
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring polymer dispersity and shape of molecular weight distributions: methods and applications
Richard
Whitfield†
 ,
Nghia P.
Truong†
,
Daniel
Messmer
,
Kostas
Parkatzidis
,
Manon
Rolland
and
Athina
Anastasaki
*
,
Nghia P.
Truong†
,
Daniel
Messmer
,
Kostas
Parkatzidis
,
Manon
Rolland
and
Athina
Anastasaki
*
Laboratory of Polymeric Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, Zurich 8093, Switzerland. E-mail: athina.anastasaki@mat.ethz.ch
First published on 28th August 2019
Abstract
The width and shape of molecular weight distributions can significantly affect the properties of polymeric materials and thus are key parameters to control. This mini-review aims to critically summarise recent approaches developed to tailor molecular weight distributions and highlights the strengths and limitations of each technique. Special emphasis will also be given to applications where tuning the molecular weight distribution has been used as a strategy to not only enhance polymer properties but also to increase the fundamental understanding behind complex mechanisms and phenomena.
 Richard Whitfield | Richard Whitfield was born in the town of Grantham in England, and received his Master's degree from Durham University. He completed his PhD under the supervision of Prof. David Haddleton at the University of Warwick, which included 2 months as a visiting scholar at the University of California Santa Barbara. He has subsequently undertaken an Early Career Innovation Fellowship in collaboration with the Institute of Advanced Study at the University of Warwick and as of 2019 is the first postdoctoral researcher in the Laboratory of Polymeric Materials at ETH Zurich. |
 Nghia P. Truong | Nghia Truong is a senior scientist in the Department of Materials at ETH Zurich. He is also an ARC DECRA research fellow and MIPS teaching fellow at Monash University. He completed bachelor and master degrees from Vietnam National University – Ho Chi Minh City and then received his PhD from the University of Queensland, Australia. His current research focuses on engineering smart polymers and nanostructured materials for a better understanding into the interface of nanomaterials with biological systems (at ETH Zurich) and translating the knowledge of the bio-nano interactions to pharmaceutical sciences and biotechnology (at Monash University). |
 Daniel Messmer | Daniel Messmer obtained his PhD in the ETH Zurich Department of Materials in 2018, working on the synthesis and characterization of very high dendritic generation dendronized polymers under the supervision of Prof. A. Dieter Schlüter. In early 2019, he joined the research group of Prof. Athina Anastasaki as a scientific collaborator, working on the chemical modification of initiators and catalysts for (photo-)ATRP. His research interests revolve around the creation of complex structures using the tools of organic chemistry and polymer synthesis. |
 Kostas Parkatzidis | Kostas Parkatzidis studied Material Science and Technology at the University of Crete, before obtaining his MSc degree in Chemistry from the same institution under the supervision of Prof. Maria Vamvakaki. During his Master's, he also joined the group of Dr Maria Farsari at the Institute of Electronic Structure and Laser at the Foundation for Research and Technology in Hellas. In 2019, he started his doctoral studies in the group of Prof. Athina Anastasaki, in the Department of Materials at ETH Zurich, working on controlled radical polymerization and block copolymer self-assembly. |
 Manon Rolland | Manon Rolland studied at CPE Lyon until 2017, where she graduated with a double Master's degree in Chemical Engineering and Innovative Materials. She then undertook a research project at Monash University, in Prof. Tom Davis's group, where she worked on controlled radical polymerization. She subsequently completed her second internship at the University of California, Santa Barbara, in the group of Professor Craig Hawker, which focused on photo-mediated polymerization. After graduation, she worked at STMicroelectronics (Grenoble) as an etch process engineer. In 2019, she joined the group of Polymeric Materials at ETH Zurich where she is currently a PhD student. |
 Athina Anastasaki | Athina Anastasaki is an Assistant Professor in the Materials Department of ETH Zurich and the head of the Polymeric Materials Lab. She previously completed her PhD studies with Professor Dave Haddleton at the University of Warwick for which she won the Jon Weaver Award for the best PhD in Polymer Chemistry in the UK. She was then awarded an Elings Fellowship and a Global Marie Curie Fellowship to pursue post-doctoral work with Professor Craig Hawker at the University of California, Santa Barbara. Her group's research currently focuses on controlled radical polymerization and polymeric self-assembly. |
Introduction
Polymers are unlike small molecules in that they do not possess a unique molecular weight, instead they exhibit a molecular weight distribution (MWD). Dispersity (Đ, formerly referred to as polydispersity index or PDI) is a measure of the width of a MWD and describes the heterogeneity (or uniformity) of the various chain molecular weights within a polymeric material.1 It can be calculated using the equation Đ = Mw/Mn, where Mw is the weight average molecular weight (ΣNiMi2/ΣNiMi) and Mn is the number average molecular weight (ΣNiMi/ΣNi). Ni and Mi are the number molecular weight of one polymeric specie within the distribution.2,3Mw and Mn can be determined experimentally utilising static light scattering and vapour pressure osmometry, respectively. The most widely used technique to measure the dispersity and shape of polymer MWDs however is size exclusion chromatography (SEC, formerly known as gel permeation chromatography, GPC), a chromatographic method in which polymer chains are separated based on their hydrodynamic volumes. This allows the molecular weight of polymers to be determined relative to standards of known molecular weight, commonly poly(methyl methacrylate) or polystyrene.4,5 While dispersity is a crude parameter for estimating the uniformity of polymer chain lengths, the shape of MWDs provide much more information about the range of chain lengths. For example, two polymeric samples with the same dispersity can have symmetric or asymmetric MWDs, (skewed towards either high or low molecular weight), which can significantly affect material properties. Đ must have a value equal to or greater than 1, but as the polymer chains approach uniform chain length, Đ approaches unity (1), which is exhibited by some natural polymers such as DNA, peptides and proteins in which only one species is present. Achieving uniformity (monodispersity, Đ = 1) is a great challenge in polymer science and apart from very limited exceptions in synthesising oligomers, has yet to be achieved.6–8 However, the discoveries of anionic9 and cationic10 polymerisation as well as controlled radical polymerisation methodologies including atom transfer radical polymerisation (ATRP),11,12 reversible addition–fragmentation chain-transfer (RAFT) polymerisation13 and nitroxide mediated polymerisation (NMP)14 have enabled the synthesis of well-defined macromolecules with controlled molecular weight, architecture, end-group fidelity and dispersity.15–23 In fact, the high end-group fidelity and the controlled nature of the polymerisation is typically confirmed by low dispersity values and as such dispersities in the range of Đ ≈ 1.01–1.20 are routinely targeted.24–32 Conversely, broader molecular weight distributions (Đ > 1.4) are often considered to be a sign of uncontrolled or “failed” polymerisation and necessitate additional optimisation to reduce the dispersity.33–38 However, the dispersity is a key parameter that impacts the physical properties of polymers, since low and high dispersity polymers can exhibit complementary properties and functions.39–41 Hence, being able to tune MWD dispersity and shape allows polymer chemists to control and advance material properties for a multitude of applications.In this mini-review we first present different methods that have been developed to tailor MWDs. For each method, the strengths and limitations are critically evaluated and discussed. Finally, we highlight the various applications whereby varying dispersity has been shown to be beneficial. Remaining challenges in the field and future directions are also discussed.
