
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-catalyzed site-selective C(sp2)–H radical acylation of phenylalanine containing peptides with aldehydes†
Marcos
San Segundo
 and
Arkaitz
Correa
*
and
Arkaitz
Correa
*
University of the Basque Country (UPV/EHU), Department of Organic Chemistry I, Joxe Mari Korta R&D Center, Avda. Tolosa 72, 20018 Donostia-San Sebastián, Spain. E-mail: arkaitz.correa@ehu.eus
First published on 7th August 2019
Abstract
The site-selective functionalization of C–H bonds within a peptide framework remains a challenging task of prime synthetic importance. Herein, the first Pd-catalyzed δ-C(sp2)–H acylation of Phe containing peptides with aldehydes is described. This oxidative coupling is distinguished by its site-specificity, tolerance of sensitive functional groups, scalability, and enantiospecificity and exhibits entire chemoselectivity for Phe motifs over other amino acid units. The compatibility of this dehydrogenative acylation platform with a number of oligopeptides of high structural complexity illustrates its ample opportunities for the late-stage peptide modification and bioconjugation.
Introduction
Driven by their enhanced biological activities and often improved pharmacokinetics compared with their native counterparts, non-natural amino acids and peptides derived thereof have lately emerged as powerful scaffolds in proteomics and drug discovery.1 As a result, recent years have witnessed tremendous interest in the site-specific chemical modification of peptides for the ultimate precise engineering of proteins.2 In this regard, transition-metal catalysis has played a critical role in bioconjugation3 and recently unlocked new paradigms for the site-selective C–H functionalization of peptides.2 The latter has altered the landscape of peptide modification strategies, thus clearly complementing classical techniques from an atom- and step-economic standpoint and allowing the sustainable manipulation of otherwise unreactive C–H bonds.4 However, despite the remarkable advances realized, the available functionalization portfolio in these endeavors primarily relies on toxic halide derivatives as coupling partners (Scheme 1, route a), hence reinforcing a change in the strategy to implement more versatile C–H counterparts.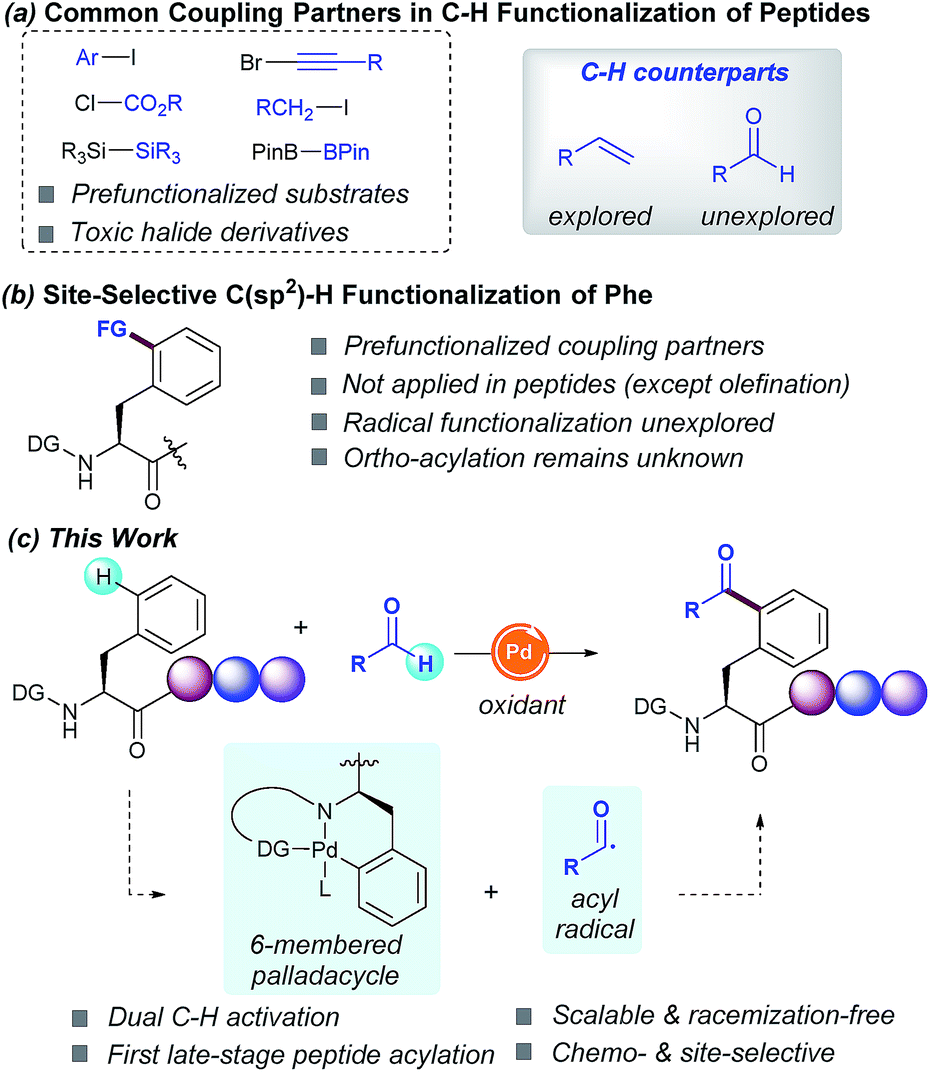 | ||
| Scheme 1 C(sp2)–H functionalization of peptides. | ||
The functionalization of C(sp3)–H bonds has been extensively studied and a number of functional groups (FG) can be selectively introduced into the α-amino acid backbone5 as well as in the β-, γ- and δ-positions within the hydrocarbon side-chains.6 In sharp contrast, relatively few methods are available for the parent C(sp2)–H functionalization of aromatic side chains of peptides. Although the modification of tryptophan-containing peptides has proven to be a rather explored avenue,7 the diversification of phenylalanine (Phe) residues remains comparatively unexplored. In fact, just a few isolated examples for the modification of simple Phe units are known to date, but they have not been applied within a challenging peptide framework. The most studied technique is the Pd-catalyzed δ-C(sp2)–H olefination introduced by Yu in simple systems,8h and recently elegantly extended to peptides and cyclopeptides by Cross9a and Wang,9c respectively (Scheme 1, route b). In this light, we envisioned that the introduction of novel, yet atom-economical C–H coupling partners could enrich our chemical toolbox for the rarely explored late-stage modification of Phe-containing peptides, thus streamlining the rapid assembly of biomolecules of paramount relevance and providing access to novel α-amino acids and peptides beyond those found in naturally occurring proteins.
