
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA lipidomic workflow capable of resolving sn- and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C location isomers of phosphatidylcholines†
C location isomers of phosphatidylcholines†
Xue
Zhao
a,
Wenpeng
Zhang
 b,
Donghui
Zhang
c,
Xinwei
Liu
c,
Wenbo
Cao
c,
Qinhua
Chen
d,
Zheng
Ouyang
c and
Yu
Xia
*a
b,
Donghui
Zhang
c,
Xinwei
Liu
c,
Wenbo
Cao
c,
Qinhua
Chen
d,
Zheng
Ouyang
c and
Yu
Xia
*a
aMOE Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biological, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: xiayu@mail.tsinghua.edu.cn
bDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
cState Key Laboratory of Precision Measurement Technology and Instruments, Department of Precision Instrument, Tsinghua University, Beijing, 100084, China
dAffiliated Dongfeng Hospital, Hubei University of Medicine, Shiyan, Hubei Province 442000, China
First published on 7th October 2019
Abstract
As a major class of mammalian lipids, phosphatidylcholines (PCs) often contain mixtures of structural isomers, resulting from different lipogenesis pathways. Profiling PCs at the isomer level, however, remains challenging in lipidomic settings, especially for characterizing the positions of fatty acyls on the glycerol backbone (sn-positions) and the locations of carbon–carbon double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cs) in unsaturated acyl chains. In this work, we have developed a workflow for profiling PCs down to sn- and CC locations at high coverage and sensitivity. This capability is enabled by radical-directed fragmentation, forming sn-1 specific fragment ions upon collision-induced dissociation (CID) of bicarbonate anion adducts of PCs ([M + HCO3]−) inside a mass spectrometer. This new tandem mass spectrometry (MS/MS) method can be simply incorporated into liquid chromatography by employing ammonium bicarbonate in the mobile phase without any instrument modification needed. It is also compatible with the online Paternò–Büchì reaction and subsequent MS/MS for the assignment of CC locations in sn-1 fatty acyl chains of unsaturated PCs. The analytical performance of the workflow is manifested by identification of 82 distinct PC molecular species from the polar extract of bovine liver, including quantification of 19 pairs of sn-isomers. Finally, we demonstrate that five pairs of PC sn-isomers show significant compositional changes in tissue samples of human breast cancer relative to controls, suggesting a potential for monitoring PC sn-isomers for biomedical applications.
Cs) in unsaturated acyl chains. In this work, we have developed a workflow for profiling PCs down to sn- and CC locations at high coverage and sensitivity. This capability is enabled by radical-directed fragmentation, forming sn-1 specific fragment ions upon collision-induced dissociation (CID) of bicarbonate anion adducts of PCs ([M + HCO3]−) inside a mass spectrometer. This new tandem mass spectrometry (MS/MS) method can be simply incorporated into liquid chromatography by employing ammonium bicarbonate in the mobile phase without any instrument modification needed. It is also compatible with the online Paternò–Büchì reaction and subsequent MS/MS for the assignment of CC locations in sn-1 fatty acyl chains of unsaturated PCs. The analytical performance of the workflow is manifested by identification of 82 distinct PC molecular species from the polar extract of bovine liver, including quantification of 19 pairs of sn-isomers. Finally, we demonstrate that five pairs of PC sn-isomers show significant compositional changes in tissue samples of human breast cancer relative to controls, suggesting a potential for monitoring PC sn-isomers for biomedical applications.
Introduction
Phosphatidylcholines (PCs) are a subclass of glycerophospholipids (GPs), accounting for more than 30% of the total lipid content in mammalian cells.1 A majority of PCs function as structural components in the cell membrane, but many of them also actively participate in cell signaling2 and gene regulation.3 Cellular PCs are initially produced by de novo synthesis (Kennedy pathway), but later dynamically modified for fatty acyl compositions via a remodeling process (Lands' cycle).4,5 The profile of PCs can serve as a readout of a certain biological state, which has found applications in tissue imaging6 and marker discovery for human disease.7,8 PCs are structurally diverse due to variations of fatty acyl chain lengths, sn-positions (relative positions of fatty acyls esterified on the glycerol backbone), or locations of carbon–carbon double bonds (CCs) in unsaturated fatty acyls. Thus, a single chemical formula of PC could contain a group of structural isomers. The number of reported PCs from a given lipidome, however, is typically less than 50, e.g., 41 PCs from human plasma9 and 14 PCs from yeast,10 much less than that has been predicted.11 One reason accounting for the small numbers of detected PCs arises from the fact that current lipid analysis methods do not differentiate structural isomers.
Mass spectrometry (MS) has become the tool of choice for lipid analysis by providing key information for lipid identification and quantitation at high sensitivity, coverage, and throughput.8,12,13 Although high-resolution mass spectrometry allows determination of the molecular formula and thus lipid sum composition, detailed structural characterization still relies on obtaining characteristic fragment ions from tandem mass spectrometry (MS/MS).14 Collision-induced dissociation (CID) is the most available and commonly used MS/MS technique for lipid identification, which can provide head group and fatty acyl composition information for GPs. Application of linked scans further enhances sensitivity and selectivity for lipid identification and quantitation from complex mixtures.15 All the above conventional MS/MS methods have been routinely incorporated into various lipidomic analysis workflows, but they fall short for the analysis of lipid isomers down to sn- or CC positions.
There has been a strong push from the MS community for developing methods capable of differentiating structural isomers, a step being considered critical to support the growth of lipidomics.16,17 Localization of CC has been recently achieved by paring CC derivatization with subsequent MS/MS via CID, such as the Paternò–Büchì (PB) reaction18,19 and epoxidation reaction.20–22 These approaches are capable of determining CC locations in various classes of lipids but they cannot provide information on the sn-positions of fatty acyls. Several MS/MS techniques utilizing ion activation/dissociation other than CID or combined with CID turn out to be quite informative for obtaining both sn-position and (or) CC location from GPs. These include electron impact excitation of ions from organics (EIEIO),23 ozone-induced dissociation (OzID),24–26 and ultraviolet photodissociation (UVPD).27,28 All these developments greatly expand the toolbox for lipid analysis; more importantly, they not only provide first-time analysis of lipidomes at the isomer level11,19,28–30 but also demonstrate the link between altered isomer compositions with disease quantitatively and spatially.31–34
Radical-directed fragmentation has attracted increasing attention in MS/MS method development due to its unique capabilities in differentiating structural isomers of biomolecules, which otherwise cannot be obtained from charge-directed fragmentations typically seen from CID.35,36 A common approach to induce radical-directed fragmentation from even-electron precursor ions, the most common ionic form derived from electrospray ionization (ESI), is to initiate homolytic cleavage of a covalent bond within the ion. Homolytic bond cleavage has been demonstrated for lipid analysis using photons,35,37 electrons,38 and radicals.39 Reid and co-workers, however, showed that it was possible to induce radical-directed fragmentation from CID of anionic adducts of PC ions, such as methylcarbonate (CH3OCO2−) and trifluoroacetate (CF3CO2−).40 We were thus motivated to explore radical-direct fragmentation via CID for PC analysis with a focus on evaluating its potential for differentiating sn-isomers. Although CID is not selective for inducing homolytic bond cleavages of unmodified bimolecular ions, we value its wide accessibility in commercial MS instruments. Such a method once developed would be MS platform independent and compatible with common existing lipidomic workflows.
In this work, we discovered that the bicarbonate anion (HCO3−) was bound strongly with the phosphocholine head group of PCs; thus, it facilitated homolytic bond cleavage (C–N bond) and formation of radical ions upon CID. These radical ions opened up an sn-specific fragmentation channel, forming diagnostic ions critical for identification and quantitation of sn-isomers. The MS/MS method allowed us to develop an LC-MS/MS workflow for high-throughput mapping of PCs within a lipidome at high sensitivity and coverage down to sn-positions and CC locations, the latter of which was accomplished from incorporating PB-MS/MS. The workflow was evaluated by analyzing a polar lipid extract from bovine liver, with 82 distinct PC molecular species being identified, including 19 pairs of sn-isomers. It was further applied to profiling changes of PCs in cancerous human breast tissue. Five pairs of PC sn-isomers were found to exhibit significant changes, suggesting that PC lipogenesis was altered in human breast cancer tissue.
