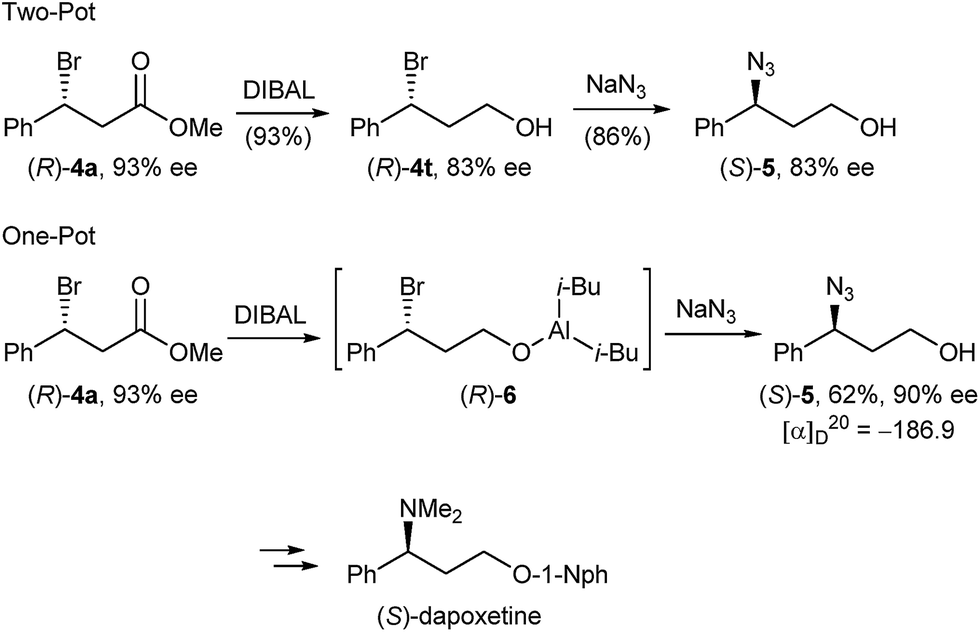 | ||
| Scheme 4 Preparation of enantiomerically enriched azido alcohol (S)-5. | ||
Given that bromo alcohol (R)-4t is racemisation sensitive, we devised an alternative one-pot protocol by submitting the in situ generated bromo aluminium alcoholate (R)-6 to sodium azide substitution and were delighted to obtain azido alcohol (S)-5 in 62% yield and 90% ee. Comparing the sign of the specific optical rotation with that of the literature-known azido alcohol (R)-530 confirmed that bromide 4a was formed with an R absolute configuration and SN2 inversion from sulphide (S)-1a. Azido alcohol (S)-5 can be considered an advanced intermediate en route to (S)-dapoxetine,31 thus showcasing that the herein described optically active β-bromo esters can serve as valuable intermediates in the transition metal free synthesis of a host of APIs having a benzylic chiral centre.
NMR study and mechanism
As outlined in the Introduction, there is uncertainty surrounding the nature of the interaction of Br2 with sulphides, with very few studies reported in solution. In order to gain insight into the mechanism operating in the presented desulphurative bromination, we conducted a series of NMR experiments. They were undertaken in light of the fact that the reaction of both PhICl232 and Cl215b,33 with sulphides leads to the oxidation of the sulphur(II) centre to sulphur(IV) forming dichlorosulphuranes.
In an initial experiment ran at an identical concentration to the actual reaction, we treated a 0.17 M solution of sulphide 1a in CD2Cl2 pre-cooled to −20 °C with 1.0 equivalent of a 1.0 M solution of Br2 in CD2Cl2 and monitored the progress of the reaction by 1H NMR spectroscopy at −20 °C (Fig. 1). After one minute of Br2 addition, and focusing on the benzylic proton, the recorded spectrum showed the total absence of the benzylic signal of sulphide 1a at 4.67 ppm with two new species appearing as double doublets at 5.43 ppm and 4.84 ppm in a ratio of 84:16. The major species was identified as bromo ester 4a, with the minor unknown species converting to bromide 4a upon further monitoring (Fig. 1). It was apparent that the new unknown species – postulated as being a sulphide bromine adduct intermediate 1a·Br2 (vide infra) – converted rapidly to bromo ester 4a, even at −20 °C.