Methods to tune dispersity
Polymer blending
Perhaps the most traditional approach to tune the dispersity is by blending (mixing) different ratios of pre-synthesised polymers (Fig. 1a).42 The individual starting polymers have either high or low dispersity values depending on whether they are prepared via free radical polymerisation or controlled polymerisation methodologies.42–46 Advantageously, this strategy requires no reaction optimisation, as one can follow published polymerisation procedures to obtain a wide selection of polymers with different molecular weights and then mix these materials in predetermined ratios to obtain materials with different dispersities. This advantage also explains why this method has been preferred not only by polymer chemists but also by physicists, engineers and material scientists in both academia and industry. Moreover, this strategy allows access to any targeted polymeric material that the chosen polymerisation strategy has access to (e.g. type of polymer or molecular weight targeted). However, blending polymers with various molecular weights can be tedious and time-consuming, as it requires the synthesis and purification of multiple materials followed by mixing in precise ratios. In addition, the vast majority of polymer blends have bimodal or multimodal MWDs which, depending on the targeted application, can be undesirable.47 A recent approach to tune MWDs through the continuous mixing of synthesised polymer fractions was developed by Boyer, Xu and co-workers.48,49 The two groups elegantly employed flow chemistry with photo-induced electron transfer (PET) RAFT polymerisation, generating polymers of different molecular weights and dispersities by adjusting flow rates, chemical concentrations and residence times. By in situ mixing of these polymer fractions, customised molecular weight distributions could be obtained (Fig. 2).48,49 The main strengths of this approach are the well-established advantages of flow over batch processes (e.g. uniform heat and mass transfer, favourable light penetration, etc.), the possibility of block formation where the dispersity of both blocks can be controlled and the broader scope of monomers compared to anionic polymerisation.50 There are challenges however, as the flow process cannot easily be adapted to the synthesis of polymer brushes (material whereby polymer chains are tethered to a surface) or other complex polymer architectures and the MWDs produced are typically bimodal or multimodal as they are in all polymer blending approaches. It should also be noted that by tuning the residence time, an increase of the dispersity values can be achieved although the breadth of the MWDs is more limited when compared to mixing.51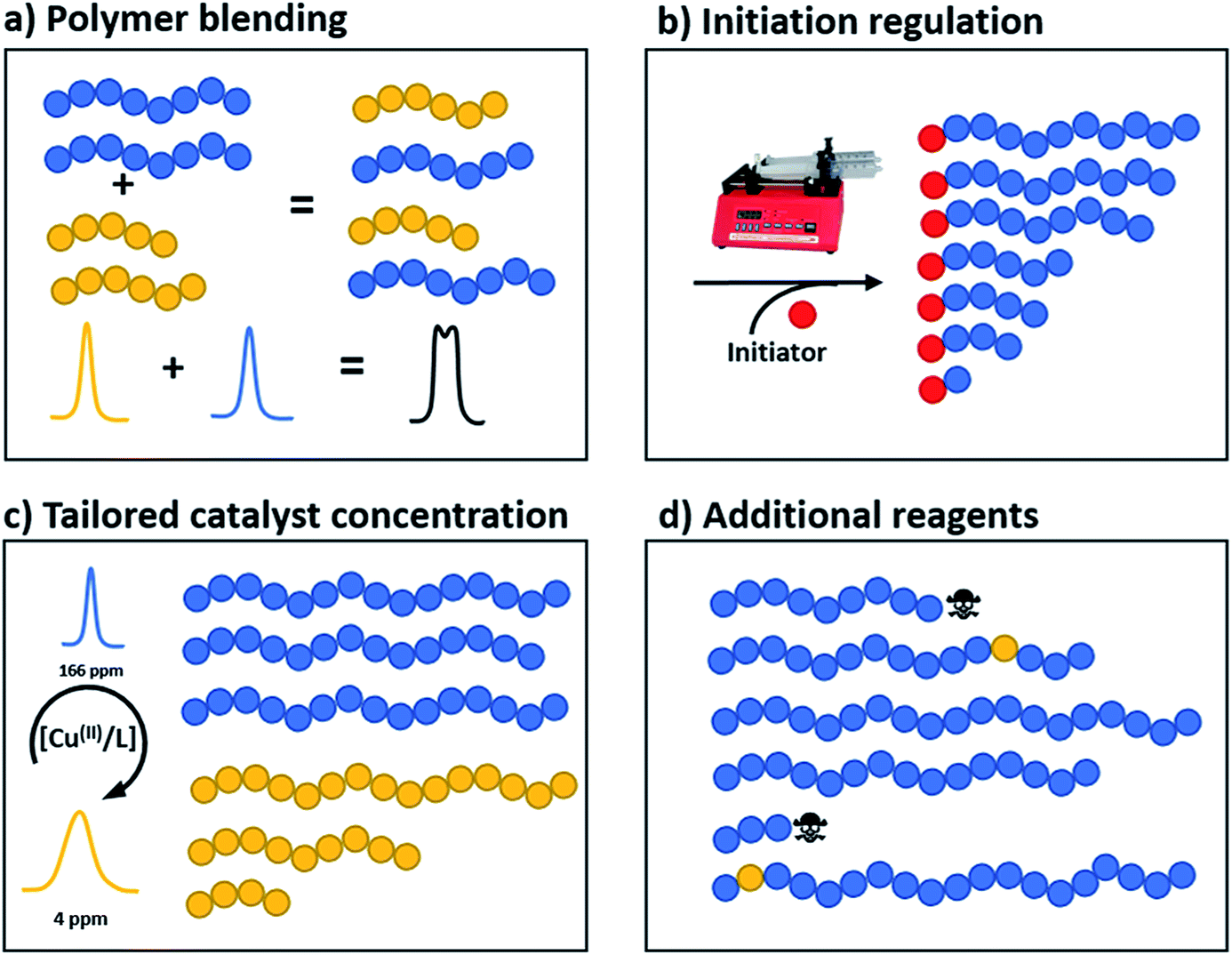 | ||
| Fig. 1 The four strategies by which the dispersity of a polymer can be tailored, namely (a) polymer blending (b) temporal regulation of initiation, (c) tailoring of catalyst concentrations and (d) the addition of extra components. | ||
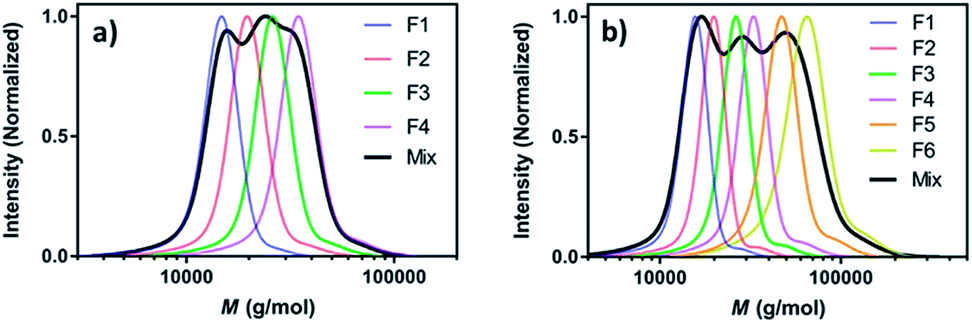 | ||
| Fig. 2 Tailored MWDs by blending pre-synthesised polymers. Illustrated is the mixing of (a) four and (b) six polymer fractions of distinct molecular weights. This figure is adapted from ref. 49 with permission from ACS publications. | ||
Temporal regulation of initiation
A more recently developed approach to tailor molecular weight distributions is via the temporal regulation of initiation (Fig. 1b). By introducing initiators into a controlled polymerisation reaction solution at a selected rate, both changes in the dispersity and the shape of the MWD have been illustrated. In 2007, Lynd and Hillmyer were the first to successfully tune the dispersity by controlling the rate of initiator addition and the temperature of an anionic polymerisation, although the shape of the MWDs was not controlled and the dispersity was limited to a maximum of 1.3.52 Fors and co-workers were the first to tune the shape as well as the dispersity, by utilising NMP as the polymerisation technique and feeding an alkyl nitroxide initiator into a styrene polymerisation.53 The MWDs were successfully broadened with SEC showing monomodal distributions as reaction time was increased, while Mn was maintained constant. In addition, through modulating the addition rate, the MWDs could also be efficiently tailed while both Mn and Đ were kept constant. The term “asymmetry factor” (As) can be used to differentiate between polymer samples with different shapes but the same molecular weight and dispersity. Values close to 1 signify symmetrical peaks, whereas values above or below 1 indicate skewness to either higher or lower molecular weights.54 Importantly, despite the broadening of the MWDs, excellent end-group fidelity could be maintained in all cases, as evidenced by the isolation of a PS macroinitiator and the efficient formation of poly(styrene-b-isoprene) block copolymers. The main strength of this technique is that any desirable dispersity can be targeted ranging from Đ ≈ 1.17–3.9. However, to make this strategy more practical and powerful, several challenges remained. For example, NMP inherently produces polymers with broader molecular weight distributions than anionic polymerisation, ATRP or RAFT, has limited monomer scope and typically has relatively low end-group fidelity at quantitative or near-quantitative conversions.55To address these limitations, the Fors group changed polymerisation methodology, selecting anionic polymerisation instead. By feeding a fixed amount of the initiator sec-butyllithium at different rates, the dispersity was increased from 1.16 to 2.47 without changing the Mn, and poly(styrene-b-isoprene) block copolymers were prepared.56 The skewness