Results and discussion
MS/MS of bicarbonate adducts of PC anions via CID
In conventional lipid analysis workflows, PCs need to be analyzed in the form of anion adduct ions ([M + A]−) in MS/MS to determine the composition of fatty acyl/alkyl, with acetate (CH3COO−) and formate (HCOO−) being the most commonly employed anions. MS2 CID of PCs in negative ion mode can resolve sn-isomers due to a higher preference to lose sn-2 relative to the sn-1 chain as neutral ketene; however it suffers from low sensitivity due to weak binding between the quaternary ammonium headgroup and the anion.41 In contrast, the bicarbonate anion (HCO3−) formed a strong adduct with the PC and thus led to more sensitive detection. Fig. 1a compares the nanoESI-MS spectra of PC 16:0/18:1 (2 μM) in negative ion mode derived from solutions containing 5 mM NH4HCO3 and NH4COOH, respectively. The [M + A]− ion signal from the bicarbonate adduct (m/z 820) is about 20 times higher than that of the formate adduct (m/z 804).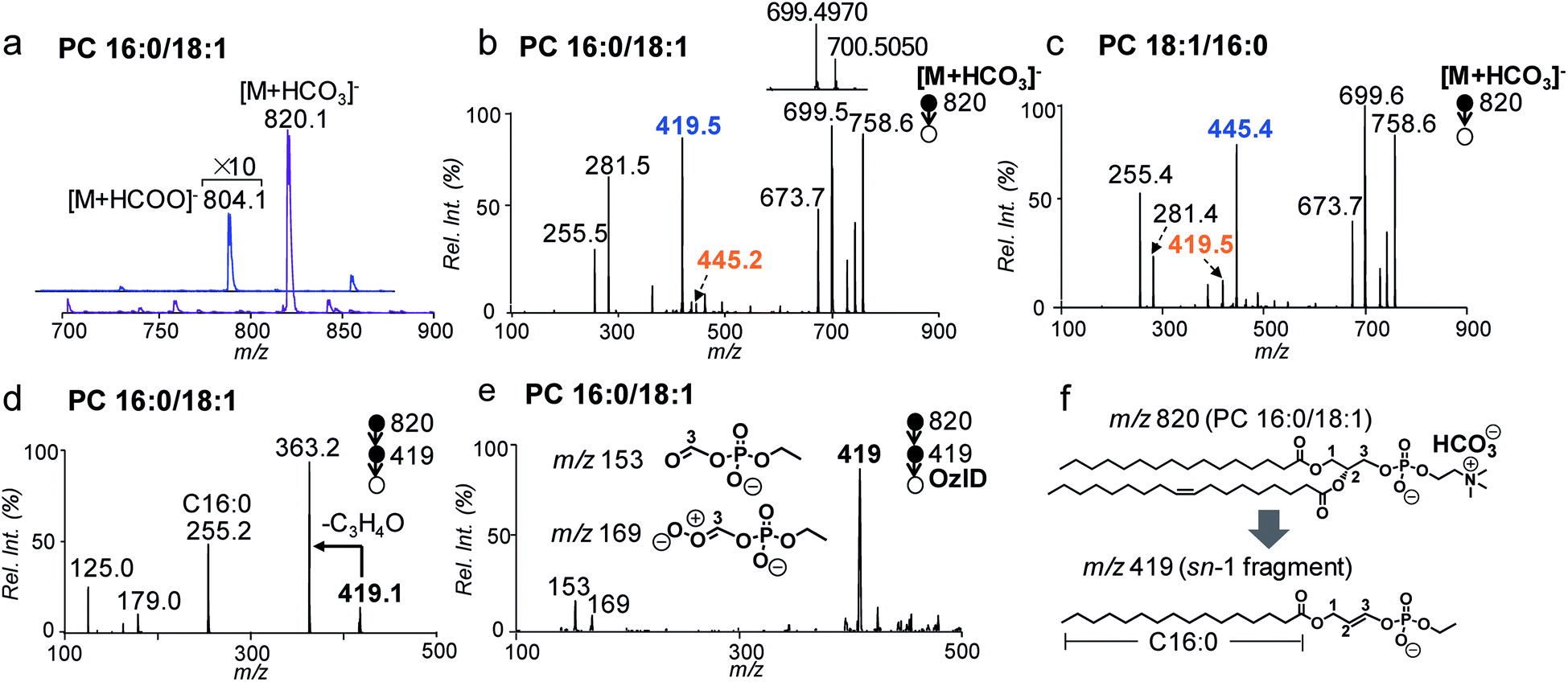 | ||
| Fig. 1 (a) Negative ion mode nanoESI-MS1 spectra of PC 16:0/18:1 (2 μM) from solutions containing 5 mM ammonium bicarbonate (NH4HCO3) and ammonium formate (NH4COOH). (b) MS2 CID of [M + HCO3]− of PC 16:0/18:1 (m/z 820), CE = 30 eV. Inset: zoomed-in spectrum of fragments in m/z 699–701. (c) MS2 CID of [M + HCO3]− of PC 18:1/16:0 (m/z 820), CE = 30 eV. (d) MS3 CID of fragment ions at m/z 419 from (b). (e) MS3 OzID spectrum of m/z 419 from PC 16:0/18:1. (f) Proposed structure of the sn-1 fragment m/z 419 derived from CID of [M + HCO3]− of PC 16:0/18:1. | ||
MS2 CID of the bicarbonate adduct ions ([M + HCO3]−, m/z 820, Fig. 1b) produced several abundant fragments in the higher m/z region due to the loss of H2CO3 (−62 Da, m/z 758), combined losses of H2CO3 and (CH3)3N (−121 Da, m/z 699), and loss of HCO3−˙(CH3)3N+ = CHCH2 (−147 Da, m/z 673). In the lower m/z region, fatty acyl anions, i.e. C16:0 (m/z 255) and C18:1 (m/z 281), were detected at lower relative abundances. The above fragmentation phenomenon is consistent with the main dissociation channels observed from CID of anion adducts of PCs reported by Reid and co-workers.40 The main difference is that the fragment ions due to neutral loss of fatty acyl as ketene (NLK), signature from CID of formate anions of PCs, were not detected; instead, a new peak at m/z 419 was formed at relatively high abundance. The above set of data suggests that bicarbonate interacts with PCs differently as compared to formate and thus significantly alters the unimolecular dissociation chemistry of the PC anion adduct ions upon collisional activation (Fig. S1†).
We further examined MS2 CID of the bicarbonate anion adduct of PC 18:1/16:0 (m/z 820, CE = 30 eV, Fig. 1c), the sn-isomer of PC 16:0/18:1. The two spectra were similar to each other, except that the peak at m/z 419 was much smaller, while the peak at m/z 445 was abundant. These differences indicate that fragment ions at m/z 419 or 445 might be correlated with the sn-positions of fatty acyls, which would be useful for structural determination. To characterize the identity of this new type of fragment ion, multi-stage tandem mass spectrometry, accurate mass measurements, and stable-isotope labeling were employed. Using the fragment peak at m/z 419 derived from PC 16:0/18:1 as an example, MS3 CID of this peak produced sn-1 fatty acyl anions at m/z 255 (C16:0), suggesting that the sn-1 chain is preserved in the fragment (Fig. 1d and possible fragmentation pathways are shown in Scheme S1†). Based on the chemical formula derived from this ion (C21H40O6P, Fig. S2†), a double bond or a ring structure should be generated. This fragment was subjected to OzID on a home-built linear ion trap mass spectrometer for structural elucidation (experimental details are provided in the ESI†). Two OzID fragments at m/z 153 and m/z 169 were observed (Fig. 1e), consistent with predicted ozonolysis products from a structure containing a double bond at C2–C3 of the glycerol backbone. Thus, a structure is proposed for the fragment ion m/z 419 (Fig. 1f), in which the sn-1 fatty acyl is connected to a dehydro-glycerol backbone while the C3-hydroxyl is esterified by ethyl phosphate. For simplicity of description, this type of ion is named as the sn-1 fragment, emphasizing that the sn-1 fatty acyl is preserved in such structures.