Radical chemistry has recently flourished into a key technique for creating molecular complexity.10 However, the radical functionalization of peptides based on inner-sphere reaction mechanisms has thus far remained elusive.11 In this respect, inspired by the emerging trends in radical reactions,10 we sought to exploit the practical use of aldehydes as versatile and cost-efficient radical sources.12 Although the metal-catalyzed directed C(sp2)–H acylation has been previously studied, its application as a late-stage functionalization tool within a peptide framework remains unknown. Based on the commonly accepted PdII/PdIV manifold,12e,f we anticipated that the judicious choice of the directing group (DG) would be crucial for achieving high positional selectivity upon the formation of a 6-membered palladacycle prone to undergo further addition of the corresponding acyl radical species. Likewise, avoidance of undesired decarbonylation12a of the transient acyl radical species poses a crucial challenge. If successful, such a conceptually simple strategy would result in the virtually unexplored carbon-centered radical acylation for the late-stage introduction of ketone motifs within peptides in a predictable and efficient manner (Scheme 1, route c). As part of our interest in sustainable catalysis, herein we report the first Pd-catalyzed site-selective C(sp2)–H acylation of Phe containing peptides with aldehydes. The salient features of our method include high chemoselectivity, broad group tolerance, scalability, retention of the native chirality, predictable site-selectivity and facile removal of the required DG.
Results and discussion
As a proof-of-concept with a simple system, we began our investigations by selecting the acylation of picolinamide (PA)-protected L–Phe–OMe (1a) with p-tolyl aldehyde (2a) as the model reaction. This auxiliary was originally introduced by Daugulis13 and has demonstrated superior directing abilities to enable a number of transformations in the realm of C–H activation, including a variety of C(sp3)–H modifications of peptides.6a–c,8c–e After systematic evaluation of all reaction parameters,14 we found that the desired transformation was feasible and the corresponding acylated compound 3a was obtained in 79% yield when a combination of Pd(OAc)2 as the catalyst, dicumyl peroxide (DCP) as the oxidant, Ag2CO3 as the additive, and DMF as the solvent was used (Table 1, entry 1). Although 3a was obtained as a mixture of mono- and diacylated products (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a′, 7:3 ratio), an optimal balance between yield and mono-selectivity was successfully achieved. Since oxidation of the aldehyde 2a to the corresponding benzoic acid was often observed, an excess of 2a was required in order to achieve full conversion. Importantly, neither undesired radical acylation on the α-C(sp3)–H bond of the Phe backbone11d nor the alkylation upon a decarbonylation reaction pathway12a was ever observed. As expected, control experiments verified the crucial role of both the Pd catalyst and oxidant as not even traces of 3a were detected in their absence (entries 2 and 3, respectively).
:3a′, 7:3 ratio), an optimal balance between yield and mono-selectivity was successfully achieved. Since oxidation of the aldehyde 2a to the corresponding benzoic acid was often observed, an excess of 2a was required in order to achieve full conversion. Importantly, neither undesired radical acylation on the α-C(sp3)–H bond of the Phe backbone11d nor the alkylation upon a decarbonylation reaction pathway12a was ever observed. As expected, control experiments verified the crucial role of both the Pd catalyst and oxidant as not even traces of 3a were detected in their absence (entries 2 and 3, respectively).