of the MWDs was also successfully tuned and a higher level of control over the shape of the distribution could be accomplished than with NMP (Fig. 3).53 Importantly due to the truly living nature of anionic polymerisation,57,58 full monomer conversions were achieved and excellent end-group fidelity could be maintained, allowing access to diblock copolymers in one pot.59 In 2019, the same group also presented a theoretical model that enables the accurate prediction of MWD composition for a range of initiator addition profiles when polystyrene was synthesised by anionic polymerisation.60 Despite the great capability of this methodology to successfully control the shape and dispersity of MWDs, all examples exhibited significant tailing at either low or high molecular weight and thus were relatively asymmetrical. Frey and co-workers could partially address this problem in continuous flow, where by varying the total flow rate they were able to achieve more symmetrical MWDs. The resulting materials retained very high end-group fidelity and allowed for the subsequent synthesis of block copolymers.61 It should be noted that an overall limitation of the temporal regulation of initiation method is that it cannot be applied to heterogeneous structures such as polymer brushes.
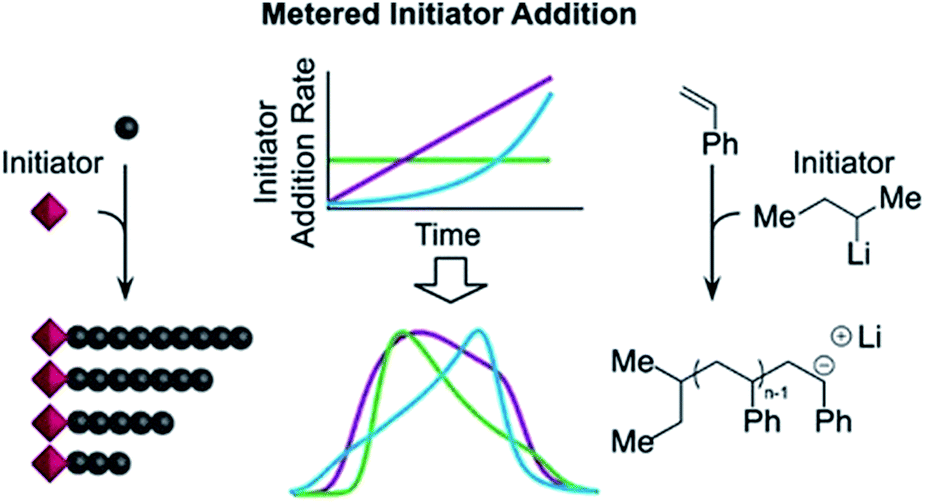 | ||
| Fig. 3 Tailored shape and dispersity of the molecular weight distribution, produced via metered addition of s-BuLi to the anionic polymerisation of styrene. This figure is adapted from ref. 56 with permission from ACS publications. | ||
Altering catalyst concentration
The first example of tuning the MWD shape and dispersity by changing the catalyst concentration was presented in the 1970s utilising Ziegler–Natta polymerisation.62 There are also reports where catalysts of different activities were mixed or where the catalyst solubility was changed, affecting the dispersity of the materials produced.63 This is an area where a significant amount of literature exists, including the effect of the dispersity on rheological properties and the application of the materials as viscosity modifiers.62,64,65 It is worthy of note, however, that these polymerisations systems typically do not have full catalyst solubility and yield dispersities in a range of 5–10, commonly with bimodal or multimodal molecular weight distributions.In 2012, Matyjaszewski and co-workers tuned the dispersity with activators regenerated by electron transfer (ARGET-)ATRP,66 selecting tin-2-ethylhexanoate as the reducing agent to produce diblock copolymers. Upon adjusting the catalyst concentration (Fig. 1c) and reaction temperature, polymers with tuneable dispersity (Đ ≈ 1.32–2.00) were obtained with high end-group fidelity, capable of undergoing block formation. Both poly(methyl acrylate) and polystyrene could be prepared with tailored MWDs without the need for the addition of reagents during polymerisation.67 In 2019, the same group further exploited ARGET-ATRP to tune the dispersity of poly(methyl methacrylate) ranging from 1.18 to 2.15.68 Also in 2019, our group reported the variation in catalyst concentration in photo-induced ATRP69–72 as a convenient and versatile technique to tune the dispersity of both homopolymers and block copolymers (Đ ≈ 1.06–1.75) where both the Mn and the Mw could be controlled (a comparison of the range of molecular weights and dispersities produced by changing the catalyst concentration and the temporal regulation of initiation is illustrated in Fig. 4).73 Significantly, very low ppm levels of copper catalyst (as low as 4 ppm) enabled perfect temporal control in photo-induced ATRP, which contrasts previous reports in organic media.69,74 The resulting materials exhibit exceptional end-group fidelity, capable of chain extension with negligible termination. Additionally, control over the MWD was also achieved in the absence of any external deoxygenation75,76 (using the so-called oxygen tolerant approach) although lower initiator efficiencies were reported.73 An equation has been derived which illustrates the relationship between the catalyst concentration and the dispersity of the polymers generated.77 The dispersity also correlates with the relative rates of propagation and deactivation, the conversion and the degree of polymerisation. This in principle permits the prediction of the dispersity of a particular polymerisation system, though for example if the monomer is changed the relevant parameters need to be found. This equation has been further refined by Zhu and Mastan to include cases where there is a significant contribution of termination.78
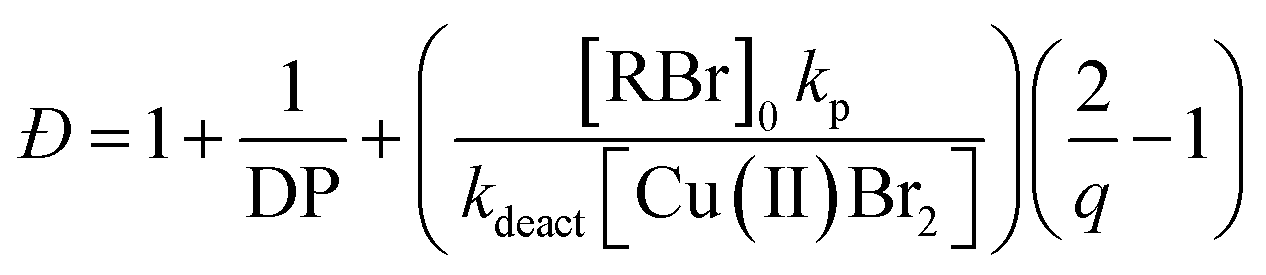 | (1) |
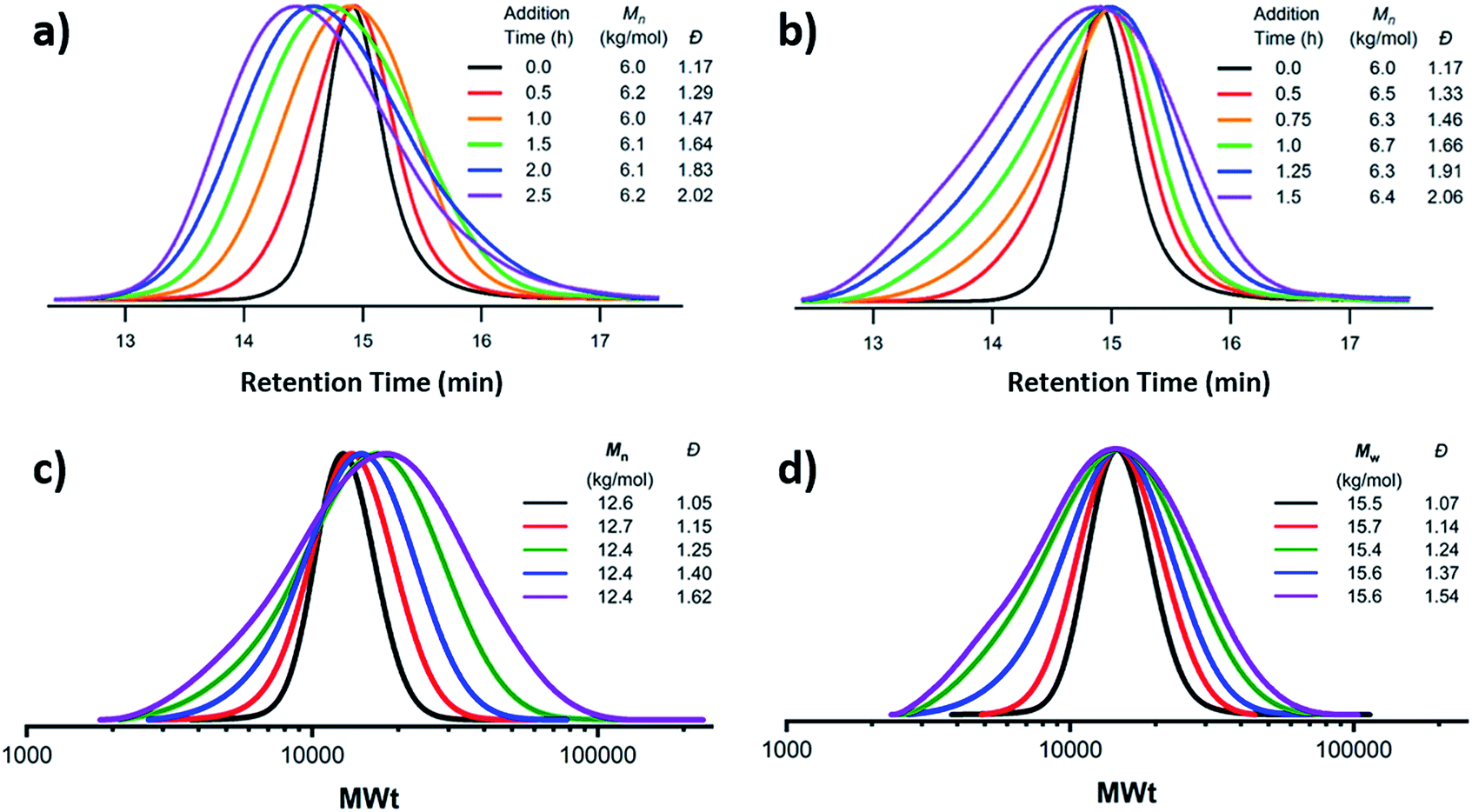 | ||
| Fig. 4 Comparison of two methods by which the molecular weight distribution is tuned. Firstly, the metered addition of initiator to the NMP of styrene with (a) at a constant rate and (b) with varying rate and secondly via the reduction of catalyst concentration in photo-induced ATRP to maintain (c) Mn or (d) Mw constant. This figure is adapted from ref. 53 and 73, permission from ACS publications and Wiley respectively. | ||
There are, however, a number of drawbacks associated with lowering the catalyst concentration, which have yet to be overcome: by lowering the catalyst concentration, there is a decrease in initiator efficiency, which results in higher molecular weight polymers than the ones targeted. Reactions also don't reach high conversions and therefore isolation of a macroinitiator is required if block copolymers are to be prepared.73 There are also no examples in the literature, where the shape of the MWD has been controlled by changing the catalyst concentration.
Additional reagents, chain coupling or terminating agents