A comparison of the sn-1 fragment to the precursor ion (Fig. 1f) suggests that it cannot be generated by simple cleavages; instead, sequential fragmentation should be involved. We performed MS3 CID of all major fragments within the m/z range 600–800 formed from MS2 CID of the bicarbonate adduct of PC 16:0/18:1. The only one that produced m/z 419 as a major fragment in MS3 CID was a radical ion at m/z 700.5 (data are provided in Fig. S3†). This species was formed at appreciable abundance right beside the fragment ion m/z 699.4970 when analyzed on a Q-TOF mass spectrometer (inset of Fig. 1b). Accurate mass measurement (m/z 700.5050) suggested an elemental composition of C39H73O8P−˙ (relative error: 1.8 ppm), likely resulting from combined losses of H2CO3 and the N,N-dimethyl aminomethyl radical ((CH3)2N–CH2˙) from the precursor ion ([PC + HCO3]−, C43H83NO11P). This process should generate a carbon centered radical ion. Indeed, when this type of radical ion was allowed to react with residual oxygen in the ion trap for 200 ms, a peak corresponding to O2 addition was clearly detected (data are provided in Fig. S4†). This ion/molecule reaction phenomenon is similar to those reported for carbon-centered peptide radical ions.42
A possible fragmentation pathway for forming the sn-1 fragment from CID of [PC + HCO3]− anions is proposed in Scheme 1. Firstly, the bicarbonate anion in the ion complex abstracts a methyl proton in choline and leaves as H2CO3, which is supported by MS2 CID of the bicarbonate adduct of d9-PC 16:0/16:0 (all nine choline hydrogen atoms are labeled with deuterium, Fig. S5†). The process is followed by homolytic cleavage of the C–N bond leading to a neutral loss of (CH3)2N–CH2˙. These two losses (120 Da) collectively lead to the formation of an ethyl radical on the remaining lipid anion (C39H73O8P˙ shown in Scheme 1). The ethyl radical then abstracts C3–H on the glycerol backbone; subsequent β-cleavage gives rise to neutral loss of sn-2 fatty acyl as ˙R2 and CO2 (ref. 43) and forms a double bond between C2 and C3 on the glycerol backbone. Given that this radical fragmentation channel is not observed from CID of formate or acetate adduct anions of PCs, we hypothesize that the –OH group in bicarbonate may form a relatively strong hydrogen bond with phosphate oxygen in PC molecules (possible interaction is indicated in Scheme 1). This hydrogen bonding may be critical to stabilize the ion complex and facilitate radical-directed fragmentation. Given the scope of this work, we did not persue molecular modeling to define the transition states involved in each fragmentation step; therefore, Scheme 1 should be viewed as a simplified pathway for the formation of the sn-1 fragment ion. An alternative fragmentation pathway involving the zwitterion intermediate is suggested in Scheme S2,† which might be favorable for lowering the barrier to homolysis of the C–N bond.
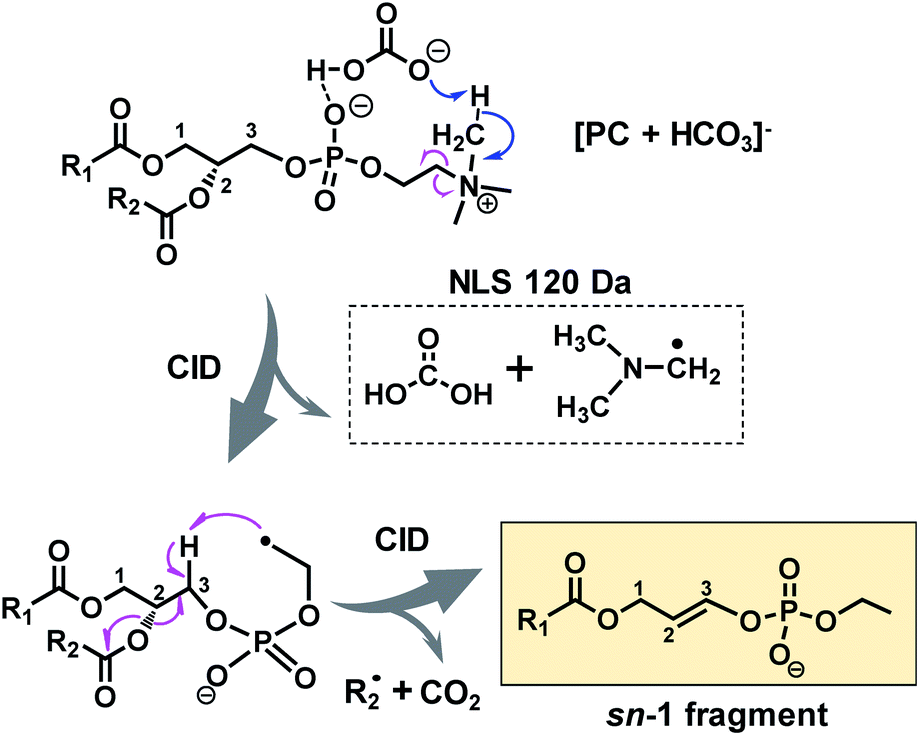 | ||
| Scheme 1 Radical-directed fragmentation pathway for forming the “sn-1 fragment” from CID of bicarbonate adducts of PCs. | ||
It is worth pointing out that the radical-directed fragmentation is sn-specific. Although ions at m/z 445 and m/z 419 were each detected at low abundances in CID of PC 16:0/18:1 (Fig. 1b) and PC 18:1/16:0 (Fig. 1c), they were later proved to be the “sn-1 fragments” resulting from corresponding sn-isomers, which are present as minor impurities in the supplied samples. We also note that this radical-directed fragmentation channel is promoted under higher energy CID conditions. For instance, no sn-1 fragments could be detected under on-resonance dipolar activation conditions (so-called ion trap CID) (Fig. S6†). This trend was observed for a series of PCs regardless of the chain lengths or the number of CCs in fatty acyls (Fig. S7†). This aspect further supports that strong H bonding between bicarbonate and the phosphate choline headgroup in the PC is key for the ions to access higher energy of activation under CID, which promotes the radical-directed fragmentation as compared to low-energy CID conditions.
Identification and quantitation of PC sn-isomers
The specificity of producing sn-1 fragments from CID of bicarbonate adducts of PCs provides a possibility of distinguishing and quantifying sn-isomers. Being aware of that the commercially obtained PC standards contain sn-isomers as minor impurities, we first measured the purity (% mol) of PC standards using the gold-standard, PLA2 hydrolysis method. For all samples tested, the sn-purity% falls in the range of 80–95%, agreeing well with previous reports.25,27 For instance, the sn-purity of the PC 16:0/18:1 sample is 93 ± 2%, meaning that it contains 7 ± 2% PC 18:1/16:0. This measurement also explains the detection of a low abundance peak at m/z 445 from the PC 16:0/18:1 sample (Fig. 1b), which is actually the sn-1 fragment derived from PC 18:1/16:0. The same discussion applies for a small peak at m/z 419 from the PC 18:1/16:0 sample (Fig. 1c), which contains 13 ± 2% sn-isomers. Using the purity information, we prepared a series of standard solutions containing different molar compositions of sn-isomers and subjected them to the analysis by MS2 CID of [M + HCO3]−. Fig. 2a shows data acquired from the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of PC 16:0/18:1 and PC 18:1/16:0 (total concentration kept at 10 μM). The relative ion abundance of the sn-1 fragment of PC 18:1/16:0 (m/z 445) was normalized to the sum of sn-1 fragments of both isomers and plotted against corresponding concentrations (% mol) (Fig. 2b). An excellent linear relationship (R2 = 0.9984) with a slope close to unity (1.02 ± 0.01) was achieved. Such a linear correlation was consistently observed for all six PC standards measured (Fig. S8†). Furthermore, the average % deviation of purities measured by the sn-1 fragment from that of the PLA2 method was within ±2% for all six PC standards (Fig. 2c and Table S1†). These values compare favorably with sn-isomer quantitation reported by Ekroos et al. via MS3 CID of [M + HCO2]− of PCs, which shows a consistent negative deviation in the range of −1% to −7% (Table S2†).41 The small system deviation associated with the sn-1 fragment ion method suggests that the relative ion abundance of the sn-1 fragment measured in a MS/MS spectrum can be directly used for quantitation of sn-isomers.