|
|
||
|---|---|---|
| Entry | Change from standard conditions | 3a (%) |
| a Reaction conditions: 1a (0.25 mmol), 2a (1.25 mmol), Pd(OAc)2 (10 mol%), DCP (2.0 equiv.), and Ag2CO3 (2.0 equiv.) in DMF (1 mL) at 100 °C for 16 h under Ar. b Conversion determined by 1H NMR analysis. c Ratio of mono- and diacylated products. d Yield of the isolated product after column chromatography. DCP = dicumyl peroxide; DTBP = di-tert-butyl peroxide; TBHP = tert-butyl hydroperoxide. | ||
| 1 | None | 79 (7:3)c,d |
| 2 | Without Pd(OAc)2 | 0 |
| 3 | Without DCP | 0 |
| 4 | Without Ag2CO3 | 56 (8:2)c |
| 5 | Under air | 66 (8:2)c |
| 6 | DMA instead of DMF | 78 (6:4)c |
| 7 | 6.0 equiv. of 2a | 88 (6:4)c |
| 8 | K2S2O8 instead of DCP | 0 |
| 9 | DTBP instead of DCP | 64 (8:2)c |
| 10 | TBHPaq instead of DCP | 63 (7:3)c |
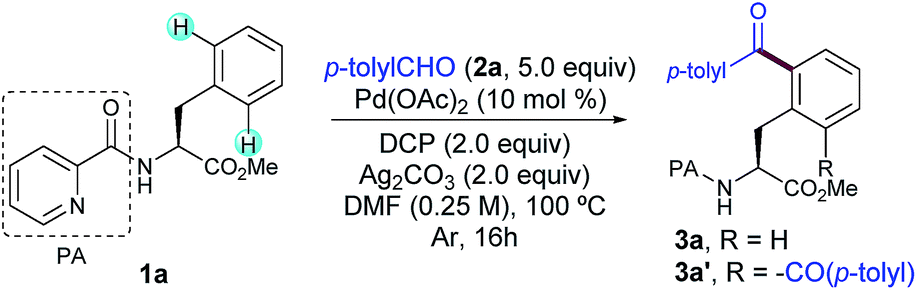
Notably, as commonly observed in related Pd-catalyzed cross-coupling techniques,6a,b,h,i the addition of Ag2CO3 proved to be highly beneficial for the process to occur (entry 4) and the reaction outcome was rather sensitive to the amount of silver carbonate.15 In order to overcome the persistent problem of regioselectivity between the mono- and diacylation reactions, the evaluation of supporting ligands, equivalents of 2a and other parameters was carefully performed.14 Unfortunately, higher selectivity toward the monoacylation product was only achieved at the expense of having much lower overall yields. Likewise, although inorganic persulfates entirely inhibited the reaction (entry 8), other peroxides such as DTBP or commonly used TBHP afforded lower yields of 3a (entries 9 and 10).14 In general terms, reactivity was favored over selectivity and preferential monoacylation (8:2) was only achieved when lower yields were obtained (up to 66%, entries 4, 5 and 9), which may underpin the tendency of the monoacylated compound 3a to undergo a subsequent acylation reaction toward the formation of 3a′. As initially anticipated, subtle modifications on the DG had a determinant impact on the reaction outcome. Although benzoyl-, tosyl- or acetyl-protected substrates devoid of an additional nitrogen-chelating unit remained unreactive, a related carboxamide bearing a 1,2,3-triazole unit could be also employed as an efficient bidentate DG, albeit in comparatively lower yields (Scheme 2).14,16
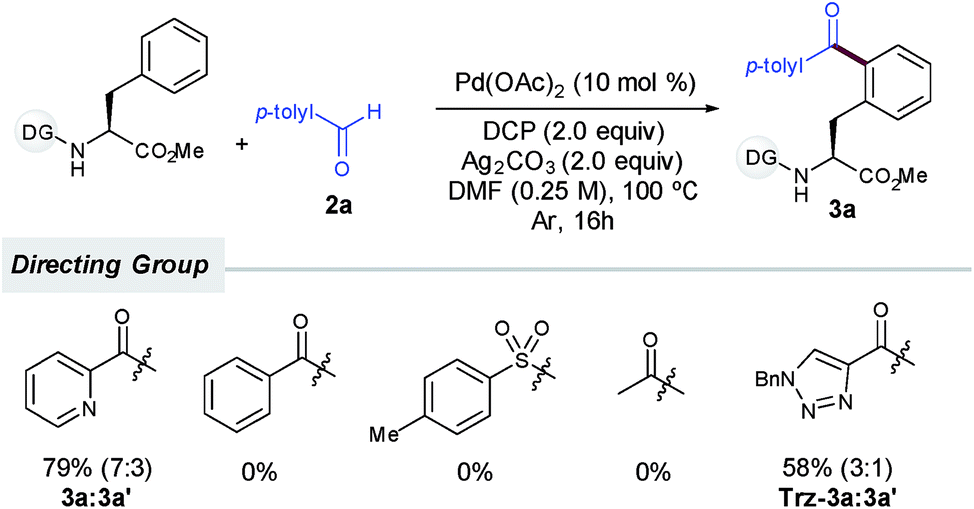 | ||
| Scheme 2 Influence of the DG. | ||
With the optimized conditions in hand, we next investigated the scope of the δ-C(sp2)–H acylation protocol with respect to the aldehyde (Table 2). Gratifyingly, a wide variety of electronically diverse aldehydes smoothly underwent the target dehydrogenative coupling in moderate to excellent yields. In general, aromatic aldehydes bearing electron-donating groups such as OMe (2b and 2e), Et2N (2c) and 2,3-dihydrofuryl (2d) provided the corresponding products 3b–e as mixtures of mono- and diacylated compounds, which were easily separated by column chromatography. In this respect, the highly electron-rich 2,4,6-trimethoxybenzaldehyde (2i) afforded selectively the diacylated compound 3i′ in 67% yield; its absolute configuration was verified by X-ray analysis. Conversely, p-hydroxybenzaldehyde 2f provided selectively the corresponding monoacylated product 3f. Likewise, the lower tendency to oxidation of benzaldehydes 2g–h bearing electron-withdrawing groups resulted in a high selectivity toward the monoacylation and furnished 3g–h in good yields. Remarkably, pharmaceutically relevant heterocyclic motifs could be also accommodated and thus N-methylindolyl (2j) and 2-thienyl carboxaldehyde (2k) selectively afforded the corresponding monoacylated products (3j and k).