All of the synthetic methods discussed thus far tailor the MWD by changing the amount of reagents essential for a successful polymerisation or the rate at which they are introduced into the reaction mixture. An alternative method to tune the dispersity is to take an existing polymerisation system that yields low dispersity polymers and introduce an additional component that can cause irreversible capping of the growing polymer chain ends, thus broadening the molecular weight distribution (Fig. 1d).79 With this in mind, Robertson, Conrad and co-workers utilised traditional ATRP with CuBr/PMDETA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) as the catalyst. By addition of varying amounts of phenylhydrazine to the ATRP of tert-butyl acrylate, dispersities from 1.08 to 1.80 could be obtained. When phenylhydrazine was replaced with an alternative reducing agent (tin-2-ethylhexanoate) under otherwise identical conditions, only low dispersity materials were obtained.79 Thus, it was concluded that phenylhydrazine does not primarily function as a reducing agent, but additionally substitutes the halogen end group, leading to dead polymer chains with different molecular weights. Although this technique offers a simple way to tune the MWD, the range of dispersities obtained was very narrow. The only dispersities reported are 1.08, 1.71 and 1.80, thus optimisation is required to tune the MWDs. In addition, the termination by nucleophilic substitution prevents the synthesis of block copolymers which further limits the applicability of this methodology.79Another interesting way to control MWDs was developed by Goto and co-workers, who exploited reversible complexation mediated polymerisation (RCMP)80,81 using alkyl iodides as dormant species and organic catalysts. By adding a small amount of butyl acrylate (4–10%) to the RCMP of methyl methacrylate the dispersity of diblock, triblock and even multiblock, star and brush polymers was efficiently tuned from 1.11 to 1.50.82 To date, this is the only reported method that can efficiently control the dispersity in such a wide range of macromolecular architectures (Fig. 5). Importantly, this method does not rely on metal catalysts, sulphur containing components or feeding, factors which are beneficial in a number of applications. A noteworthy limitation, however, is that in all cases the addition of small amounts of an acrylate monomer is required to broaden the MWD, thus resulting in the formation of random copolymers, for example polymethacrylates with up to 10% of acrylate monomer incorporated. From this latter point, it is also clear that this method cannot be used to tune the dispersity polyacrylates and polystyrene.82
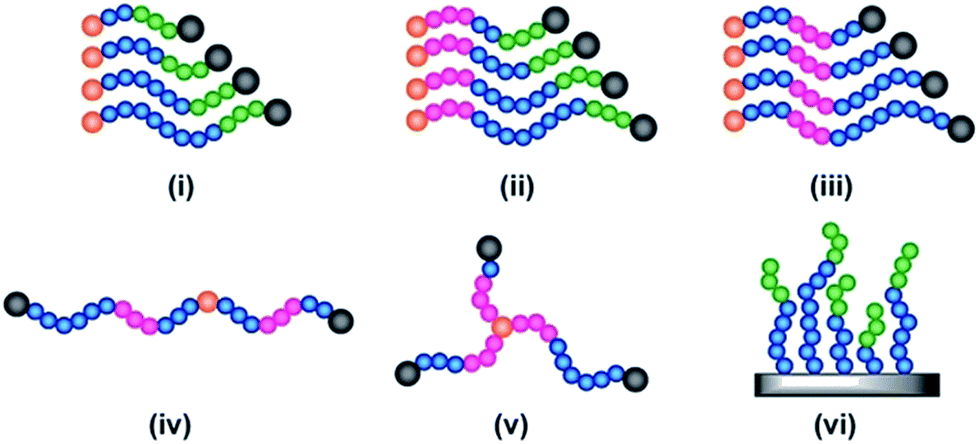 | ||
| Fig. 5 The range of complex materials synthesised via organocatalysed LRP, illustrating a variety of architectures, namely (i) diblock copolymers, (ii) and (iii) triblocks with variation of dispersity of different blocks, (iv) multiblock (v) 3 arm star and (vi) a brush. This figure is adapted from ref. 82 with permission from Wiley. | ||
The final method we discuss is to tune the dispersity is by controlling the coupling of pre-synthesised polymers. For example, utilising the process of atom transfer radical coupling, whereby radicals are generated on macroinitiator chain ends by an atom transfer radical process. Rather than moderating the concentration of radicals for controlled radical polymerisation, the concentration is maximised to accelerate coupling in the absence of monomer. The Yagci group illustrated the high efficiency of this coupling reaction utilising monobrominated polystyrene to form predominantly double molecular weight polymers with dispersities in the range of 1.17–1.35.83 The Matyjaszewski group successfully increased the dispersity to values as high as 36.4 by chain-end coupling dibrominated polymers. Importantly, with this approach the amount of coupling can be tuned by varying the concentration and ratio of mono- or dibrominated species. However high end group fidelity must be achieved in the initial polymer synthesis for this method to be effective and the molecular weight distributions, in particular at higher dispersity values, are commonly multimodal.84
Applications
Control of physical properties
Being able to understand and tune the thermal and viscoelastic properties of polymeric materials is useful in many industrial applications such as injection-moulding, 3D printing and hot-melt extrusion.43,85–87 Early work showed that both thermal and rheological properties depend not only on the chemical structure and molecular weight of a polymeric material, but also on the dispersity. For example, Sridhar and Ye compared low and high dispersity polystyrene, the latter produced by artificially blending 18 low dispersity polymers (Đ = 3.2). The high dispersity mixture showed a much more gradual and broad relaxation spectrum, slightly lower zero-shear-rate viscosity, an earlier onset of shear thinning, much larger extensional viscosity and more pronounced strain hardening behaviour.43 Although these results clearly demonstrate the effect of dispersity on rheological properties, the symmetry and shape of the MWDs were not taken into account. In recent work, Fors and co-workers found that the shape of MWDs had a considerable influence on the stiffness of the materials (Fig. 6a).56 This was further highlighted in a subsequent collaboration between the Fors and Connal groups, who showed that when the MWDs are skewed to higher molecular weights, a higher glass transition temperature, increased stiffness, thermal stability and higher apparent viscosities are obtained.88 Significantly, this suggests that the shape of the molecular weight distributions as well as the dispersity can influence fundamental properties of polymers, and that these parameters can be utilised to optimise materials properties.62,64,65,88–90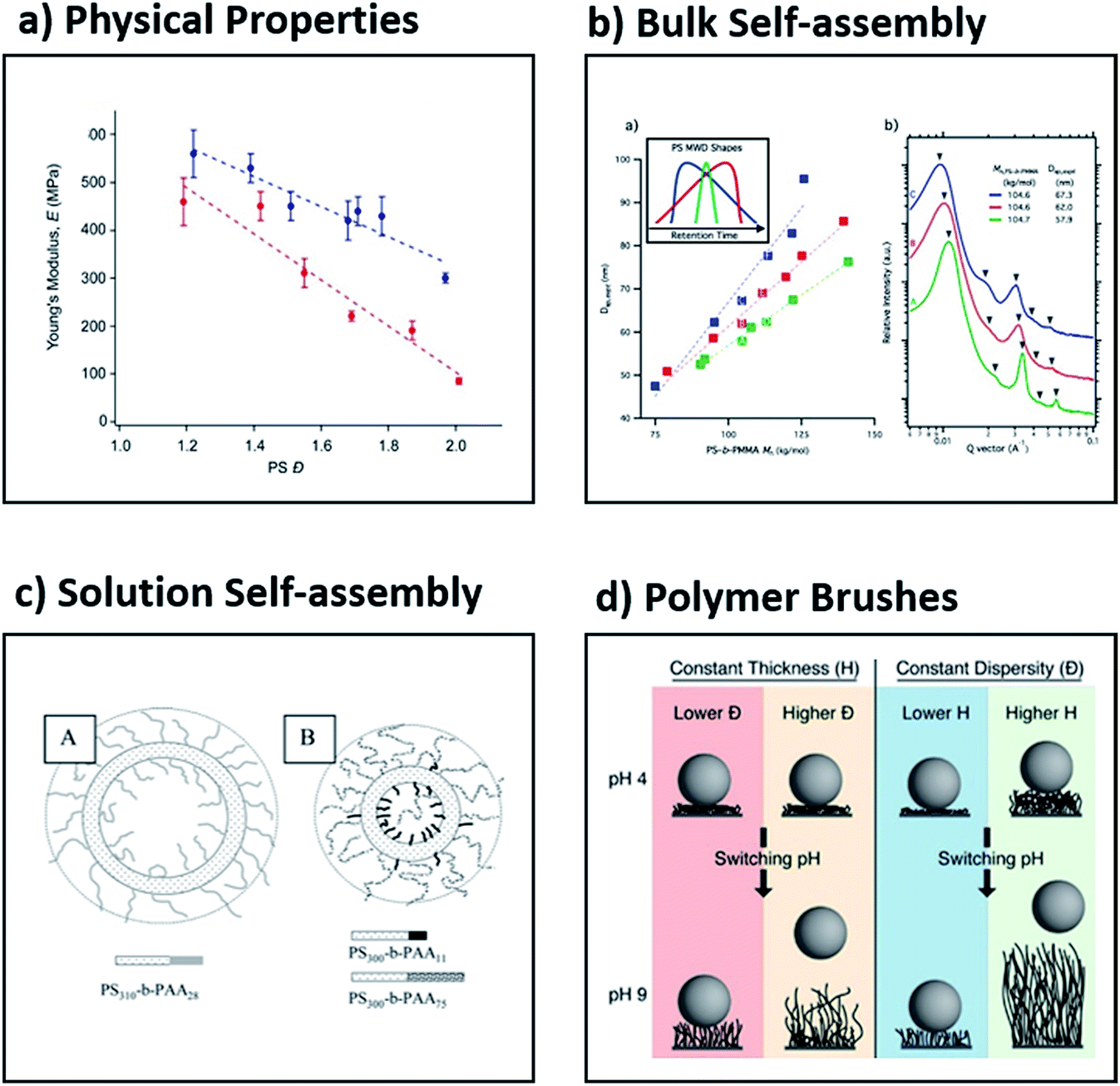 | ||