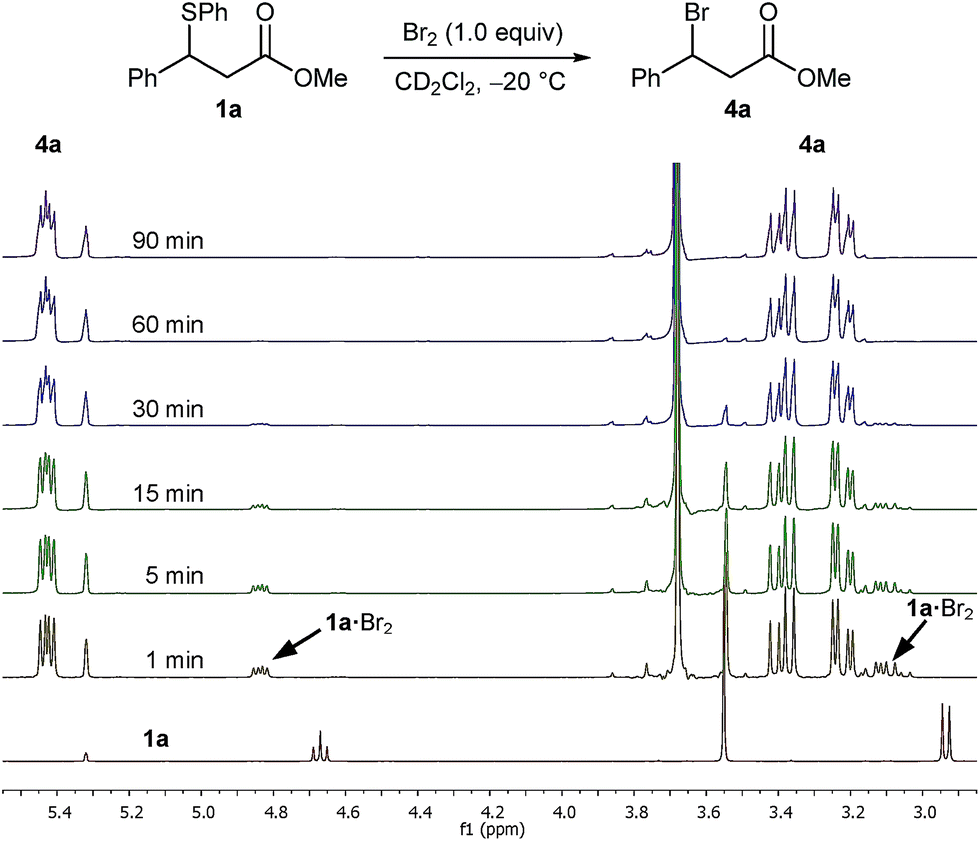 | ||
| Fig. 1 1H NMR spectra (400 MHz) of the reaction of Br2 (1.0 M) with sulphide 1a (0.17 M) in CD2Cl2 at −20 °C over a period of 90 min. | ||
Given the high rate of bromination for 1a, we turned our attention to the slow-reacting sulphide 1e (Fig. 2). Thus, adding a 1.0 M solution of Br2 in CD2Cl2 to a pre-cooled 0.17 M solution of sulphide 1e and recording a 1H NMR spectrum at −20 °C showed, after 2 min, the complete disappearance of 1e and its conversion to a single new species, identified by the complete disappearance of the benzylic proton signal at 4.69 ppm and the appearance of a new double doublet at 4.82 ppm (Fig. 2b). Other significant shifts included downfield shifts of the diastereotopic Hβ hydrogen atoms at 2.94 and 2.99 ppm to 3.05 and 3.14 ppm, respectively, and a downfield shift of the signal of hydrogens ortho to the sulphur by 0.14 ppm (not shown).