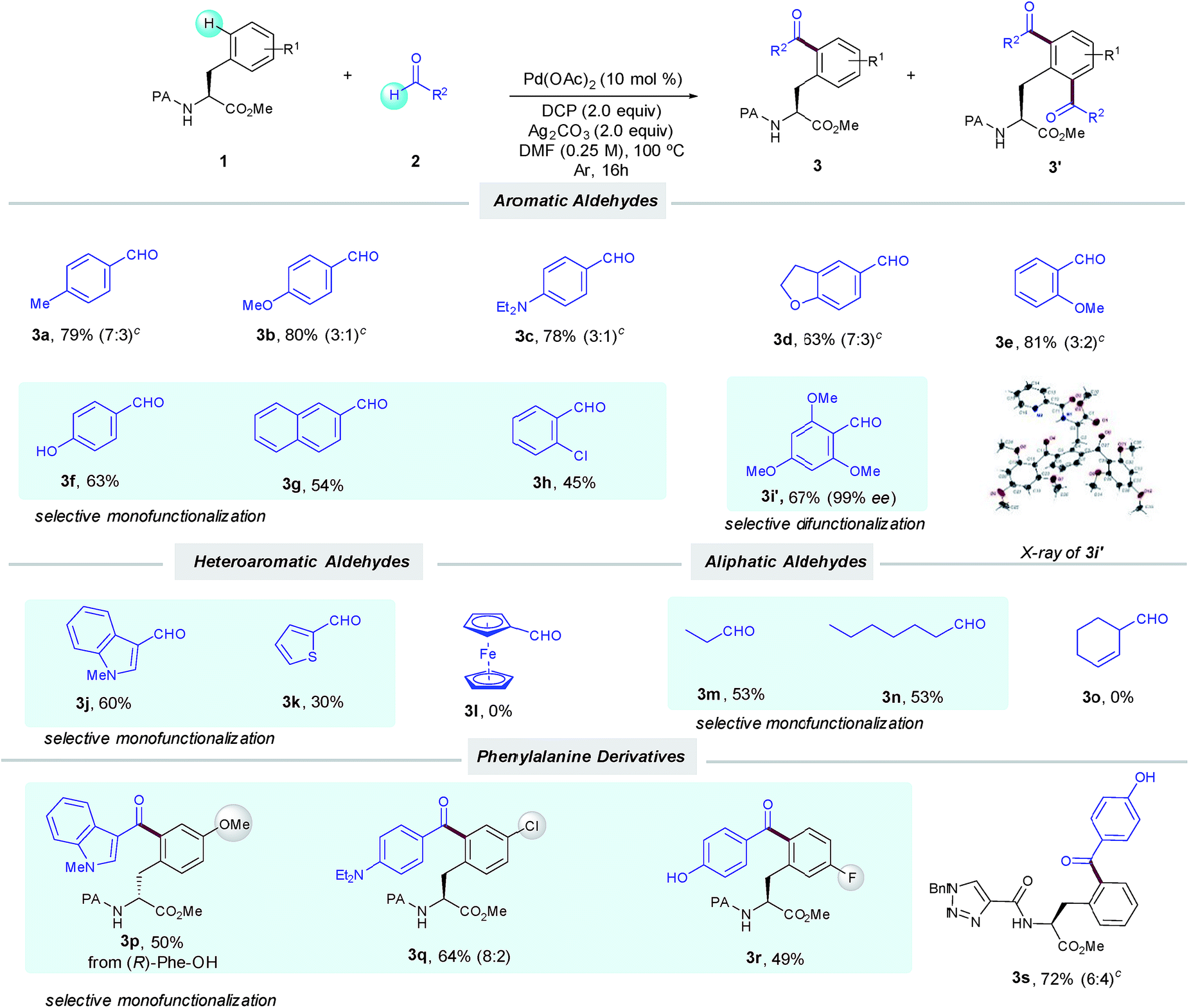
Additionally, aliphatic aldehydes could also be employed toward the selective monoacylation of 3m,n, albeit in moderate yields. The latter selectivity could be related to their lower reactivity since full conversion was not achieved. Of remarkable importance are 3c and 3d, where high chemoselectivity was achieved toward the preferential activation of the aldehyde motif versus the C(sp3)–H bonds adjacent to nitrogen and oxygen atoms.5a Moreover, Phe substituted derivatives smoothly furnished monoacylated products 3p–r in moderate to good yields. As verified by HPLC analysis,14 no racemization occurred along our oxidative process. It is important to note that the method was found incompatible with the use of aldehydes incorporating alkenes or a ferrocene motif, which could be tentatively attributed to competitive radical functionalization reactions.
Encouraged by these results, we next evaluated our oxidative acylation in the more complex setting of dipeptides, which are known to undergo oxidative fragmentations upon the formation of α-carbon radicals17 and hence their selective acylation could be a challenging task to tackle. Notably, dipeptides containing Phe (5a,b), Leu (5c–e) and Pro (5g) units selectively underwent the PA-directed acylation with a variety of benzaldehydes on the terminal Phe unit (Table 3). Of particular importance is the tolerance to the oxidizable side-chain hydroxyl group in Thr of dipeptide 5f, which remained intact along the oxidative process. Likewise, the C–H acylation could be also efficiently directed by a triazole-containing group (5h). The preservation of the native chirality of the substrates was underpinned by NMR analysis. The robustness of our method was further demonstrated by the site-selective functionalization of tri- (5i,j), tetra- (5k) and even pentapeptides (5l′–n) in moderate to good yields. It is known that the additional amide bonds within oligopeptides can reasonably deactivate the metal catalyst by the formation of N,N-chelated complexes.6g,18 Indeed, by comparison of pentapeptide 5l′ with 5m,n bearing Pro residues, where such an undesired catalyst deactivation is avoided, the site-selective acylation was achieved in excellent yields. The latter underscores the high potential for Pro residues as key elements at the late-stage functionalization in peptide settings. Noteworthy, the acylation exclusively occurred at the N-terminal Phe unit and other residues bearing oxidizable aliphatic chains with reactive secondary C–H bonds such as Leu, Ile, Ala, and Val remained intact.19 Collectively, the small library of oligopeptides rapidly assembled illustrates the vast potential of our catalytic manifold to introduce ketone motifs in a late-stage fashion to furnish densely decorated peptides. The reaction represents an innovative, yet challenging dehydrogenative radical technique, which offers previously unrecognized opportunities in the field of peptide chemistry. In this respect, although good to excellent yields could be obtained with certain peptides (up to 78% yield), it is important to note that the sometimes obtained low to moderate yields were due to incomplete conversion of the starting material; the reactions were very clean and the only side-product was derived from the oxidation of the aldehyde to the corresponding carboxylic acid, which was easily removed upon the reaction work-up.