:1 mixture of PC 16:0/18:1 and PC 18:1/16:0 (total concentration kept at 10 μM). The relative ion abundance of the sn-1 fragment of PC 18:1/16:0 (m/z 445) was normalized to the sum of sn-1 fragments of both isomers and plotted against corresponding concentrations (% mol) (Fig. 2b). An excellent linear relationship (R2 = 0.9984) with a slope close to unity (1.02 ± 0.01) was achieved. Such a linear correlation was consistently observed for all six PC standards measured (Fig. S8†). Furthermore, the average % deviation of purities measured by the sn-1 fragment from that of the PLA2 method was within ±2% for all six PC standards (Fig. 2c and Table S1†). These values compare favorably with sn-isomer quantitation reported by Ekroos et al. via MS3 CID of [M + HCO2]− of PCs, which shows a consistent negative deviation in the range of −1% to −7% (Table S2†).41 The small system deviation associated with the sn-1 fragment ion method suggests that the relative ion abundance of the sn-1 fragment measured in a MS/MS spectrum can be directly used for quantitation of sn-isomers.
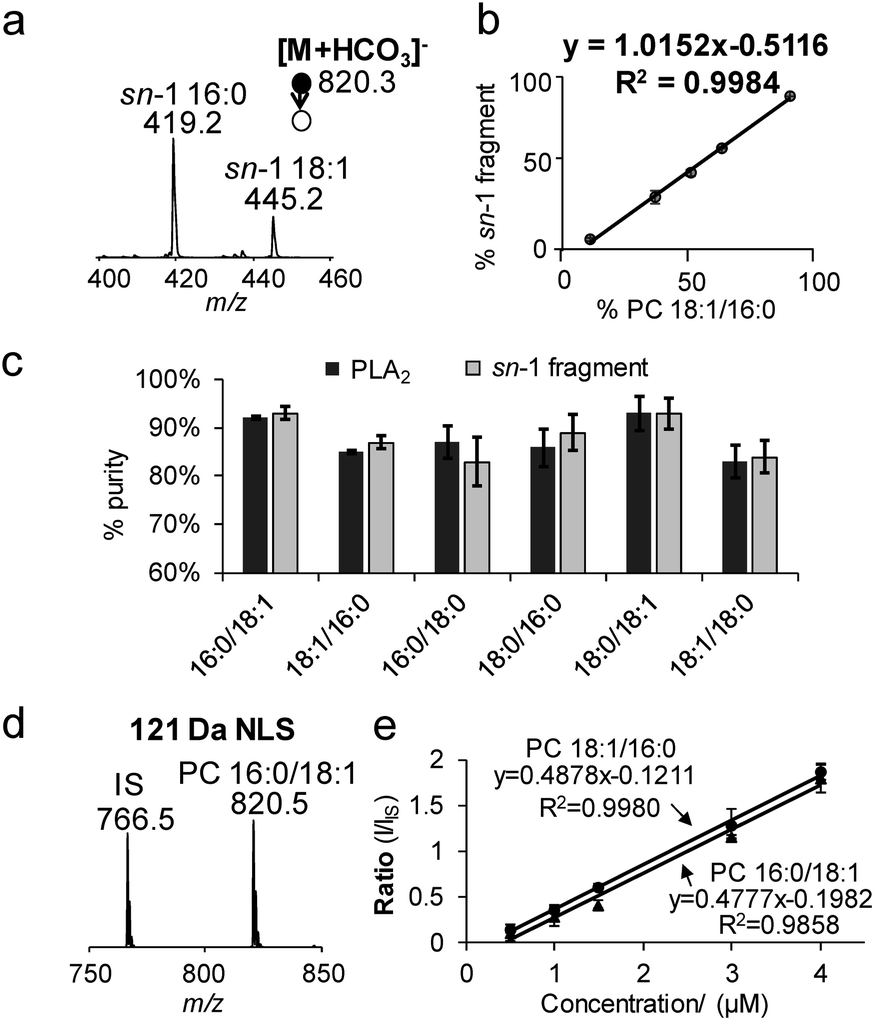 | ||
| Fig. 2 (a) MS2 CID spectrum of [M + HCO3]− derived from a 2:1 mixture of PC 16:0/18:1 and PC 18:1/16:0. Only the m/z region of sn-1 fragment ions is shown. (b) Correlations between % sn-1 18:1 ions (I445/(I445 + I419) ×100) as a function of mol% of PC 16:0_18:1 with Ctotal kept at 10 μM. (c) Comparisons of the purity of six PC standards measured by the sn-1 fragment and PLA2 method. (d) 121 Da NLS of a mixture containing 3 μM PC 16:0/18:1 and 2.5 μM IS, PC 15:0/15:0. (e) Quantitation for PC 16:0/18:1 and PC 18:1/16:0 from 121 Da NLS with IS (PC 15:0/15:0) kept at 0.5 μM. | ||
121 Da NLS for profiling and quantitation of PCs
Neutral loss of 121 Da (combined losses of H2CO3 and N(CH3)3) was a facile fragmentation channel from CID of PC bicarbonate adduct anions. It also showed high specificity to the choline head group as compared to other fragments, i.e. loss of 62 Da (H2CO3); therefore, it was tested for profiling of PCs from complex mixtures. The limit of detection (LOD) was estimated to be 10 pM from 121 Da NLS of the bicarbonate adduct of PC 16:0/18:1, based on a signal-to-noise ratio (S/N) greater than 3 (Fig. S9a†). Although this LOD was about 5 times higher than that obtained from PIS m/z 184 (phosphocholine head group ion) of protonated PC 16:0/18:1 (Fig. S9b†), it was significantly more sensitive than the use of formate or acetate anion adduct ions (Fig. S9c†).Neutral loss of 121 Da (Fig. 2d) also allowed achieving quantitation of PCs and their sn-isomers in a simple fashion. Fig. 2e compares calibration curves resulting from 121 Da NLS for PC 16:0/18:1 and PC 18:1/16:0 with PC 15:0/15:0 used as the internal standard (IS, 0.5 μM). Besides the excellent linearity of each plot, one most important feature is that the calibration curves of the two sn-isomers almost overlay with each other. These results suggest that the sn-positions of fatty acyls do not have a significant impact on the 121 Da NLS fragmentation channel for PCs. Moreover, hypothesis testing of the covariances and slopes of the two calibration curves suggest that they are not statistically different (testing details are provided in the ESI†). Thus, a calibration curve generated from 121 Da NLS of any sn-isomer can be used for quantitation of the sum concentration of a mixture of the two sn-isomers, greatly simplifying the overall procedure. Once the total quantity of the sn-isomers is found, quantitation of the isomers can be obtained by measuring their corresponding % sn-1 fragment as described earlier in Fig. 2b. We also noticed that the identity of the fatty acyl chains in PCs, i.e. length or degrees of unsaturation, had an impact on ion response in 121 Da NLS (spectrum of an equimolar mixture of PC 15:0/15:0, PC 16:0/18:0, and PC 18:0/20:4 is shown in Fig. S10†). Therefore, calibration curve based on 121 Da NLS should be established for each individual PC species.
Pinpointing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C locations in sn-specific fatty acyl chains of PCs
C locations in sn-specific fatty acyl chains of PCs
The Paternò–Büchì reaction coupled with subsequent MS/MS (PB-MS/MS) allows for pinpointing CC locations in fatty acyls of PCs; however, it cannot differentiate sn-positions.18,19 Therefore, we tested pairing CID of bicarbonate adducts of PCs with PB-MS/MS for the determination of CCs in sn-1 fatty acyls of PCs. The presence of ammonium bicarbonate did not have an obvious impact on the conversion rates of the PB reactions nor did the PB reactions show any preference to sn-positions (Fig. S11†). The conversions typically ranged between 30% and 40% within 10 s, similar to that obtained without bicarbonate addition. Using PC 18:1/16:0 as a model compound (10 μM, 50/50, v/v, acetone/H2O with 5 mM NH4HCO3), the solution was subjected to offline PB reaction and collected for nanoESI in negative ion mode (experimental details are provided in the ESI†). The PB product at m/z 878 ([PBM + HCO3]−) can be clearly detected (Fig. 3a). MS2 CID of the PB product (m/z 878) (Fig. 3b) produced the PB modified sn-1 fragment at m/z 503, which had a characteristic mass increase of 58 Da due to acetone addition to the intact sn-1 fragment (m/z 445). MS3 CID of m/z 503 produced the PB modified C18:1 at m/z 339 (Fig. 3c). Further activation (MS4 CID) of this ion produced CC diagnostic ions at m/z 171 and 197, providing definitive evidence that CC is located at Δ9 of the sn-1 chain (C18:1) (Fig. 3d).