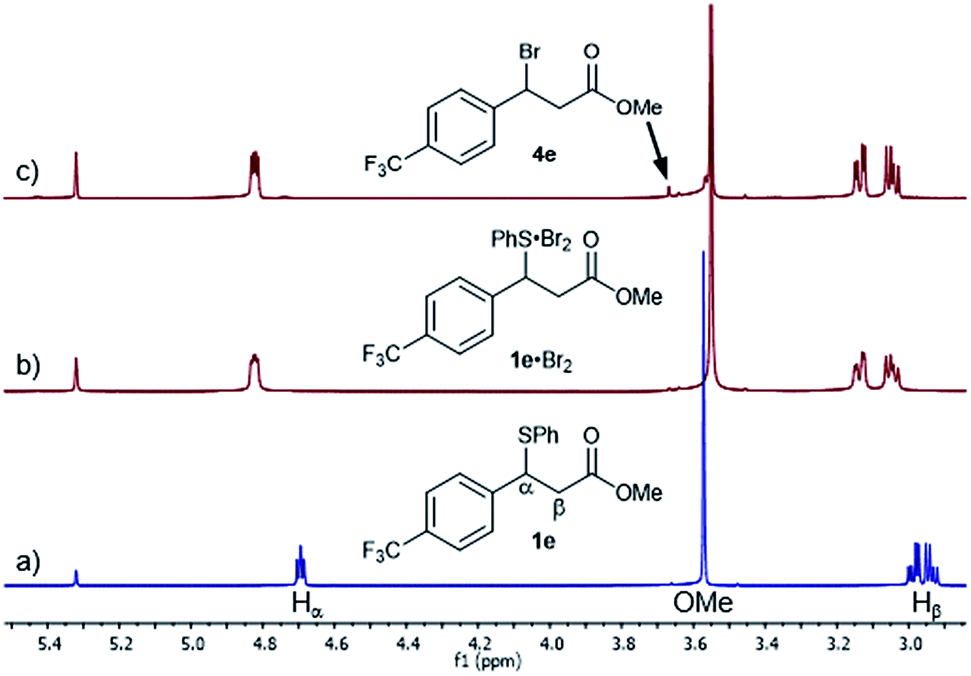 | ||
| Fig. 2 1H NMR spectra (800 MHz) of the Br2 (1.0 M) addition to sulphide 1e (0.17 M) in CD2Cl2 at −20 °C: (a) sulphide 1e at −20 °C before Br2 addition, (b) 2 minutes after Br2 addition and formation of the 1e·Br2 adduct and (c) 1 hour after Br2 addition showing the appearance of bromide 4e. | ||
Gratifyingly, the new species was sufficiently stable at −20 °C to allow its full characterisation by 1D and 2D NMR spectroscopy. The 13C NMR spectrum showed significant shifts of signals compared to 1e (Fig. 3). These included large shifts for carbons flanking the sulphur atom corresponding to an upfield shift of the resonance of the Ci of 1e to 129.0 ppm (Δδ = −3.7 ppm) and a downfield shift of the benzylic carbon Cα to 52.9 ppm (Δδ = +4.6 ppm), as well as an upfield shift for Ci′ to 141.2 ppm (Δδ = −3.7 ppm) (Fig. 3b). In addition, notable shifts were observed for Cp of 1e to 130.6 ppm (Δδ = +2.3 ppm) and Cβ to 38.4 ppm (Δδ = −1.6 ppm).
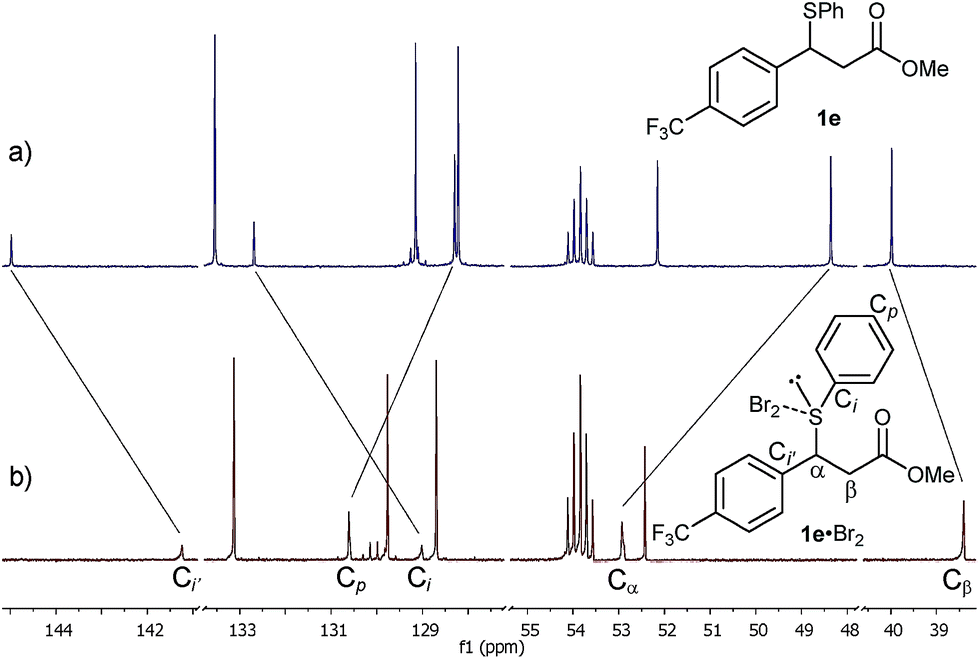 | ||
| Fig. 3 13C NMR spectra (201 MHz) of the Br2 (1.0 M) addition to sulphide 1e (0.17 M) in CD2Cl2 at −20 °C: (a) sulphide 1e at −20 °C before Br2 addition and (b) after Br2 addition, displaying significant shifts of the formed 1e·Br2 adduct. | ||
All of the above 1H and 13C NMR Δδs are less pronounced at 20 °C.15a Hence, taking into account the NMR spectroscopic criteria proposed by Nakanishi to distinguish between MCs and TBs of R1R2S·X2 adducts in solution,15b the observed shifts – most notably the upfield shift of Ci of the SPh group – are generally consistent in terms of magnitude, direction and temperature behaviour with the presence of a MC structure for the 1e·Br2 adduct.15a These findings provide, to the best of our knowledge, strong evidence for the first observation of a benzylic sulphide Br2 adduct in solution.