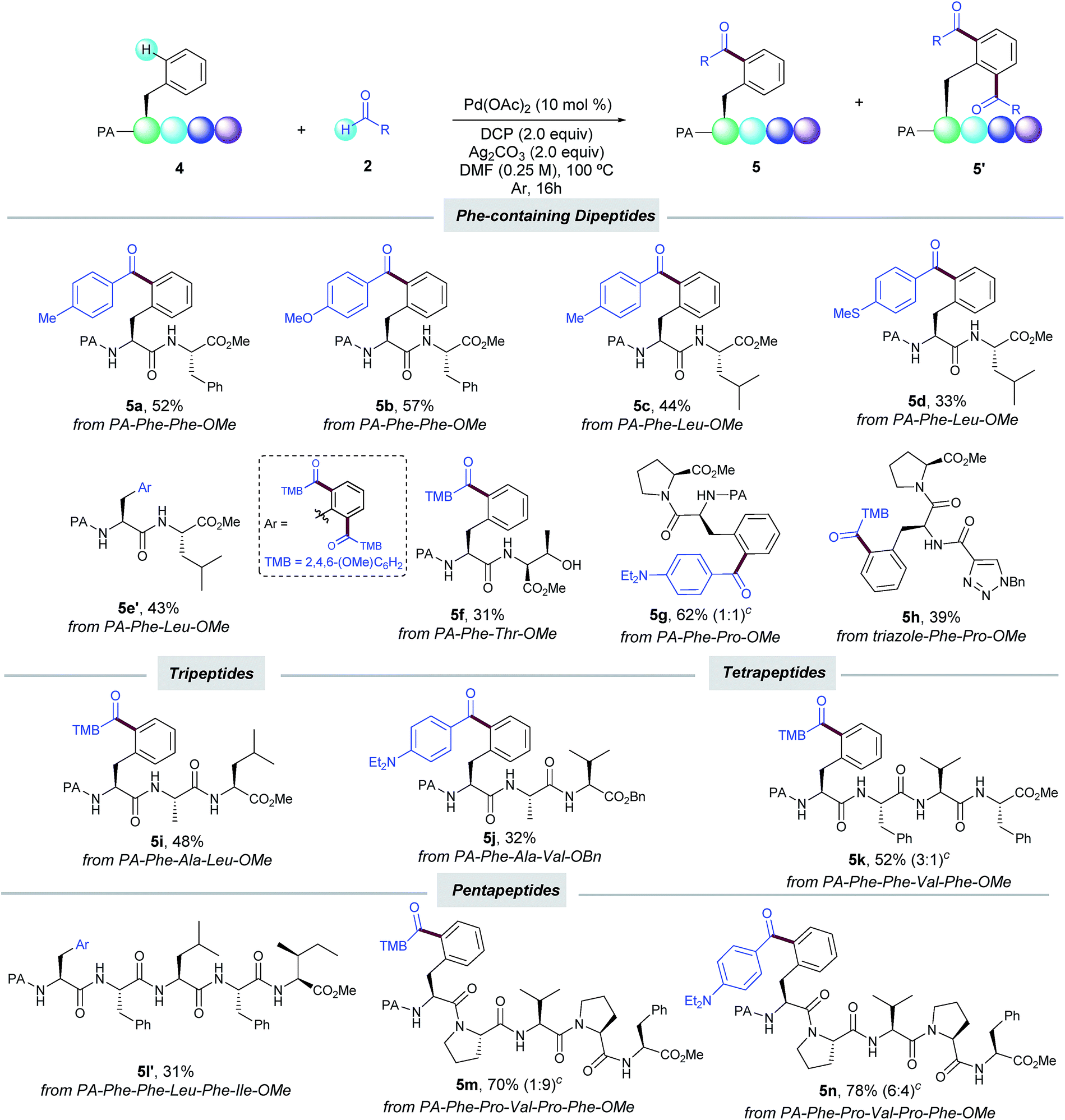
The synthetic utility and robustness of our site-selective functionalization manifold were highlighted by scaling up the acylation reaction to the gram level and 3a was obtained in a remarkable 82% yield. However, the extended reaction time to reach completion resulted in a lower selectivity than that of the experiment at 0.25 mmol (1:1 vs. 7:3). The facile removal of the PA group6a,11d showcased its practicality to ultimately deliver highly functionalized peptide molecules bearing a synthetically versatile free-amino group (Scheme 3).
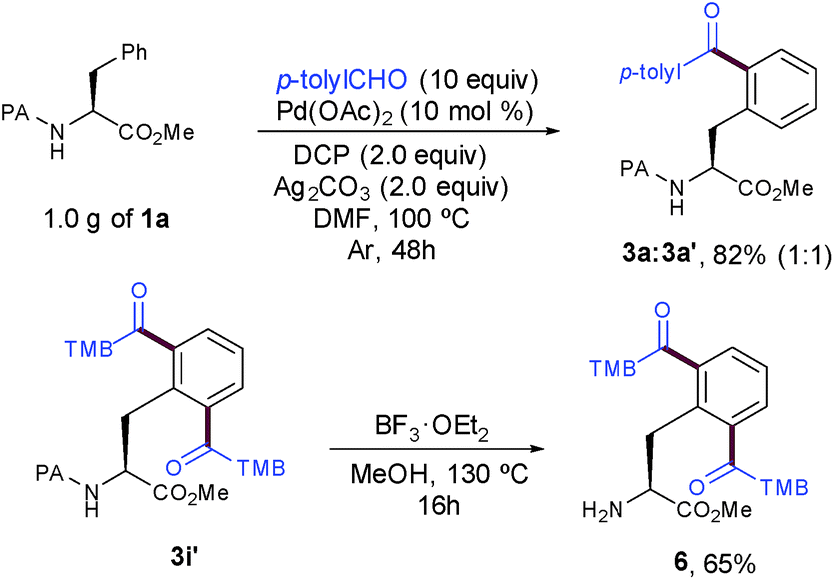 | ||
| Scheme 3 Cleavage of the DG and gram scale synthesis. | ||
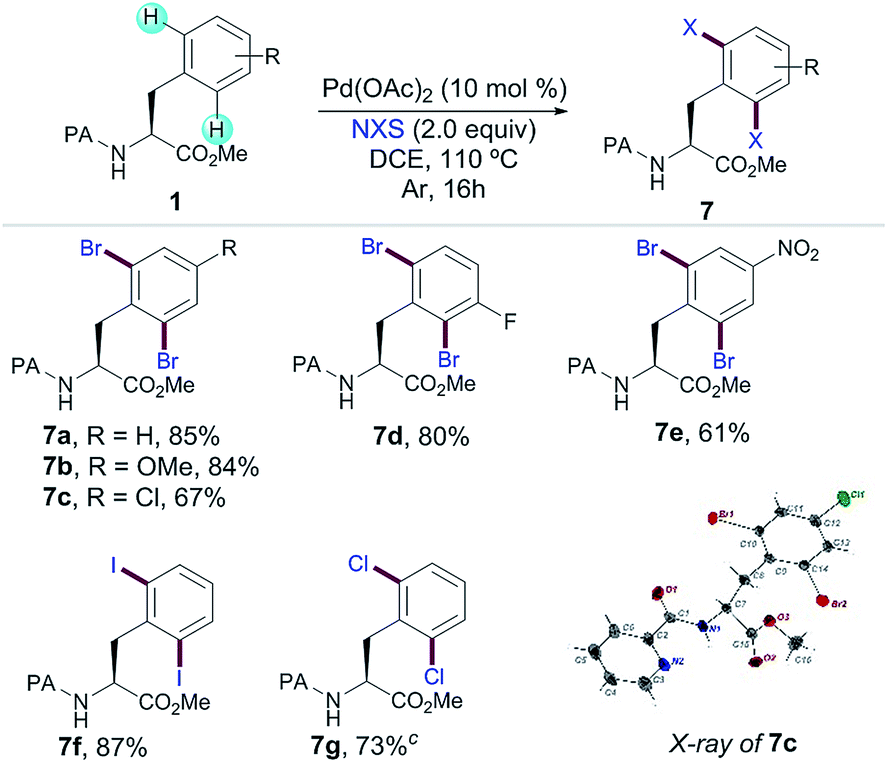
In order to expand the potential of Phe as a fully diversifiable unit through the formation of a 6-membered palladacycle, we next studied the PA-directed Pd-catalyzed C–halogen bond-forming processes upon a related Pd(II)/Pd(IV) regime. Yu and co-workers have developed iodination8h reactions with a combination of PhI(OAc)2 and I2 using triflamide as the DG. Owing to the more practical features of non-halogenated and easily removable PA, we successfully accomplished a variety of dihalogenation reactions of Phe derivatives;14,20 the corresponding dibromination with N-bromosuccinimide was efficiently applied to the assembly of a small family of substituted Phe derivatives 7a–e in excellent yields (Table 4). The structure of 7c was unambiguously assigned by X-ray analysis verifying that the bromination proceeded with enantiospecificity. Importantly, the use of related halosuccinimides afforded iodinated (7f) and chlorinated (7g) products in good to excellent yields. The latter illustrated that PA can be an efficient auxiliary for performing not only C–H acylations but also relevant C–H halogenation reactions in Phe derivatives.
To shed light on the reaction mechanism, we carried out several control experiments with 1a as a simple model system (Scheme 4). On the one hand, we found that the acylation of 1a with aldehyde 2a was entirely inhibited in the presence of radical traps such as TEMPO, BHT, and diphenylethylene,14 which indicated that a radical scenario may be operative. On the other hand, in order to support the intermediacy of a palladacycle intermediate the following experiments were performed. First, a stoichiometric reaction of 1a with Pd(OAc)2 provided IntA in 80% yield, which was characterized by NMR spectroscopy. Second, when IntA was subjected to the optimized acylation conditions, 3a was obtained in 45% yield as the monoacetylated compound, and in lower yield in the absence of silver carbonate.6a,21 Likewise, IntA efficiently catalyzed the formation of 3a from 1a in 58% isolated yield, which underpinned its key role as a viable precatalyst.