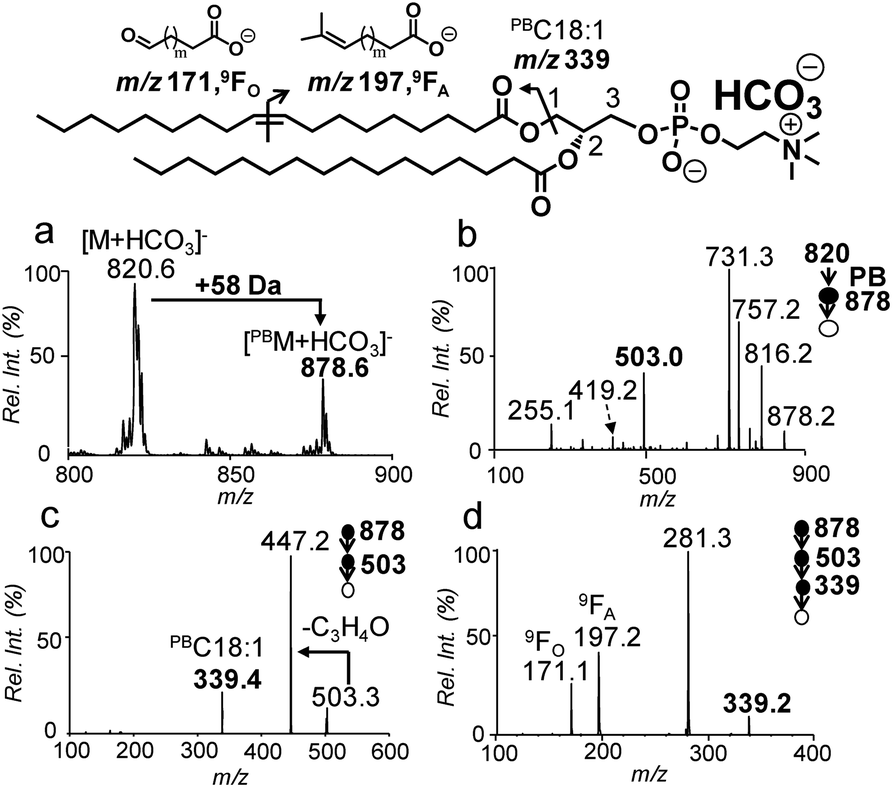 | ||
| Fig. 3 (a) PB-MS of 10 μM PC 18:1/16:0 after 8 s UV irradiation. (b) PB-MS2via beam-type CID of ions at m/z 878. (c) PB-MS3 ion trap CID of ions at m/z 503 from (b). (d) PB-MS4 ion trap CID of [PBC 18:1]− (m/z 339) from (c). | ||
An LC-MS/MS workflow for the analysis of PCs down to sn- and CC positions
The bicarbonate chemistry can be readily incorporated into an LC-MS/MS system. The platform consisted of HILIC-ESI-MS, in which an online photochemical reactor was installed post-column and right before ESI-MS.30 The acetone/ACN/H2O mobile phase system was used with NH4HCO3 added as buffer to assist the formation of anion adducts of PCs. Regarding lipid separation, use of NH4HCO3 as buffer in the mobile phase showed very similar performance to use of NH4HCO2 as buffer; however, detection of PCs in negative ion mode was significantly improved in the former case. Such a comparison can be found in Fig. S12,† using a polar lipid extract of bovine liver as an example. Data collection consisted of three LC-MS/MS runs (Fig. 4a). The first run employed 121 Da NLS in negative ion mode for PC subclass identification and relative quantitation. This run also provided a list of precursor m/z values of PCs ([M + HCO3]−), which was used to guide the second LC-MS/MS run in a targeted fashion. The second run (MS2 CID of [M + HCO3]−) led to identification of PCs for fatty acyl composition, sn-position, and sn-isomer composition. Based on the above information, a list of precursor ions of unsaturated PCs was generated for the last run, LC-PB-MS2 CID, for CC location assignment. Note that we choose to perform LC-PB-MS2 CID in positive ion mode because it is more sensitive on chromatographic timescale as compared to performing PB-MS3 or MS4 CID in negative ion mode. Consequently, the CC location cannot be determined with sn-specificity. However, with known fatty acyl and sn-composition information, the CC location can be assigned for PCs which do not contain major fatty acyl or sn-isomers. If the sn-1 unsaturated fatty acyl needs to be characterized, PB-MS4 CID in negative ion mode needs to be performed in separate runs.
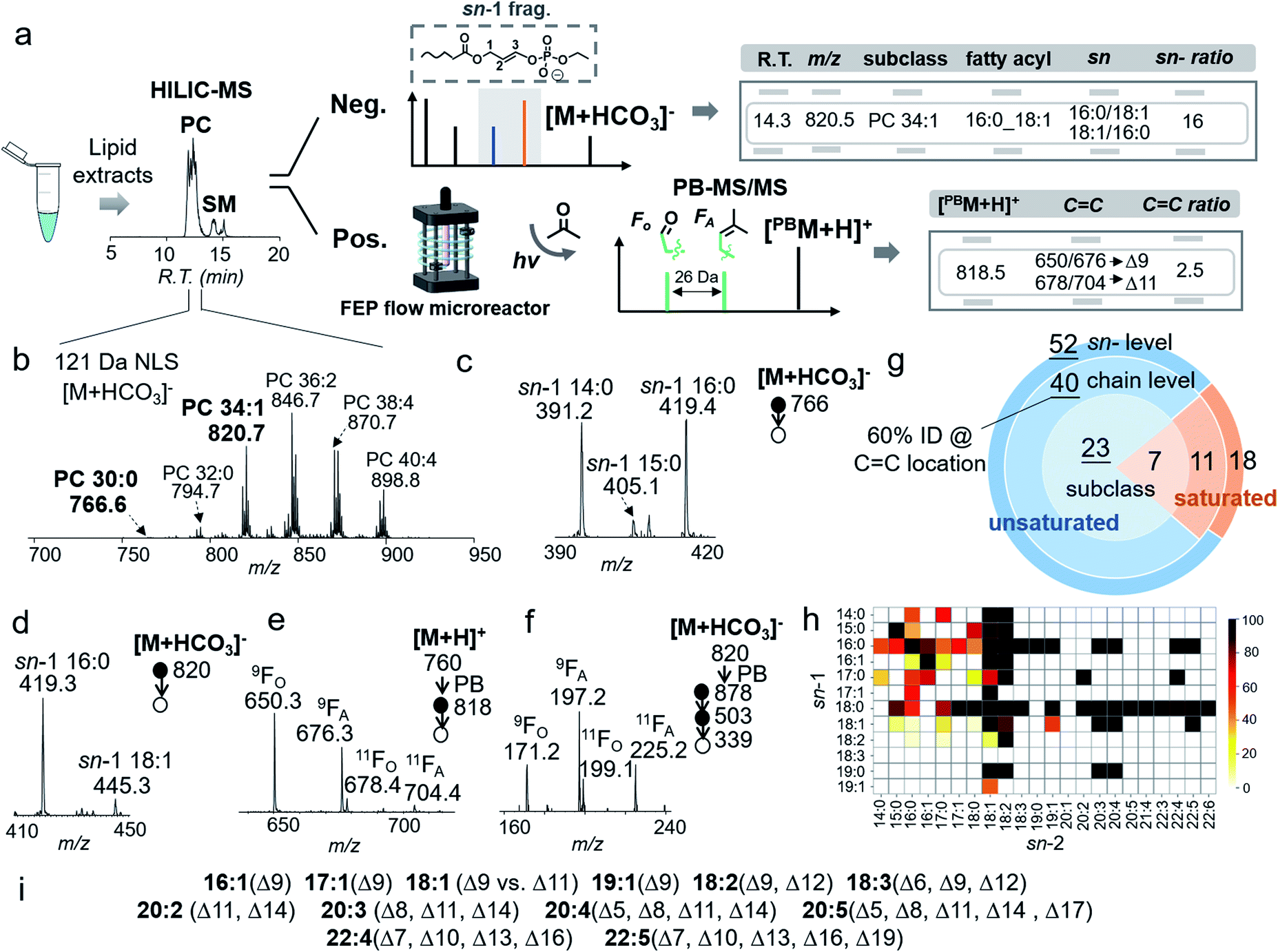 | ||
| Fig. 4 (a) A HILIC-MS workflow for structural identification of PC in the bovine liver extract. (b) NLS 121 Da of [M + HCO3]− of PCs from retention time 10.5–12.0 min in (a). MS2 CID of [M + HCO3]− of (c) PC 30:0 (m/z 766) and (d) PC 34:1 (m/z 820). (e) PB-MS2 CID of PC 34:1 (m/z 818) in positive ion mode. (f) PB-MS4 CID of [PBC 18:1]− derived from PC 18:1/16:0. (g) The numbers of identified PCs from the bovine liver extract at different structural levels. (h) Heatmap of fatty acyls at sn-1 and sn-2 positions of identified PCs. (i) Identified CC locations in unsaturated fatty acyls of PCs. | ||
The analytical performance of the system was evaluated using a polar lipid extract from bovine liver (0.4 μg lipid injection per run). HILIC provided separations of PCs from other classes of lipids (i.e., PEs and SMs), while 121 Da NLS in negative ion mode provides the profile of PCs ([M + HCO3]−), leading to identification of 30 PCs at the subclass level (Fig. 4b). The coverage is the same as using PIS m/z 184 Da (Fig. S13†). Targeted MS/MS via beam-type CID identified PCs with the fatty acyl composition and sn-position. Taking PC 30:0 ([PC + HCO3]−, m/z 766) as an example, albeit at relatively low ion abundance (0.5%, normalized to PC 36:2, the most abundant PC in the bovine liver extract), HILIC-MS2 CID produced high quality data for structural characterization. In the m/z region of sn-1 fragment ions (Fig. 4c), detection of ions at m/z 391 (sn-1 14:0), m/z 405 (sn-1 15:0), and m/z 419 (sn-1 16:0) proved that PC 30:0 contained three molecular species: PC 14:0/16:0, PC 16:0/14:0, and PC 15:0/15:0. The composition of the three species can be deduced as 47%, 47%, and 6%, respectively, according to the corresponding fractions of ion abundances.