After 1 hour, signals assigned to bromide 4e were visible, with further conversion to 4e being very slow at −20 °C.34 We thus raised the temperature of the experiment to 20 °C and continued monitoring the progress of the reaction for a further 14.5 hours, and observed that 1e·Br2 progressively converted to bromide 4e (Fig. 4). Repeating this experiment at 20 °C for 60 h showed conversion of 1e·Br2 to 4e slowing markedly down to 87% after approximately 48 hours (see the ESI† for spectra). It is important to note that the actual bromination of 1e was carried out at double the concentration (0.33 M) requiring 48 h for complete conversion to 4e.
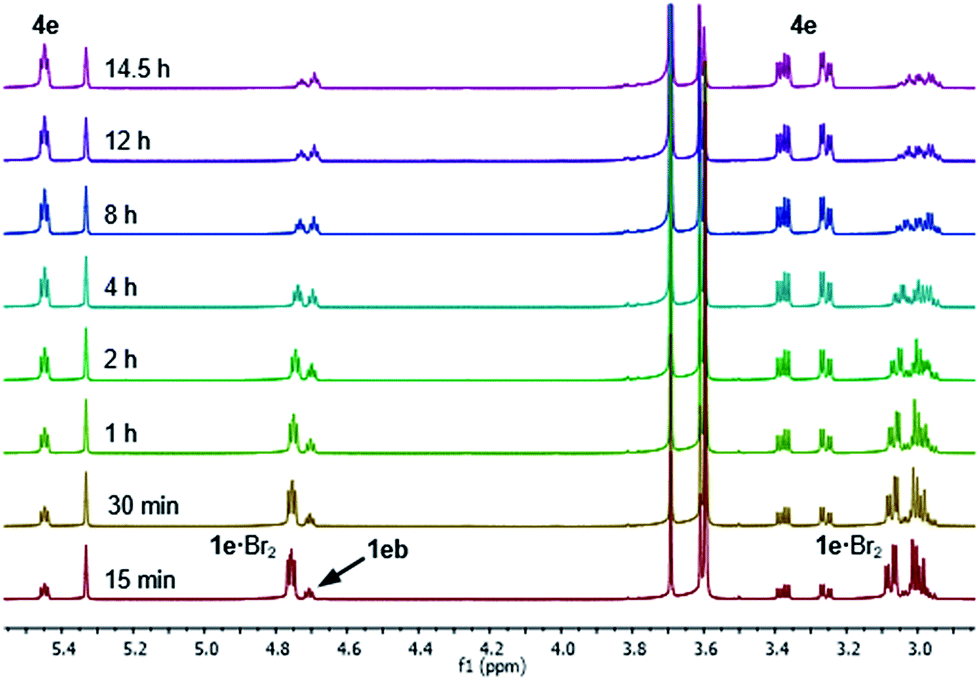 | ||
| Fig. 4 1H NMR spectra (800 MHz) of the Br2 (1.0 M) addition to sulphide 1e (0.17 M) in CD2Cl2 after raising the temperature to 20 °C, showing the progressive conversion of the 1e·Br2 adduct into bromide 4e and the presence of side product sulphide 1eb. | ||
The reaction of 1e also showed the formation of a side product observed with experiments at −20 °C and 20 °C, which started to form after the appearance of bromide 4e. This side product was proposed as being sulphide 1eb derived from the para bromination of the SPh group in 1e (Fig. 4), which was confirmed after comparison with an authentic sample. In an NMR experiment conducted at 20 °C, sulphide 1eb converted cleanly to bromide 4e upon addition of 1.0 equivalent of Br2 (see the ESI† for spectra). However, the bromination was slower than that with sulphide 1e, stopping at 82% conversion after 60 h, with complete conversion reached only after a further addition of 0.2 equivalents of Br2.