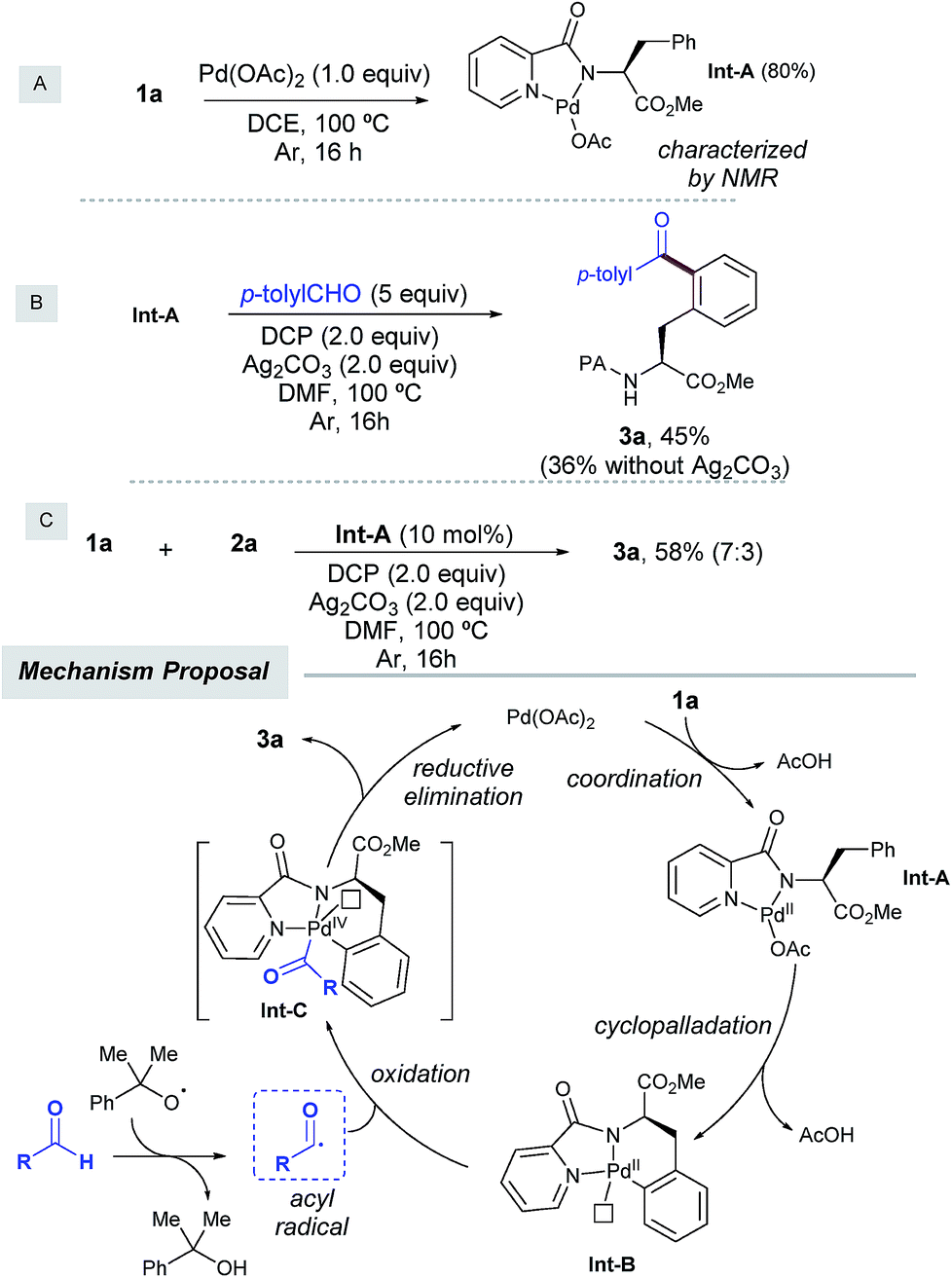 | ||
| Scheme 4 Control experiments and the proposed mechanism. | ||
On the basis of the above results and previous literature reports,12 a plausible monomeric reaction mechanism is proposed in Scheme 4. Complexation of 1a with Pd(OAc)2 would initially afford Pd(II) complex IntA,6c which would next undergo a directed ortho-selective cyclometallation to provide the six-membered palladacycle IntB.9a,b The latter would next react with the acyl radical,12 which was in situ generated upon hydrogen atom abstraction by cumyl peroxyl radical species to provide transient Pd(III) species12e,f which would be subsequently oxidized to deliver IntC.22 This species has been proposed to exist as either Pd(IV)23 or dimeric Pd(III)24 intermediates and would furnish the acylated product 3a through C–C bond forming reductive elimination, thereby regenerating the active Pd(II) catalyst. Importantly, a competitive intramolecular C–N bond forming reductive elimination8f was never observed. At this stage, the involvement of polynuclear Pd complexes6a or heterodimeric Pd–Ag21 intermediates cannot be ruled out within our catalytic cycle.
Conclusions
In summary, we have developed a practical protocol for the assembly of non-proteogenic acylated Phe-containing oligopeptides via a novel Pd-catalyzed δ-C(sp2)–H functionalization reaction with abundant and readily available aldehydes. From a fundamental point of view, this transformation represents a robust, yet innovative means for the radical functionalization of a wide range of Phe-containing compounds, thus expanding the landscape of peptide synthesis to provide heavily substituted peptide analogues containing aryl, heteroaryl and even aliphatic ketone residues. The important features of our strategy are the widespread availability of aldehydes, the broad functional group tolerance, the retention of the chiral integrity of the existing stereocenters in peptide settings, the site-selectivity toward the functionalization of the Phe unit assisted by the N-terminal PA group, and the facile removal of the required PA group. Moreover, the process can be extended to the use of medicinally relevant 1,2,3-triazoles as alternative bidentante DGs. Therefore, we anticipate that our Pd-catalyzed oxidative acylation method could become a powerful platform technology of tremendous importance in drug discovery and protein engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to MINECO (CTQ2016-78395-P; RTI2018-093721-B-I00) and Basque Government (IT1033-16) for financial support. A. C. thanks MINECO for a Ramón y Cajal research contract (RYC-2012-09873). We acknowledge technical and human support provided by SGIker of UPV/EHU and European funding (ERDF and ESF). Cost-CHAOS action (CA15106) is also acknowledged.Notes and references