HILIC-PB-MS/MS allowed determination of the CC location in unsaturated PCs, which accounted for more than 80% of the identified PCs. Using PC 34:1 as an example, MS2 CID of [M + HCO3]− (m/z 820) suggested that it contained 94% PC 16:0/18:1 and 6% PC 18:1/16:0 (Fig. 4d). By applying the quantitation procedure described earlier, PC 16:0/18:1 was determined to be 1.7 ± 0.1 ng/(0.4 μg extract), while PC 18:1/16:0 was 0.1 ng/(0.4 μg extract). Given that PC 16:0/18:1 was the dominant component (94%), HILIC-PB-MS2 CID in positive ion mode readily provided CC location information, viz. Δ9 (major) and Δ11 (minor) for C18:1 at the sn-2 position (Fig. 4e). In order to pinpoint the CC location of the minor sn-isomer (4%), PB-MS4 CID was conducted. The zoomed-in region in Fig. 4f shows two pairs of CC diagnostic ions (m/z 171, 197; m/z 199, 225) of C18:1, suggesting that PC 18:1/16:0 contains two CC isomers, PC 18:1(Δ9)/16:0 and PC 18:1(Δ11)/16:0. Combining all the above information, it is evident that PC 16:0_18:1 contains four distinct molecular species, including two sn-isomers and two CC location isomers of the C18:1. This level of detailed structural information would not be available from the conventional analysis workflow relying just on CID.
Using the workflow described above, 70 PCs were identified for sn-positions, including 19 pairs of sn-isomers (Fig. 4g). This is a significant increase as compared to 51 PCs identified at the fatty acyl level due to the improved capability of structural identification. Fig. 4h shows the heatmap of identified PCs down to sn-positions; the color codes represent mol% of sn-isomers within the pair. It is evident that fatty acyls at the sn-1 position show lower diversity than those at the sn-2 position, with C16:0 and C18:0 as two high frequency groups. Although it has been suggested that the sn-2 position is preferred by polyunsaturated acyl chains,29,44 our data quantitatively show that many of them still consist of minor sn-isomers, such as PC 20:4/16:0 (4%) and PC 22:4/18:0 (9%). For unsaturated PCs, about 60% were identified for CC locations. The results for CC determination in unsaturated fatty acyls are summarized in Fig. 4i. The most frequent CC isomers are Δ9 and Δ11 isomers of C18:1 fatty acyl, while PUFAs are dominant by either ω-6 or ω-3 CC locations, which are consistent with a previous report.30 Counting identified CC location isomers, a total of 82 PC molecular species were identified. The complete identification list is provided in ESI Data 1.†
Analysis of isomeric PCs from human breast cancerous tissue
Several recent MS imaging studies clearly demonstrated that lipid isomers, including fatty acyl compositional isomers, sn-, and CC location isomers, were distributed heterogeneously in tissue sections or among different types of tissues, likely carrying distinct functions.31–33,45 In an earlier study, we reported that changes of CC location isomers were more sensitive to pathological conditions than lipid profile changes.19 In this study, we were interested to test if changes of PC sn-isomers would differentiate lipidomes of diseased and healthy states, which may be potentially used as markers in biomedical and clinical studies.46 Using the HILIC-LC-MS workflow described earlier, we thus evaluated this idea by analyzing PCs in cancerous human breast tissue samples (N = 3), while para-carcinoma tissue samples (N = 3) were used as controls (three technical repeats were acquired for each sample). Four levels of structural and quantitative analysis were performed for PCs, including subclass, fatty acyl composition, sn-composition, and CC location. At the subclass level, 42 PC species were identified from NLS 121 Da of their bicarbonate adduct anions (Fig. S14a†). Among these, only PC 32:1 and PC 34:2 showed significant changes in relative ion abundances (IS: PC 15:0/15:0). This result was consistent with previous findings in which relative quantitation at the subclass level was obtained from PIS m/z 184.30
Application of HILIC-MS/MS led to identification of 80 PCs down to fatty acyl compositions and sn-positions (complete identification list is in ESI Data 2†). The identified PCs were found the same between normal and cancerous tissue, while their relative abundances and sn-isomer compositions varied. Among 26 pairs of PC sn-isomers, five showed significant changes in isomeric ratios (P < 0.001, average RSD = 22 ± 11%). These include PC 16:0_18:1, PC 16:0_16:1 (data are provided in Fig. S14b†), PC 15:0_18:1, PC 16:0_17:1, and PC 16:0_17:0. For instance, PC 16:0_17:0 was found to exhibit significant changes in sn-ratios (P = 4.6 × 10−7), accompanied by a large increase of PC 16:0/17:0 (65%) in cancerous tissue relative to control (15%) (Fig. 5d). Note that subclass profiling is blind to isomers and thus cannot differentiate fluctuations of each isomer when the summed abundances of the isomer pair are not significantly altered (Fig. 5a). Moreover, because PC 16:0_17:0 is a saturated lipid, it cannot be analyzed for CC location isomers. A more challenging example is given by PC 33:1 (Fig. 5b). Beam-type CID revealed that it contained two fatty acyl compositional isomers: PC 16:0_17:1 (major) and PC 15:0_18:1 (minor). The compositional changes of the C18:1 Δ9 and Δ11 isomers of PC 15:0_18:1 were not statistically different between the normal and cancerous tissue samples.30 In contrast, sn-position analysis revealed that both PC 16:0_17:1 and PC 15:0_18:1 contained sn-isomers and each pair of isomers showed significant changes between cancerous and normal breast tissues (Fig. 5b, e, and 5f). There were also cases that sn-isomers and CC location isomers were both altered in cancerous tissue samples. PC 16:0_18:1 was one of the most abundant species in breast tissue and it was identified to consist of sn-isomers and CC location isomers (Δ9 vs. Δ11 in C18:1). Relative to the control, the major sn-isomer PC 16:0/18:1 and the CC location isomer C18:1(Δ9) were found to increase significantly in the cancerous tissue samples (P = 3.5 × 10−7 and P = 2 × 10−4, respectively, Fig. 5c, g and 5h). Notably, the same pair of sn-isomers was found to distribute distinctly in the tumor region relative to surrounding tissue in a mouse brain section, as recently revealed by OzID MALDI-imaging31 and DESI-UVPD-imaging.33,45 Our data clearly suggest that PC 16:0_18:1 may contain four structural isomers from combinations of two sn-isomers and two CC location isomers of the C18:1. Quantitative comparison of each individual isomer, however, is challenging for the current workflow because CC isomeric ratios of the sn-2 fatty acyl chain cannot be measured independently. Pham et al. demonstrated monitoring of relative compositions of the same four isomers of PC 16:0_18:1 individually from MS/MS experiments containing different sequences of OzID and CID.26 Another interesting aspect unveiled from sn-isomer measurements is that for the sn-isomers which demonstrate significant changes in cancerous tissue they all showed selective enrichment of sn-isomers containing unsaturated or odd carbon number fatty acyls at sn-2 positions. Although this trend needs to be verified with larger sampling size, it provides a new insight into the lipogenesis of PCs which is dynamically regulated by de novo synthesis and remodeling processes.47,48 Collectively, the capability of monitoring changes of a large number of sn-isomers enables more sensitive detection of changes of PCs in diseased tissue samples, which is highly complementary to data obtained from subclass analysis (Fig. S14a†) or CC location isomer analysis (Fig. S14c†).