A preliminary mechanistic picture is emerging from the above NMR experiments (Scheme 5). We propose that the addition of Br2 to sulphide (S)-1 results in the formation of sulphur(II) adduct 1·Br2-MC. Based on the facts that (i) PhSBr is formed as a by-product and (ii) 1 equivalent of bromide ions must be generated from Br2 for SN2 C–Br bond formation to occur, we propose that 1·Br2-MC must be in equilibrium with trigonal bipyramidal sulphur(IV) dibromosulphurane intermediate 1·Br2-TB.15 However, due to its high reactivity, it is very likely that 1·Br2-TB is present in very low concentrations. Invertive bromide ion attack on 1·Br2-TB produces bromide (R)-4 with concomitant release of PhSBr and one equivalent of bromide ions. Therefore, only catalytic amounts of bromide ions are required to initiate the reaction. Bromide ions could be derived from the ionisation of 1·Br2-TB to form, in equilibrium, trigonal pyramidal bromosulphonium bromide [1-Br]Br. The fact that the reaction is significantly faster in coordinating MeCN (Table 1, entry 7) supports such a hypothesis.35
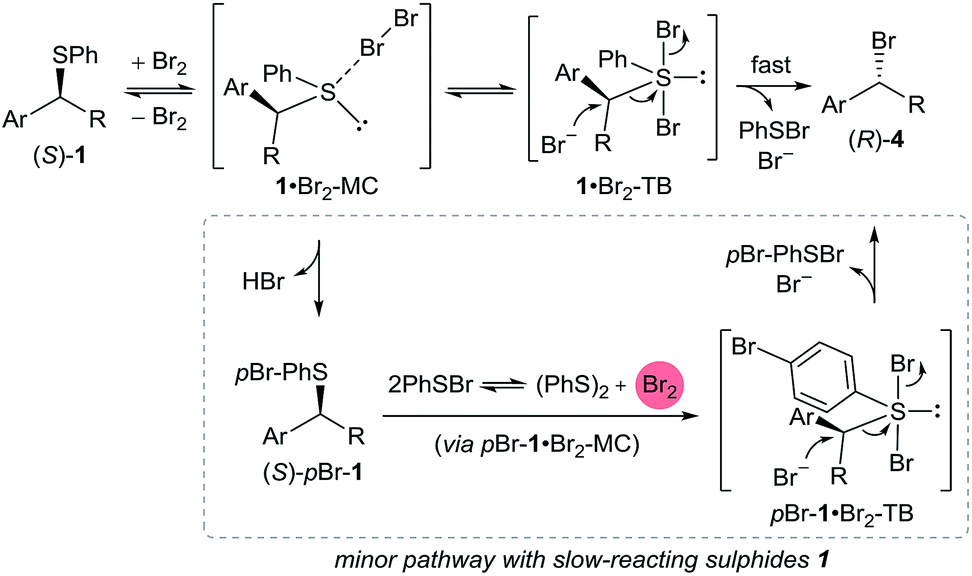 | ||
| Scheme 5 Proposed mechanism taking into account stereochemical and NMR evidence, as well as side-product formation. | ||
As seen with sulphide 1e, slow-reacting sulphides can form the para-brominated side product (S)-pBr-1, presumably via reaction with generated 1·Br2-TB or possibly PhSBr3 formed in situ (in equilibrium) from the generated PhSBr and Br2 from 1·Br2-MC.36 The exclusive para bromination of activated aromatics with bromodimethylsulphonium bromide has been reported.17b Under optimised reaction conditions, the reductive coupling of two PhSBr molecules generates one molecule of Br2 that could oxidatively brominate (S)-pBr-1 in an analogous manner to (S)-1 (Scheme 5).37
Finally, a pathway involving n → σ* type activation of the C–S bond in 1·Br2-MC towards bromide ion attack cannot be ruled out at present,38 and will be the subject of a detailed mechanistic analysis in the future.