- (a) K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122 CrossRef CAS PubMed; (b) L. Pollegioni and S. Servi, Non-natural Amino Acids: Methods and Protocols, Springer, New York, 2012 CrossRef; (c) A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry, Wiley-VCH, Weinheim, 2011, vol. 1 CrossRef.
- For reviews, see: (a) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700 CrossRef CAS PubMed; (b) L. R. Malins, Pept. Sci., 2018, 110, e24049 CrossRef; (c) X. Lu, S.-J. He, W.-M. Cheng and J. Shi, Chin. Chem. Lett., 2018, 29, 1001 CrossRef CAS; (d) S. Mondal and S. Chowdhury, Adv. Synth. Catal., 2018, 360, 1884 CrossRef CAS; (e) L. R. Malins, Curr. Opin. Chem. Biol., 2018, 46, 25 CrossRef CAS PubMed; (f) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS PubMed; (g) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS PubMed; (h) A. M. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1 CrossRef CAS PubMed; (i) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed.
- (a) J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., Int. Ed., 2019, 58, 6176 CrossRef CAS PubMed; (b) M. Jbara, S. K. Maity and A. Brik, Angew. Chem., Int. Ed., 2017, 56, 10644 CrossRef CAS PubMed; (c) J. M. Chalker, Metal-mediated Bioconjugation, in Chemoselective and Bioorthogonal Ligation Reactions: Concepts and Applications, ed. I. Medintz, W. R. Algar and P. Dawson, Wiley, 2017, p. 231 Search PubMed.
- For selected reviews on the late-stage C–H functionalization, see: (a) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234 CrossRef CAS PubMed; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (c) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- (a) M. San Segundo and A. Correa, Synthesis, 2018, 50, 2853 CrossRef CAS; (b) T. Brandhofer and O. García Mancheño, Eur. J. Org. Chem., 2018, 6050 CrossRef CAS.
- For selected examples, see: (a) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633 CrossRef CAS PubMed; (b) B.-B. Zhan, J. Fan, L. Jin and B.-F. Shi, ACS Catal., 2019, 9, 3298 CrossRef CAS; (c) B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858 CrossRef CAS PubMed; (d) W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10554 CrossRef CAS PubMed; (e) Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, J. Am. Chem. Soc., 2018, 140, 6797 CrossRef CAS PubMed; (f) M. Bauer, W. Wang, M. M. Lorion, C. Dong and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 203 CrossRef CAS PubMed; (g) T. Liu, J. X. Qiao, M. A. Poss and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10924 CrossRef CAS PubMed; (h) A. F. M. Noisier, J. García, I. A. Ionut and F. Albericio, Angew. Chem., Int. Ed., 2017, 56, 314 CrossRef CAS PubMed; (i) G. Liao, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, Nat. Commun., 2016, 7, 12901 CrossRef PubMed; (j) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed.