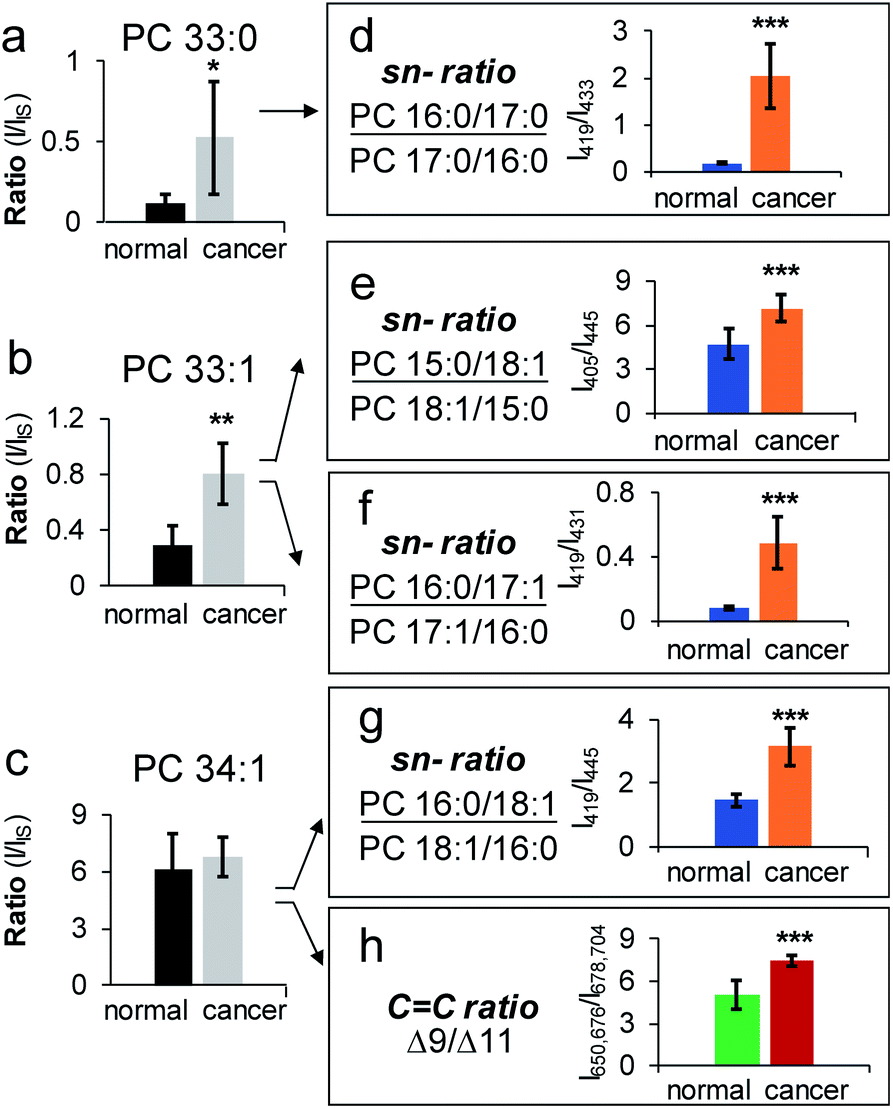 | ||
| Fig. 5 Analysis of isomeric PCs in human breast tissue samples, cancerous vs. normal. Relative quantitation of (a) PC 33:0, (b) PC 33:1, and (c) PC 34:1 at the subclass level. Changes of sn-isomeric ratios of (d) PC 16:0_17:0, (e) PC 15:0_18:1, (f) PC 16:0_17:1, and (g) PC 16:0_18:1. (h) Relative ratio changes of CC location isomers (Δ9/Δ11) of PC 16:0_18:1. Differences between the two groups of samples were evaluated for statistical significance using the two-tailed student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001). Error bar represents ± s.d. (N = 3). | ||
Conclusions
In this study we have developed an LC-MS/MS workflow for identification and quantitation of PCs down to sn-positions with high lipid coverage and sensitivity (10 pM LOD). This unique capability is enabled by radical-directed fragmentation, forming sn-1 specific fragment ions upon CID of the bicarbonate anion adducts of PCs ([M + HCO3]−). It is worth noting that identities of fatty acyls, their sn-positions, and the quantity of sn-isomers can all be directly obtained from a single MS2 CID spectrum. More importantly, PB-MS/MS can be merged into the LC-MS workflow, allowing determination of CC locations in unsaturated fatty acyls of PCs. Using a polar lipid extract from bovine liver as a model system, 82 molecular species of PCs have been identified for sn-positions while a majority of unsaturated PCs have been identified for the locations of CCs. The capability of quantifying a variety of sn-isomers of PCs is an important addition for lipid biomarker discovery and it is highly complementary to monitoring compositional changes of CC location isomers. Five pairs of PC sn-isomers were found to exhibit significant changes from human breast cancer tissue relative to the control. Interestingly, the changes of isomers showed a preference to sort unsaturated fatty acyls and odd numbers of carbons at the sn-2 position, which might be linked with altered PC biosynthetic pathways in human breast cancer. Regarding accessibility, the LC-MS/MS workflow should be highly compatible with existing LC and shotgun lipid analysis workflows, because it only requires an addition of ammonium bicarbonate to either the mobile phase or the lipid extract. Limited by the mechanism of forming sn-1 specific fragment ions, lipids which do not contain phosphocholine headgroups cannot be analyzed for sn-positions. Nevertheless, the success for PC analysis highlights the potential of using radical-directed fragmentation for lipid isomer analysis by mass spectrometry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21722506 and No. 21621003) and NIH R01GM118484 is greatly appreciated.References
- K. Koichi, F. Michiya and N. Makoto, BBA, Biochim. Biophys. Acta, Lipids Lipid Metab., 1974, 369, 222–233 CrossRef.
- A. Koeberle, H. Shindou, S. C. Koeberle, S. A. Laufer, T. Shimizu and O. Werz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2546–2551 CrossRef CAS PubMed.
- M. V. Chakravarthy, I. J. Lodhi, L. Yin, R. R. Malapaka, H. E. Xu, J. Turk and C. F. Semenkovich, Cell, 2009, 138, 476–488 CrossRef CAS PubMed.
- H. Shindou and T. Shimizu, J. Biol. Chem., 2009, 284, 1–5 CrossRef CAS PubMed.
- H. Shindou, D. Hishikawa, T. Harayama, K. Yuki and T. Shimizu, J. Lipid Res., 2009, 50, S46–S51 CrossRef PubMed.
- E. Marien, M. Meister, T. Muley, T. G. del Pulgar, R. Derua, J. M. Spraggins, R. Van de Plas, F. Vanderhoydonc, J. Machiels, M. M. Binda, J. Dehairs, J. Willette-Brown, Y. L. Hu, H. Dienemann, M. Thomas, P. A. Schnabel, R. M. Caprioli, J. C. Lacal, E. Waelkens and J. V. Swinnen, Oncotarget, 2016, 7, 12582–12597 CrossRef PubMed.