| Fig. 6 Examples of the effect of dispersity on four different application areas, namely (a) the Young's modulus as determined by dynamic mechanical analysis, (b) the effect of skew on self-assembly in bulk, (c) the formation of vesicles in solution and (d) the release of adherent bacteria by polymer brushes. This figure is adapted from ref. 56, 59, 108 and 116 with permission from ACS publications. | ||
Self-assembly in bulk
Block copolymers can be assembled in bulk into well-ordered nanostructures.39,89,91 Owing to the great utility of these materials in both fundamental and applied polymer science, several theories have been developed to predict the formation of interesting new morphologies. Most theories are based on the assumption of perfectly monodisperse materials (Đ = 1). The groups of Hawker and Meijer independently reported the synthesis of a range of discrete low molecular weight diblocks (Đ = 1) and demonstrated that such discrete materials exhibit a pronounced decrease in domain spacing and sharper scattering reflections when compared to their high dispersity analogues.92–94 The order–disorder transition temperature also decreased with increased dispersity, which suggested the stabilisation of the disordered phase. In general, however, the effect of dispersity on the stabilisation or destabilisation of the ordered or disordered phases is complex and depends on both the relative volume fractions and the dispersities of the two blocks.39 Although such studies are of great importance to increase our fundamental understanding of self-assembly, they are very few in number and are further limited to the synthesis of low molecular weight block copolymers. In contrast, block copolymers with a range of dispersity values constitute the vast majority of literature reports in the self-assembly of block copolymers in bulk.43,45–47,52,95,96 In all these cases, it has been demonstrated that dispersity plays a very significant role. For instance, Mahanthappa and co-workers showed that high dispersity triblock copolymers self-assemble into periodic structures with unexpectedly enhanced stability in comparison to their low dispersity analogues.97 The same group reported that when the dispersity of the B block in an ABA poly(lactide-b-1,4-butadiene-b-lactide) triblock copolymer was varied, the free energy of the microphase-separated melt increased with increased dispersity. This dispersity-induced destabilisation was attributed to amplified fluctuations in these low molecular weight, high dispersity materials.98 Another example is from Lynd and Hillmyer, who demonstrated that upon increasing the dispersity in the minority component of a diblock copolymer, a decrease in the segregation strength at the order–disorder transition is observed, whereas increasing dispersity in the majority component resulted in an increase in the segregation strength at the order–disorder transition.52 Moreover, the groups of Bockstaller and Matyjaszewski discovered that “metastable” morphologies (e.g. hexagonally perforated layers) can be stabilised with higher dispersity polymers.99 More recently, Fors and co-workers reported a method to tune the domain spacing in block copolymers over a large range of lamellar periods by modifying where both the breadth and the skew of the molecular weight distributions (Fig. 6b).59 These examples show that recently established methods to control molecular weight distributions discussed in this review will enable further understanding and discoveries in the self-assembly of block copolymers in the solid state.Self-assembly in solution
Amphiphilic block copolymers can self-assemble into well-defined nanostructures of different shapes (e.g. spheres, worms, micelles, etc.) in selected solvents.100–103 Such polymeric nanoparticles can find use in a wide range of applications ranging from catalysis, organic electronics and energy storage to immunotherapy and drug delivery.104–107 It can be hypothesised that by changing the dispersity of either the solvophobic or the solvophilic block, the final morphology of the polymeric nanoparticle may also be varied.41 This was demonstrated by Eisenberg and co-workers, who studied the effect of the dispersity of the corona block in poly(styrene-b-acrylic acid) (PS–PAA) using the traditional self-assembly technique.108,109 By using the blending technique, they mixed diblock copolymers of low dispersity PS and PAA of varied dispersity, achieving overall dispersities ranging from 1.1 to 2.1. Interestingly, with a low dispersity PAA block (Đ = 1.1), large and highly disperse vesicles were observed, while with a high dispersity PAA block, the size of the vesicles decreased and small spheres were also observed. It was rationalised qualitatively that diblocks with longer PAA blocks segregate on the outer surface, while shorter chains prefer the inner surface of the vesicle (Fig. 6c).108,109 Similarly, Mahanthappa and co-workers studied the effect of the dispersity of the hydrophobic block using poly(ethylene oxide-b-1,4-butadiene-b-ethylene oxide) bolaamphiphiles. When the hydrophobic block had a low dispersity, a pure vesicular morphology was observed, but by increasing the dispersity, the authors could obtain a mixture of vesicles, spheres and worms. This suggests that the increased dispersity provides a facile methodology to modify the desired morphology, but the preparation of pure morphologies via this method has yet to be achieved.97 Shi, Liang and co-workers utilised two-dimensional real-space self-consistent-field theory to study the effect of dispersity on the structure of vesicles assembled by amphiphilic diblock copolymers. The authors found that when higher dispersity block copolymers were selected, the formation of smaller vesicles or quasi-vesicles was favoured.110 Despite these great developments and given the large number of publications in the area of solution self-assembly, the effect of dispersity in the block copolymers needs to be further clarified.Polymer brushes