Conclusions
We have disclosed a novel nucleophilic bromination reaction that employs easily accessible alkyl aryl sulphides as starting materials and basic elemental Br2 as an oxidative brominating agent. Reaction conditions are exceptionally mild, allowing the isolation of otherwise difficult to access and highly versatile benzylic β-bromo esters and nitriles in generally good to excellent yields. The reaction tolerates various functionalities and, remarkably, proceeds in the presence of protic functionalities such as alkyl acids and alcohols; a transformation incompatible with the vast majority of deoxybromination procedures. Optically active benzylic β-sulphido esters could be converted into the corresponding inverted β-bromo esters with high stereoselectivities. These bromides are configurationally stable at −20 °C, which should pave the way for their exploitation as highly useful chiral synthons in organic and medicinal chemistry. Their utility was demonstrated by the preparation of γ-azido alcohol (S)-5, an advanced intermediate en route to dapoxetine, in 90% ee. The developed one-pot sequence from bromo ester (R)-4a, consisting of a DIBAL ester reduction in the presence of a benzylic bromide and subsequent invertive nucleophilic azidation, proceeded with high stereochemical fidelity. Significantly, the required stereochemistry was introduced into sulphide precursor (S)-1avia an asymmetric sulpha-Michael reaction, thus bypassing the dependency on optically active benzylic alcohols.Low temperature NMR spectroscopic studies pointed to an initial MC adduct formation between the starting sulphide and Br2 en route to the bromide product. This was observed for adducts 1a·Br2 and 1e·Br2, with the latter, derived from slow-reacting sulphide 1e, being sufficiently stable at −20 °C to allow its full characterisation. Subsequent stereoinvertive C–Br bond formation is postulated to occur from the highly reactive isomeric dibromosulphurane TB adduct, proposed to be present in equilibrium with the MC adduct.
Given the very recent progress made in the synthesis of enantiomerically enriched benzylic aryl sulphides,39 we anticipate that the nucleophilic desulphurative bromination reported herein will find wide utility in the synthesis of optically active benzylic bromides relevant to drug discovery, agrochemicals, and materials.
Conceptually, the in situ oxidative activation of sulphides towards SN2 nucleophilic substitution should provide a platform for the development of other C–X and C–C bond forming reactions, and could prove to be a general, practical and viable alternative to oxygen based leaving groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by IRC (grants GOIPG/2015/3942 and EPSPG/2016/185 to D. C. and C. F.), the H2020 Programme (MSCA-RISE Halo 734361) and the Enterprise-Ireland CF Fund (CF20144034). We thank Dr Gary Hessman (TCD) for HRMS analysis.Notes and references
- For example, see: (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 137, 11562 Search PubMed; (b) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624 CrossRef CAS PubMed; (c) C. Li, Y. Zhang, Q. Sun, T. Gu, H. Peng and W. Tang, J. Am. Chem. Soc., 2016, 138, 10774 CrossRef CAS PubMed; (d) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562 CrossRef CAS PubMed; (e) A. López-Pérez, J. Adrio and J. C. Carretero, Org. Lett., 2009, 11, 5514 CrossRef PubMed and references therein; (f) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490 CrossRef CAS PubMed; (g) X. Lu, Y. Wang, B. Zhang, J.-J. Pi, X.-X. Wang, T.-J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632 CrossRef CAS PubMed.
- For recent examples, see: (a) A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693 CrossRef CAS PubMed; (b) C. K. Chu, Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 6404 CrossRef CAS PubMed; (c) D. M. Peacock, C. B. Roos and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 647 CrossRef CAS PubMed.
- (a) W.-J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396 CrossRef CAS PubMed; (b) G. W. Gribble, Mar. Drugs, 2015, 13, 4044 CrossRef CAS PubMed.
- For a recent method via C–H bromination, see: S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100 RSC and references therein.
- R. Bohlmann, in Comprehensive Organic Transformations, ed. R. C. Larock, Wiley-VCH, New York, 2nd edn, 1999, pp. 689–702 Search PubMed.
- (a) C. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2011, 3, 140 CrossRef CAS PubMed; (b) T. V. Nguyen and A. Bekensir, Org. Lett., 2014, 16, 1720 CrossRef CAS PubMed; (c) J. P. Moerdyk and C. W. Bielawski, Chem.–Eur. J., 2014, 20, 13487 CrossRef CAS PubMed; (d) L. De Luca, G. Giacomelli and A. Porcheddu, Org. Lett., 2002, 4, 553 CrossRef CAS PubMed.
- (a) J. Chen, J.-H. Lin and J.-C. Xiao, Org. Lett., 2018, 20, 3061 CrossRef CAS PubMed and references therein; (b) J.-R. Dormoy, B. Castro and T. P. Bobinski, Triphenylphosphine Dibromide, in e-EROS, 2018, DOI:10.1002/047084289X.rt370.pub2; (c) B. P. Mundy and C. A. Stewart, Phosphorus(III) Bromide, in e-EROS, 2008, DOI:10.1002/047084289x.rp154.pub2.