- For selected examples, see: (a) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476 CrossRef CAS PubMed; (b) A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576 CrossRef CAS PubMed; (c) Y. Zhu, M. Bauer and L. Ackermann, Chem.–Eur. J., 2015, 21, 9980 CrossRef CAS PubMed; (d) L. Mendive-Tapia, S. Preciado, J. García, R. Ramón, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- (a) W. Zeng, M. Nukeyeva, Q. Wang and C. Jiang, Org. Biomol. Chem., 2018, 16, 598 RSC; (b) L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899 CrossRef CAS PubMed; (c) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed; (d) S.-Y. Zhang, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313 CrossRef CAS PubMed; (e) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS PubMed; (f) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944 CrossRef CAS PubMed; (g) R. B. Bedford, M. F. Haddow, R. L. Webster and C. J. Mitchell, Org. Biomol. Chem., 2009, 7, 3119 RSC; (h) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed.
- (a) M. J. Terrey, C. C. Perry and W. B. Cross, Org. Lett., 2019, 21, 104 CrossRef PubMed; (b) Y. Zheng and W. Song, Org. Lett., 2019, 21, 3257 CrossRef CAS PubMed; (c) Z. Bai, C. Cai, Z. Yu and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 13912 CrossRef CAS PubMed; (d) F. Zhao, X. Jia, J. Zhao, C. Fei, L. Liu, G. Liu, D. Wang and F. Chen, RSC Adv., 2017, 7, 25031 RSC. For a related olefination of α-Phe–Gly–OH, (e) N. Dastbaravardeh, T. Toba, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877 CrossRef CAS PubMed.
- For selected reviews, see: (a) W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654 RSC; (b) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed; (d) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (e) Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed; (f) S. Zard, Radical Reactions in Organic Synthesis, Oxford University Press, Oxford, U.K., 2003 Search PubMed; (g) B. Giese, Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon, Oxford, 1986 Search PubMed.
- For selected C–H functionalizations of peptides based on an outer-sphere mechanism, see: (a) M. San Segundo and A. Correa, ChemSusChem, 2018, 11, 3893 CrossRef CAS PubMed; (b) C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 1584 Search PubMed; (c) M. San Segundo, I. Guerrero and A. Correa, Org. Lett., 2017, 19, 5288 CrossRef CAS PubMed; (d) K. Li, Q. Wu, J. Lan and J. You, Nat. Commun., 2015, 6, 8404 CrossRef CAS PubMed.
- For selected reviews, see: (a) W.-C. Yang, J.-G. Feng, L. Wu and Y.-Q. Zhang, Adv. Synth. Catal., 2019, 361, 1700 CrossRef CAS; (b) A. Banerjee, Z. Lei and M.-Y. Nga, Synthesis, 2019, 51, 303 CrossRef CAS PubMed; (c) X.-F. Wu, Chem.–Eur. J., 2015, 21, 12252 CrossRef CAS PubMed; (d) C. Pan, X. Jia and J. Cheng, Synthesis, 2012, 44, 677 CrossRef CAS. For selected examples, see: (e) X. Jia, S. Zhang, W. Wang, F. Lou and J. Cheng, Org. Lett., 2009, 11, 3120 CrossRef CAS PubMed; (f) O. Baslé, J. Bidange, Q. Shuai and C.-J. Li, Adv. Synth. Catal., 2010, 352, 1145 CrossRef.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- For more details, see the ESI.†.
- A. Maji, A. Dahiya, G. Lu, T. Bhattacharya, M. Brochetta, G. Zanoni, P. Liu and D. Maiti, Nat. Commun., 2018, 9, 3582 CrossRef PubMed.
- Triazoles have been elegantly used by Ackermann as DGs in the late-stage diversification of peptides, see for examples ref. 6c and e. For a general review on the role of triazoles in the field of C–H functionalization, see: I. Guerrero and A. Correa, Eur. J. Org. Chem., 2018, 6034 CrossRef CAS.
- R. T. Dean, S. Fu, R. Stocker and M. J. Davies, Biochem. J., 1997, 324, 1 CrossRef CAS PubMed.
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef CAS PubMed.
- T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Nature, 2016, 537, 214 CrossRef CAS PubMed.
- Related C–H halogenation reactions with α-phenyl glycine derivatives upon the formation of a kinetically more favored 5-membered palladacycle have been recently described (see ref. 8a). However, the halogenation of Phe-derivatives remained unexplored.
- The mode of action of Ag2CO3 as a non-innocent additive within the reaction pathway still remains to be established. In this regard, a dual role has been proposed in related metal-catalyzed C–H functionalization reactions and besides acting as an oxidant to effectively turnover the catalytic cycle, it may also form heterodimeric Pd–Ag complexes, thus lowering the activation barriers of oxidative addition/reductive elimination steps: (a) M. Anand, R. B. Sunoj and H. F. Schaefer III, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef PubMed; (b) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef PubMed; (c) A. L. Mudarra, S. Martínez de Salinas and M. H. Pérez-Temprano, Org. Biomol. Chem., 2019, 17, 1655 RSC.
- According to the common square planar geometry of Pd complexes, AcOH or a molecule of chelating DMF could occupy the remaining vacant coordination sites both in IntB and IntC. Likewise, dimeric species may come into play.
- (a) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed; (b) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS PubMed; (c) J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed.
- (a) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed; (b) D. C. Powers, M. A. L. Geibel, J. E. M. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, data for new compounds, and 1H and 13C NMR spectra. CCDC 1939205 and 1939206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03425k |
| This journal is © The Royal Society of Chemistry 2019 |