Polymer brushes are an established class of materials that are extensively investigated due to their unique applications in a number of research areas including catalysis, sensing, controlled cell-adhesion, chromatography and particle/surface modification.111–113 Thus, being able to efficiently tune the dispersity of polymer brushes in an analogous way to polymers in solution can significantly alter the brush properties. Early work by Jayaraman and co-workers showed that high dispersity polymers were more effective in stabilising suspensions of polymer-grafted nanoparticles.114 Furthermore, the groups of Conrad, Robertson and co-workers observed both higher low-pH contact angles and lower degrees of hysteretic memory with higher dispersity poly(acrylic acid) brushes. This was attributed to the higher dispersity polymer's ability to increase solvent uptake, thus resulting in an increase in brush swelling. Atomic Force Microscopy (AFM) was used to measure the surface roughness and illustrated that the hysteric memory behaviour in contact angles is not explained simply by the variation in brush length.115 The same groups illustrated that these high dispersity brushes promoted bacterial detachment in comparison to their low dispersity analogues which exhibited negligible detachment. It should be noted, however, that bacterial adhesion (rather than detachment) was independent of dispersity, but strongly correlated to the brush thickness (Fig. 6d).116 In a similar vein, Goto and co-workers found that concentrated poly(trifluoroethyl methacrylate) brushes with high dispersity values are not uniform in thickness, which enhances their flexibility in conformation, allowing large solute molecules to diffuse into the brush layer.82 In contrast, analogous materials with low dispersity prevent large solute molecules from diffusing into the brush layer, owing to the tighter packing of the neighbouring chains. Such observations may open up new directions for biological and sensing applications. The Matyjaszewski and Bockstaller groups in collaboration demonstrated that the initial concentration of the copper catalyst significantly affected the dispersity of poly(methyl methacrylate) brushes formed via surface-initiated ATRP, in a similar manner to polymers synthesised in solution. Importantly, both the dispersity and the grafting density could be efficiently tuned using a facile and scalable approach.68 In another important contribution from the two groups, it was shown that by having bimodal rather than monomodal MWDs, the mechanical toughness of films (produced from polymer grafted particles) is significantly increased.117 This finding highlights that monomodal MWDs should not be the only target of new synthetic protocols developed.Current challenges and future outlook
In summary, recent progress in polymer science has allowed for the development of versatile and facile methodologies that can control both the dispersity and the shape of MWDs. The resulting polymers have numerous potential applications in both the academic and industrial arenas. In addition to new applications, being able to efficiently tune the MWDs in a given polymerisation system can open up new avenues to enhance our understanding of polymerisation mechanisms and can result in further optimisation of polymerisation protocols. Despite the developments in this area, there are still several limitations from a synthetic standpoint that need to be overcome. For example, many of the reported methods suffer from significant amounts of termination, thus prohibiting efficient block copolymer formation, and feeding is limited to homogeneous systems (e.g. polymer brushes are not accessible). Other weaknesses of the current methodologies include low initiator efficiencies for ATRP-based systems73 or adulterated polymer chains.82 Additionally, with very few exceptions,82 the vast majority of the reports are limited to homopolymers and block copolymers rather than more complex architectures such as multiblocks, stars and protein–polymer conjugates. Another important consideration is the efficient purification of high dispersity materials which can be challenging when a wide range of polymer chain lengths are present. For example, precipitation can result in the loss of lower molecular weight species. Moreover, most of the currently available methods are either limited to fairly asymmetric MWDs and a narrow dispersity range, or conversely to fairly symmetric MWDs. In addition, very few simulation studies have been conducted that allow for better prediction of MWDs without the need for multiple polymerisations and system optimisation.Finally, a further potentially interesting method to tune the dispersity would be to utilise exchange between polymerisation mechanisms.42 For example, there are reports of interconversion between cationic and radical polymerisation mechanisms, either utilising a chemical stimulus or by changing the wavelength of light,118 and between ATRP and RAFT mechanisms utilising an ATRP-functionalised RAFT chain transfer agent.119,120 However, these methods are yet to be applied to the synthesis of materials with a range of molecular weight distribution dispersities and shapes.