- (a) R. M. Denton, J. An, B. Adeniran, A. J. Blake, W. Lewis and A. M. Poulton, J. Org. Chem., 2011, 76, 6749 CrossRef CAS PubMed; (b) H. A. van Kalkeren, S. H. Leenders, C. R. Hommersom, F. P. Rutjes and F. L. van Delft, Chem.–Eur. J., 2011, 17, 11290 CrossRef CAS PubMed; (c) H. A. van Kalkeren, F. P. Rutjes and F. L. van Delft, Pure Appl. Chem., 2013, 86, 817 Search PubMed.
- (a) J. An, R. M. Denton, T. H. Lambert and E. D. Nacsa, Org. Biomol. Chem., 2014, 12, 2993 RSC; (b) C. M. Vanos and T. H. Lambert, Angew. Chem., Int. Ed., 2011, 50, 12222 CrossRef CAS PubMed; (c) P. H. Huy, S. Motsch and S. M. Kappler, Angew. Chem., Int. Ed., 2016, 55, 10145 CrossRef CAS PubMed; (d) T. Stach, J. Dräger and P. H. Huy, Org. Lett., 2018, 20, 2980 CrossRef CAS PubMed.
- (a) R. O. Hutchins, D. Masilamani and C. A. Maryanoff, J. Org. Chem., 1976, 41, 1071 CrossRef CAS; (b) R. Stein, R. D. Dawe and J. R. Sweet, Can. J. Chem., 1985, 63, 3442 CrossRef; (c) Y. Chen, W. L. Tang, J. Mou and Z. Li, Angew. Chem., Int. Ed., 2010, 49, 5278 CrossRef CAS PubMed.
- D. Canestrari, S. Lancianesi, E. Badiolaý, C. Strinna, H. Ibrahim and M. F. A. Adamo, Org. Lett., 2017, 19, 918 CrossRef CAS PubMed.
- (a) T. Zincke and W. Frohneberg, Ber. Dtsch. Chem. Ges., 1910, 43, 837 CrossRef CAS; (b) H. Meerwein, K. F. Zenner and R. Gipp, Justus Liebigs Ann. Chem., 1965, 688, 67 CrossRef CAS.
- (a) H. F. Askew, P. N. Gates and A. S. Muir, J. Raman Spectrosc., 1991, 22, 265 CrossRef CAS; (b) H. Böhme and E. Boll, Z. Anorg. Allg. Chem., 1957, 290, 17 CrossRef.
- (a) G. Allegra, G. E. Wilson, E. Benedetti, C. Pedone and R. Albert, J. Am. Chem. Soc., 1970, 92, 4002 CrossRef CAS and references therein; ; (b) H. Bock, Z. Havlas, A. Rauschenbach, C. Näther and M. Kleine, Chem. Commun., 1996, 1529 RSC; (c) B. Regelmann, K. W. Klinkhammer and A. Schmidt, Z. Anorg. Allg. Chem., 1997, 623, 1633 CrossRef CAS.
- (a) W. Nakanishi, S. Hayashi, H. Tukada and H. Iwamura, J. Phys. Org. Chem., 1990, 3, 358 CrossRef CAS; (b) A. Tanioku, T. Nakai, S. Hayashi and W. Nakanishi, Heteroat. Chem., 2011, 22, 446 CrossRef CAS.
- (a) S. A. Snyder and D. S. Treitler, Angew. Chem., Int. Ed., 2009, 48, 7899 CrossRef CAS PubMed; (b) A. P. Brucks, S.-A. Treitler, D. S. Liu and S. A. Snyder, Synthesis, 2013, 45, 1886 CrossRef CAS.
- For examples on the reactivity of bromodimethylsulphonium bromide (BDMB), see: (a) L. H. Choudhury, Synlett, 2006, 1619 CrossRef; (b) G. A. Olah, L. Ohannesian and M. Arvanaghi, Synthesis, 1986, 868 CrossRef CAS; (c) A. T. Khan, M. A. Ali, P. Goswami and L. H. Choudhury, J. Org. Chem., 2006, 71, 8961 CrossRef CAS PubMed; (d) G. A. Olah, M. Arvanaghi and Y. D. Vankar, Synthesis, 1979, 721 CrossRef CAS.
- (a) E. F. Perozzi and J. C. Martin, J. Am. Chem. Soc., 1972, 94, 5519 CrossRef CAS; (b) G. W. Astrologes and J. C. Martin, J. Am. Chem. Soc., 1977, 99, 4400 CrossRef CAS.
- (a) J. C. Martin, R. J. Arhart, J. A. Franz, E. F. Perozzi and L. J. Kaplan, Org. Synth., 1988, 6, 163 Search PubMed; (b) S. S. Pooppanal, Synlett, 2009, 850 CrossRef CAS.