- M. Mapstone, A. K. Cheema, M. S. Fiandaca, X. G. Zhong, T. R. Mhyre, L. H. MacArthur, W. J. Hall, S. G. Fisher, D. R. Peterson, J. M. Haley, M. D. Nazar, S. A. Rich, D. J. Berlau, C. B. Peltz, M. T. Tan, C. H. Kawas and H. J. Federoff, Nat. Med., 2014, 20, 415–418 CrossRef CAS.
- K. Yang and X. Han, Trends Biochem. Sci., 2016, 41, 954–969 CrossRef CAS.
- P. Zacek, M. Bukowski, T. A. Rosenberger and M. Picklo, J. Lipid Res., 2016, 57, 2225–2234 CrossRef CAS PubMed.
- M. Bhuiyan, D. Tucker and K. Watson, J. Microbiol. Methods, 2014, 105, 1–15 CrossRef CAS PubMed.
- D. L. Marshall, A. Criscuolo, R. S. E. Young, B. L. J. Poad, M. Zeller, G. E. Reid, T. W. Mitchell and S. J. Blanksby, J. Am. Soc. Mass Spectrom., 2019, 30, 1621–1630 CrossRef CAS.
- M. Wang, C. Wang, R. H. Han and X. Han, Prog. Lipid Res., 2016, 61, 83–108 CrossRef CAS PubMed.
- T. Hu and J. L. Zhang, J. Sep. Sci., 2018, 41, 351–372 CrossRef CAS PubMed.
- X. Han, K. Yang and R. W. Gross, Mass Spectrom. Rev., 2012, 31, 134–178 CrossRef CAS PubMed.
- K. Ekroos, I. V. Chernushevich, K. Simons and A. Shevchenko, Anal. Chem., 2002, 74, 941–949 CrossRef CAS PubMed.
- Y. H. Rustam and G. E. Reid, Anal. Chem., 2018, 90, 374–397 CrossRef CAS PubMed.
- T. Porta Siegel, K. Ekroos and S. R. Ellis, Angew. Chem., Int. Ed., 2019, 58, 6492–6501 CrossRef CAS PubMed.
- X. Ma and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 2592–2596 CrossRef CAS PubMed.
- X. Ma, L. Chong, R. Tian, R. Shi, T. Y. Hu, Z. Ouyang and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2573–2578 CrossRef CAS PubMed.
- Y. Feng, B. Chen, Q. Yu and L. Li, Anal. Chem., 2019, 91, 1791–1795 CrossRef CAS PubMed.
- Y. Zhao, H. Zhao, X. Zhao, J. Jia, Q. Ma, S. Zhang, X. Zhang, H. Chiba, S. P. Hui and X. Ma, Anal. Chem., 2017, 89, 10270–10278 CrossRef CAS PubMed.
- W. Cao, X. Ma, Z. Li, X. Zhou and Z. Ouyang, Anal. Chem., 2018, 90, 10286–10292 CrossRef CAS PubMed.
- J. L. Campbell and T. Baba, Anal. Chem., 2015, 87, 5837–5845 CrossRef CAS PubMed.
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, J. R. Nealon and S. J. Blanksby, Anal. Chem., 2008, 80, 303–311 CrossRef CAS PubMed.
- R. L. Kozlowski, T. W. Mitchell and S. J. Blanksby, Sci. Rep., 2015, 5, 9243 CrossRef CAS PubMed.
- H. T. Pham, A. T. Maccarone, M. C. Thomas, J. L. Campbell, T. W. Mitchell and S. J. Blanksby, Analyst, 2014, 139, 204–214 RSC.
- P. E. Williams, D. R. Klein, S. M. Greer and J. S. Brodbelt, J. Am. Chem. Soc., 2017, 139, 15681–15690 CrossRef CAS.
- D. R. Klein and J. S. Brodbelt, Anal. Chem., 2017, 89, 1516–1522 CrossRef CAS PubMed.
- T. Baba, J. L. Campbell, J. C. L. Blanc, P. R. S. Baker and K. Ikeda, J. Lipid Res., 2018, 59, 910–919 CrossRef CAS.
- W. Zhang, D. Zhang, Q. Chen, J. Wu, Z. Ouyang and Y. Xia, Nat. Commun., 2019, 10, 79 CrossRef.
- S. R. Ellis, M. R. L. Paine, G. B. Eijkel, J. K. Pauling, P. Husen, M. W. Jervelund, M. Hermansson, C. S. Ejsing and R. M. A. Heeren, Nat. Methods, 2018, 15, 515–518 CrossRef CAS.
- A. Bednarik, S. Bolsker, J. Soltwisch and K. Dreisewerd, Angew. Chem., Int. Ed., 2018, 57, 12092–12096 CrossRef CAS PubMed.
- M. R. L. Paine, B. L. J. Poad, G. B. Eijkel, D. L. Marshall, S. J. Blanksby, R. M. A. Heeren and S. R. Ellis, Angew. Chem., Int. Ed., 2018, 57, 10530–10534 CrossRef CAS PubMed.
- M. Stahlman, H. T. Pham, M. Adiels, T. W. Mitchell, S. J. Blanksby, B. Fagerberg, K. Ekroos and J. Boren, Diabetologia, 2012, 55, 1156–1166 CrossRef CAS PubMed.
- H. T. Pham and R. R. Julian, Int. J. Mass Spectrom., 2014, 370, 58–65 CrossRef CAS.
- F. Tureček and R. R. Julian, Chem. Rev., 2013, 113, 6691–6733 CrossRef PubMed.
- H. T. Pham, T. Ly, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Anal. Chem., 2012, 84, 7525–7532 CrossRef CAS PubMed.
- T. Baba, J. L. Campbell, J. C. Le Blanc, J. W. Hager and B. A. Thomson, Anal. Chem., 2015, 87, 785–792 CrossRef CAS.
- H. Takahashi, Y. Shimabukuro, D. Asakawa, S. Yamauchi, S. Sekiya, S. Iwamoto, M. Wada and K. Tanaka, Anal. Chem., 2018, 90, 7230–7238 CrossRef CAS PubMed.
- X. Zhang and G. E. Reid, Int. J. Mass Spectrom., 2006, 252, 242–255 CrossRef CAS.
- K. Ekroos, C. S. Ejsing, U. Bahr, M. Karas, K. Simons and A. Shevchenko, J. Lipid Res., 2003, 44, 2181–2192 CrossRef CAS PubMed.
- Y. Xia, P. A. Chrisman, S. J. Pitteri, D. E. Erickson and S. A. McLuckey, J. Am. Chem. Soc., 2006, 128, 11792–11798 CrossRef CAS PubMed.
- B. L. J. Poad, B. B. Kirk, P. I. Hettiarachchi, A. J. Trevitt, S. J. Blanksby and T. Clark, Angew. Chem., Int. Ed., 2013, 52, 9301–9304 CrossRef CAS PubMed.
- H. Martinez-Seara, T. Rog, M. Karttunen, I. Vattulainen and R. Reigada, J. Phys. Chem. B, 2009, 113, 8347–8356 CrossRef CAS.
- D. R. Klein, C. L. Feider, K. Y. Garza, J. Q. Lin, L. S. Eberlin and J. S. Brodbelt, Anal. Chem., 2018, 90, 10100–10104 CrossRef CAS.
- M. Sans, J. Zhang, J. Q. Lin, C. L. Feider, N. Giese, M. T. Breen, K. Sebastian, J. Liu, A. K. Sood and L. S. Eberlin, Clin. Chem., 2019, 65, 674–683 CrossRef CAS.
- T. Harayama, M. Eto, H. Shindou, Y. Kita, E. Otsubo, D. Hishikawa, S. Ishii, K. Sakimura, M. Mishina and T. Shimizu, Cell Metab., 2014, 20, 295–305 CrossRef CAS PubMed.
- H. Kawana, K. Kano, H. Shindou, A. Inoue, T. Shimizu and J. Aoki, BBA, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019, 1864, 1053–1060 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03521d |
| This journal is © The Royal Society of Chemistry 2019 |