With respect to applications, we have just started to scratch the surface. For instance, concerning self-assembly in solution there are multiple publications that attribute interesting observations (such as mixed morphologies) to broad molecular weight distributions, but very little has been done to comprehensively prove such hypotheses.107–109 It is noted that the shape of MWDs has been significantly less explored than the dispersity. We thus envisage that continued synthetic advances in tailoring the molecular weight distribution together with associated applications will further enhance the impact of controlled polymerisation strategies across multiple research areas.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. A. gratefully acknowledges ETH Zurich for financial support. N. P. T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076).References
- R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351 Search PubMed
.
- R. F. T. Stepto, Polym. Int., 2010, 59, 23 CrossRef CAS
- S. Harrisson, Polym. Chem., 2018, 9, 1366 RSC
- H. Benoit, Z. Grubisic, P. Rempp, D. Decker and J. G. Zilliox, Journal de Chimie Physique, 1966, 63, 1507 CrossRef CAS
- Y. Guillaneuf and P. Castignolles, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 897 CrossRef CAS
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094 CrossRef CAS PubMed
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376 CrossRef CAS PubMed
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182 CrossRef CAS PubMed
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747 CrossRef CAS
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245 CrossRef CAS PubMed
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559 CrossRef CAS
-
D. H. Solomon, E. Rizzardo and P. Cacioli, US Pat., 4581429, 1986
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835 CrossRef CAS PubMed
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed
- J. Tanaka, A. S. Gleinich, Q. Zhang, R. Whitfield, K. Kempe, D. M. Haddleton, T. P. Davis, S. Perrier, D. A. Mitchell and P. Wilson, Biomacromolecules, 2017, 18, 1624 CrossRef CAS PubMed
- A. B. Cook and S. Perrier, Adv. Funct. Mater., 2019, 1901001 CrossRef
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365 CrossRef CAS
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743 CrossRef CAS PubMed
- J. Tanaka, P. Gurnani, A. B. Cook, S. Häkkinen, J. Zhang, J. Yang, A. Kerr, D. M. Haddleton, S. Perrier and P. Wilson, Polym. Chem., 2019, 10, 1186 RSC
- R. Whitfield, A. Anastasaki, N. P. Truong, P. Wilson, K. Kempe, J. A. Burns, T. P. Davis and D. M. Haddleton, Macromolecules, 2016, 49, 8914 CrossRef CAS
- A. Kuroki, I. Martinez-Botella, C. H. Hornung, L. Martin, E. G. L. Williams, K. E. S. Locock, M. Hartleib and S. Perrier, Polym. Chem., 2017, 8, 3249 RSC
- N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848 CrossRef CAS PubMed
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692 RSC
- G. Lligadas, S. Grama and V. Percec, Biomacromolecules, 2017, 18, 2981 CrossRef CAS PubMed
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. Van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483 CrossRef CAS PubMed
- A. Keyes, H. E. Basbug Alhan, E. Ordonez, U. Ha, D. B. Beezer, H. Dau, Y. S. Liu, E. Tsogtgerel, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2019, 131, 2 CrossRef
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS PubMed
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS PubMed
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73 CrossRef CAS
- V. Sciannamea, R. Jerome and C. Detrembleur, Chem. Rev., 2008, 108, 1104 CrossRef CAS PubMed
- M. Rubens, J. H. Vrijsen, J. Laun and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 3183 CrossRef CAS PubMed
- J. J. Haven, E. Baeten, J. Claes, J. Vandenbergh and T. Junkers, Polym. Chem., 2017, 8, 2972 RSC
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003 CrossRef CAS PubMed
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382 RSC
- R. Whitfield, A. Anastasaki, G. R. Jones and D. M. Haddleton, Polym. Chem., 2018, 9, 4395 RSC
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875 CrossRef CAS
- D. Nichetti and I. Manas-Zloczower, Polym. Eng. Sci., 1999, 39, 887 CAS
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119 RSC
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2005, 38, 8803 CrossRef CAS
- X. Ye and T. Sridhar, Macromolecules, 2005, 38, 3442 CrossRef CAS
- T. Hashimoto, T. Tanaka and H. Hasegawa, Macromolecules, 1985, 18, 1864 CrossRef CAS
- A. Noro, M. Iinuma, J. Suzuki, A. Takano and Y. Matsushita, Macromolecules, 2004, 37, 3804 CAS
- S. C. Schmidt and M. A. Hillmyer, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2364 CrossRef CAS
- M. Rubens and T. Junkers, ChemRxiv.org, 2019, DOI:10.26434/chemrxiv.8233460.