- For example, see: J. Drabowicz, W. Midura and M. Mikołajczyk, Synthesis, 1979, 39 CrossRef CAS.
- PhICl2 dissolved in chlorinated solvents precipitates upon cooling. For example, see: H. J. Lucas, E. R. Kennedy and M. W. Formo, Org. Synth., 1955, 3, 483 Search PubMed.
- In comparison, the crude material from the deoxybromination of methyl 3-hydroxy-3-phenylpropanoate with PPh3/CBr4 under Appel conditions contained 18% alkene 3a.
- For applications of β-chloronitriles, see for example: T. J. Senter, M. C. O'Reilly, K. M. Chong, G. A. Sulikowski and C. W. Lindsley, Tetrahedron Lett., 2015, 56, 1276 CrossRef CAS PubMed.
- For chlorodehydration, see: (a) U. Münch and W. Pfleiderer, Helv. Chim. Acta, 2001, 84, 1504 CrossRef; (b) T. N. Majid, M. C. P. Yeh and P. Knochel, Tetrahedron Lett., 1989, 30, 5069 CrossRef CAS.
- B. Chen, C. Fang, P. Liu and J. M. Ready, Angew. Chem., Int. Ed., 2017, 56, 8780 CrossRef CAS PubMed.
- R. J. Van Hoveln, S. C. Schmid, M. Tretbar, C. T. Buttke and J. M. Schomaker, Chem. Sci., 2014, 5, 4763 RSC.
- X. Fang, J. Li and C.-J. Wang, Org. Lett., 2013, 15, 3448 CrossRef CAS PubMed.
- V. V. Thankur, M. D. Nikalje and A. Sudalai, Tetrahedron: Asymmetry, 2003, 14, 581 CrossRef . This paper reported the synthesis of the corresponding optically active bromo ethyl ester (S)-4da from the alcohol precursor by deoxybromination with PBr3. In our hands, the crude material from this procedure (racemic alcohol) gave a mixture containing ca. 42% 4da (see the ESI†).
- Enantiomerically enriched bromo alcohol 4t (21% ee) racemised completely within a few days: A. Isleyen, C. Tanyeli and O. Dogan, Tetrahedron: Asymmetry, 2006, 17, 1561 CrossRef CAS.
- The (R)-5 enantiomer is literature known: K. Sahasrabudhe, V. Gracias, K. Furness, B. T. Smith, C. E. Katz, D. S. Reddy and J. Aube, J. Am. Chem. Soc., 2003, 125, 7914 CrossRef CAS PubMed.
- K. Venkatesan and K. V. Srinivasan, ARKIVOC, 2008, XVI, 302 Search PubMed , and references therein.
- M. Cartwright and A. A. Woolf, J. Fluorine Chem., 1981, 19, 101 CrossRef CAS.
- (a) N. C. Baenziger, R. E. Buckles, R. J. Maner and T. D. Simpson, J. Am. Chem. Soc., 1969, 91, 5749 CrossRef CAS; (b) G. E. Wilson Jr and M. M. Y. Chang, J. Am. Chem. Soc., 1974, 96, 7533 CrossRef.
- An NMR experiment ran at −20 °C showed 8% conversion to 4e after 10 hours. See the ESI† for further information.
- HBr generated from the para bromination of the SPh group in slow-reacting substrates is unlikely the source of bromide ions, because with sulphide 1e, side-product sulphide 1eb appears after initial formation of 4e. Furthermore, sulphide 1eb undergoes clean bromination to 4e.
- T. G. Back, S. Collins and M. V. Krishna, Can. J. Chem., 1987, 65, 38 CrossRef CAS.
- For reductive self-coupling of PhSX, see for example: (a) Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188 RSC; (b) G. W. Kabalka, M. S. Reddy and M.-L. Yao, Tetrahedron Lett., 2009, 50, 7340 CrossRef CAS; (c) G. A. Olah, S. C. Narang, L. D. Field and R. Karpeles, J. Org. Chem., 1981, 46, 2408 CrossRef CAS.
- W. Nakanishi, S. Hayashi and H. Kihara, J. Org. Chem., 1999, 64, 2630 CrossRef CAS PubMed.
- G. Liu, Z. Han, X.-Q. Dong and X. Zhang, Org. Lett., 2018, 20, 5636 CrossRef CAS PubMed and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic characterization for all new compounds, including details of NMR experiments and racemisation study. See DOI: 10.1039/c9sc03560e |
| This journal is © The Royal Society of Chemistry 2019 |