- N. Corrigan, A. Almasri, W. Taillades, J. Xu and C. Boyer, Macromolecules, 2017, 50, 8438 CAS
- N. Corrigan, R. Manahan, Z. T. Lew, J. Yeow, J. Xu and C. Boyer, Macromolecules, 2018, 51, 4553 CrossRef CAS
- N. Zaquen, J. Yeow, T. Junkers, C. Boyer and P. B. Zetterlund, Macromolecules, 2018, 51, 5165 CrossRef CAS
- M. H. Reis, T. P. Varner and F. A. Leibfarth, Macromolecules, 2019, 52, 3551 CrossRef CAS
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2007, 40, 8050 CrossRef CAS
- D. T. Gentekos, L. N. Dupuis and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 1848 CAS
- J. J. Kirkland, W. W. Yau, H. J. Stoklosa and C. H. J. J. Dilks, J. Chromatogr. Sci., 1977, 15, 303 CAS
- R. B. Grubbs, Polym. Rev., 2011, 51, 104 CrossRef CAS
- V. Kottisch, D. T. Gentekos and B. P. Fors, ACS Macro Lett., 2016, 5, 796 CrossRef CAS
- J. Blankenburg, E. Kersten, K. Maciol, M. Wagner, S. Zarbakhshd and H. Frey, Polym. Chem., 2019, 10, 2863 RSC
- E. Grune, J. Bareuther, J. Blankenburg, M. Appold, L. Shaw, A. H. E. Müller, G. Floudas, L. R. Hutchings, M. Gallei and H. Frey, Polym. Chem., 2019, 10, 1213 CAS
- D. T. Gentekos, J. Jia, E. S. Tirado, K. P. Barteau, D.-M. Smilgies, R. A. DiStasio and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 4639 CAS
- S. Domanskyi, D. T. Gentekos, V. Privman and B. Fors, Polym. Chem., 2019 10.1039/c9py00074g
- J. Morsbach, A. H. E. Müller, E. Berger-Nicoletti and H. Frey, Macromolecules, 2016, 49, 5043 CAS
- Z. Umberto and C. Giuliano, Adv. Polym. Sci., 1983, 51, 101 CrossRef
- G. A. Mortimer, J. Appl. Polym. Sci., 1976, 20, 55 CrossRef CAS
- A. Ahlers and W. Kaminsky, Makromol. Chem., Rapid Commun., 1988, 9, 457 CrossRef CAS
- T. O. Ahn, S. C. Hong, J. H. Kim and D. Lee, J. Appl. Polym. Sci., 1998, 67, 2213 CAS
- K. Min, H. F. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789 CrossRef CAS
- A. Plichta, M. Zhong, W. Li, A. M. Elsen and K. Matyjaszewski, Macromol. Chem. Phys., 2012, 213, 2659 CrossRef CAS
- Z. Wang, J. Yan, T. Liu, Q. Wei, S. Li, M. Olszewski, J. Wu, J. Sobieski, M. Fantin, M. R. Bockstaller and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 859 CrossRef CAS
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox and V. Percec, J. Am. Chem. Soc., 2014, 136, 1141 CrossRef CAS PubMed
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219 CrossRef CAS
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58 CrossRef CAS PubMed
- V. Nikolaou, A. Anastasaki, F. Alsubaie, A. Simula, D. J. Fox and D. M. Haddleton, Polym. Chem., 2015, 6, 3581 RSC
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201906471
- N. D. Dolinski, Z. A. Page, E. H. Discekici, D. Meis, I. H. Lee, G. R. Jones, R. Whitfield, X. Pan, B. G. McCarthy, S. Shanmugam, V. Kottisch, B. P. Fors, C. Boyer, G. M. Miyake, K. Matyjaszewski, D. M. Haddleton, J. Read de Alaniz, A. Anastasaki and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 268 CrossRef CAS PubMed
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998 CrossRef CAS PubMed
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963 RSC
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309 CrossRef CAS PubMed
- E. Mastan and S. Zhu, Macromolecules, 2015, 48, 6440–6449 CAS
- V. Yadav, N. Hashmi, W. Ding, T. H. Li, M. K. Mahanthappa, J. C. Conrad and M. L. Robertson, Polym. Chem., 2018, 9, 4332 RSC
- A. Goto, H. Zushi, N. Hirai, T. Wakada, Y. Tsujii and T. Fukuda, J. Am. Chem. Soc., 2007, 129, 13347 CrossRef CAS PubMed
- A. Goto, T. Suzuki, H. Ohfuji, M. Tanishima, T. Fukuda, Y. Tsujii and H. Kaji, Macromolecules, 2011, 44, 8709 CrossRef CAS
- X. Liu, C. Wang and A. Goto, Angew. Chem., 2019, 131, 5654 CrossRef
- S. Yurteri, I. Cianga and Y. Yagci, Macromol. Chem. Phys., 2003, 204, 1771 CrossRef CAS
- T. Sarbu, K. Y. Lin, J. Ell, D. J. Siegwart, J. Spanswick and K. Matyjaszewski, Macromolecules, 2004, 37, 3120 CAS
- X. Wang, M. Jiang, Z. Zhou, J. Gou and D. Hui, Composites, Part B, 2017, 110, 442 CrossRef CAS
- Y. Xu, Q. Zhang, W. Zhang and P. Zhang, Int. J. Adv. Manuf. Technol., 2015, 76, 2199 CrossRef
- M. Stanković, H. W. Frijlink and W. L. J. Hinrichs, Drug Discovery Today, 2015, 20, 812 CrossRef PubMed
- M. Nadgorny, D. T. Gentekos, Z. Xiao, S. P. Singleton, B. P. Fors and L. A. Connal, Macromol. Rapid Commun., 2017, 38, 1700352 CrossRef PubMed
- R. A. Register, Nature, 2012, 483, 167 CrossRef CAS PubMed
- J. J. Haven and T. Junkers, Polym. Chem., 2019, 10, 679 RSC
- I. Kim and S. Li, Polym. Rev., 2019, 59, 561 CrossRef CAS
- B. Oschmann, J. Lawrence, M. W. Schulze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pester, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6, 668 CAS
- B. Van Genabeek, B. F. M. De Waal, M. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 4210 CrossRef CAS PubMed
- B. Van Genabeek, B. F. M. De Waal, A. R. A. Palmans and E. W. Meijer, Polym. Chem., 2018, 9, 2746 RSC
- P. D. Hustad, G. R. Marchand, E. I. Garcia-Meitin, P. L. Roberts and J. D. Weinhold, Macromolecules, 2009, 42, 3788 CrossRef CAS
- H. Xu, E. M. Greve and M. K. Mahanthappa, Macromolecules, 2019, 52, 5722 CrossRef CAS
- J. M. Widin, A. K. Schmitt, A. L. Schmitt, K. Im and M. K. Mahanthappa, J. Am. Chem. Soc., 2012, 134, 3834 CrossRef CAS PubMed
- A. K. Schmitt and M. K. Mahanthappa, Macromolecules, 2017, 50, 6779 CrossRef CAS
- J. Listak, W. Jakubowski, L. Mueller, A. Plichta, K. Matyjaszewski and M. R. Bockstaller, Macromolecules, 2008, 41, 5919 CrossRef CAS
- N. P. Truong, M. R. Whittaker, C. W. Mak and T. P. Davis, Expert Opin. Drug Delivery, 2015, 12, 129 CrossRef CAS PubMed
- H. T. Ta, N. P. Truong, A. K. Whittaker, T. P. Davis and K. Peter, Expert Opin. Drug Delivery, 2018, 15, 33 CrossRef CAS PubMed
- S. Y. Khor, M. N. Vu, E. H. Pilkington, A. P. R. Johnston, M. R. Whittaker, J. F. Quinn, N. P. Truong and T. P. Davis, Small, 2018, 14, e1801702 Search PubMed
- N. P. Truong, J. F. Quinn, A. Anastasaki, D. M. Haddleton, M. R. Whittaker and T. P. Davis, Chem. Commun., 2016, 52, 4497 RSC
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746 RSC
- L. Zhou, Z. Zhuang, H. Zhao, M. Lin, D. Zhao and L. Mai, Adv. Mater., 2017, 29, 1602914 CrossRef PubMed
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CAS
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187 CAS
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601 CrossRef CAS
- O. Terreau, C. Bartels and A. Eisenberg, Langmuir, 2004, 20, 637 CrossRef CAS PubMed
- Y. Jiang, T. Chen, F. Ye, H. Liang and A. Shi, Macromolecules, 2005, 38, 6710 CrossRef CAS
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437 CrossRef CAS PubMed
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105 CrossRef CAS PubMed
- M. Badoux, M. Billing and H.-A. Klok, Polym. Chem., 2019, 10, 2925 CAS
- T. B. Martin, P. M. Dodd and A. Jayaraman, Phys. Rev. Lett., 2013, 110, 018301 Search PubMed
- V. Yadav, A. V. Harkin, M. L. Robertson and J. C. Conrad, Soft Matter, 2016, 12, 3589 RSC
- V. Yadav, Y. A. Jaimes-Lizcano, N. K. Dewangan, N. Park, T. Li, M. L. Robertson and J. C. Conrad, ACS Appl. Mater. Interfaces, 2017, 9, 44900 CAS
- J. Yan, T. Kristufek, M. Schmitt, Z. Wang, G. Xie, A. Dang, C. Ming Hui, J. Pietrasik, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2015, 48, 8208 CAS
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 10655 CrossRef
- B. M. Peterson, V. Kottisch, M. J. Supej and B. P. Fors, ACS Cent. Sci., 2018, 4, 1228 CrossRef CAS
- R. Nicolaÿ, Y. Kwak and K. Matyjaszewski, Macromolecules, 2008, 41, 4585 CrossRef
Footnote |
| † Both authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |