
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of quaternary 3,4-dihydroisoquinolinones via Heck carbonylation reactions: development and application to the synthesis of Minalrestat analogues†
Cang
Cheng
,
Bin
Wan
,
Bo
Zhou
 ,
Yichao
Gu
and
Yanghui
Zhang
*
,
Yichao
Gu
and
Yanghui
Zhang
*
School of Chemical Science and Engineering, Shanghai Key Laboratory of Chemical Assessment and Sustainability, Tongji University, 1239 Siping Road, Shanghai 200092, China. E-mail: zhangyanghui@tongji.edu.cn
First published on 3rd September 2019
Abstract
Minalrestat and its analogues represent structurally novel aldose reductase inhibitors, and the asymmetric synthesis of such pharmaceutically privileged molecules has not been reported yet. We have developed a palladium-catalyzed enantioselective intramolecular carbonylative Heck reaction by using formate esters as the source of CO, which represents the first enantioselective synthesis of quaternary 3,4-dihydroisoquinolines. The reaction provides a facile and efficient method for the synthesis of enantiopure nitrogen-containing heterocyclic compounds bearing an all-carbon quaternary stereocenter. The reaction has been successfully applied to the first asymmetric synthesis of Minalrestat analogues.
Introduction
Diabetes mellitus is a major health concern and affects millions of people worldwide.1 Aldose reductase inhibitors (ARIs) are attractive therapeutic targets for designing drugs to prevent or slow the progression of diabetic complications.2 Although various ARIs have been discovered, almost all of them have been withdrawn due to adverse side effects or low efficacy.3 Minalrestat is an important ARI that shows appreciable activity and safety profiles and is a promising drug candidate.4 Minalrestat is a 3,4-dihydroisoquinolinone derivative bearing a spirosuccinimide moiety at the 4-position (Fig. 1). Actually, the isoquinolinone backbone in Minalrestat represents a structurally novel framework for designing potent ARIs, and a range of derivatives derived from the backbone exhibit intrinsic activity and good oral potency.4 Notably, these bioactive isoquinolinone derivatives usually contain a quaternary carbon stereocenter, and the stereocenter plays crucial roles in the bioactivities and oral potency. Unfortunately, the stereocenters tend to racemize via enolization.4a The asymmetric synthesis of this isoquinolinone skeleton is not available,5 and the enantiopure compounds were obtained by the resolution of racemates.4a Therefore, it is a formidable task to develop enantioselective reactions for the construction of such six-membered isoquinolinones containing an all-carbon quaternary stereocenter, which would allow us to not only obtain enantiopure compounds for drug discovery but also modify the structures to prevent racemization.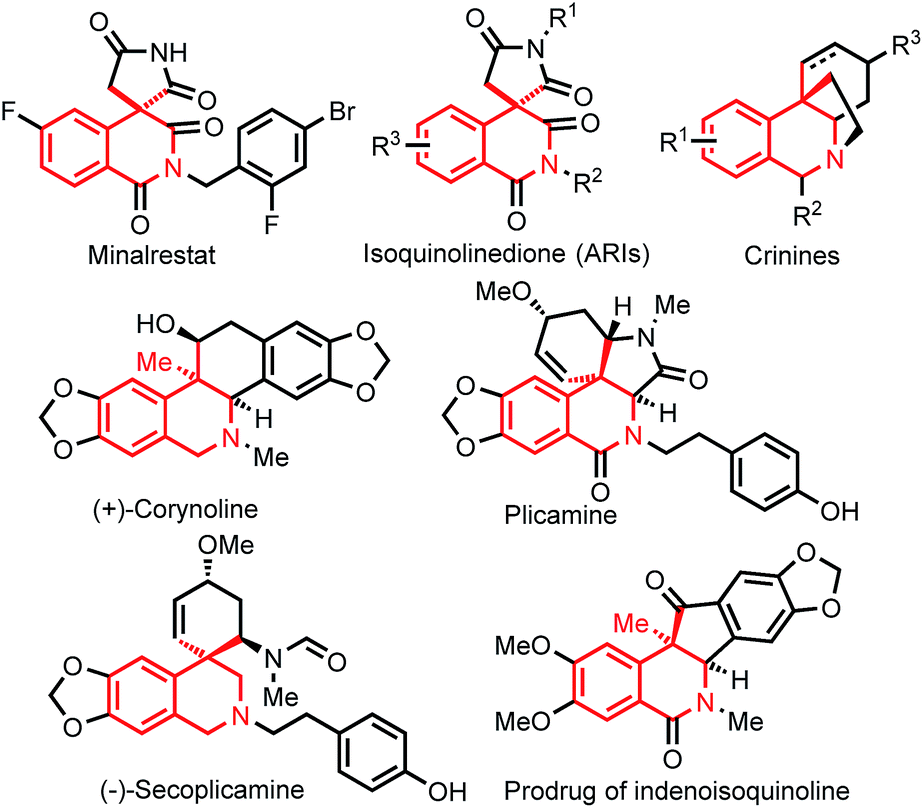 | ||
| Fig. 1 Bioactive 3,4-dihydroisoquinolinone derivatives containing all-carbon quaternary stereocenters at the 4-position. | ||
3,4-Dihydroisoquinolinone skeletons are widely found in natural products and are pivotal structural motifs in drug molecules,6 and many of the bioactive 3,4-dihydroisoquinolinone derivatives contain an all-carbon quaternary stereocenter at the 4-position7 (Fig. 1). As such, asymmetric reactions for the construction of such isoquinolinone skeletons would find wide applications in organic synthesis. Although diastereoselective formation of 3,4-dihydroisoquinolinones by using enantiorich substrates has been reported by the Lautens group (Fig. 2b),8 catalytic enantioselective reactions remain to be developed.
The asymmetric intramolecular Heck reaction has been intensively studied since its discovery by Shibasaki and Overman,9 and has found extensive applications in the synthesis of natural products.10 Notably, the alkylpalladium species formed by intramolecular carbopalladation in the intramolecular Heck reactions can be captured by external reagents. This enantioselective carbopalladation/trapping process provides a straightforward synthetic method for complex chiral heterocyclic compounds, in particular for those bearing all-carbon quaternary stereocenters. This domino intramolecular Heck reaction has gained considerable attention very recently, and a range of capturing reagents have been developed, including cyanide,11 iodide,12 azoles,13 isocyanides,14 terminal alkynes,15 and CO.16 The alkylpalladium species can also be captured by hydrides to form hydroarylated products,17 and the reactions can be catalyzed by other transition metals in addition to palladium.18 We envisioned that such asymmetric domino intramolecular Heck reaction should be a desirable method for the construction of the six-membered isoquinolinone skeleton in Minalrestat. However, almost all the current domino intramolecular Heck reactions involve the formation of five-membered rings, and six-membered cyclization reactions are scarce (Fig. 2).19,20
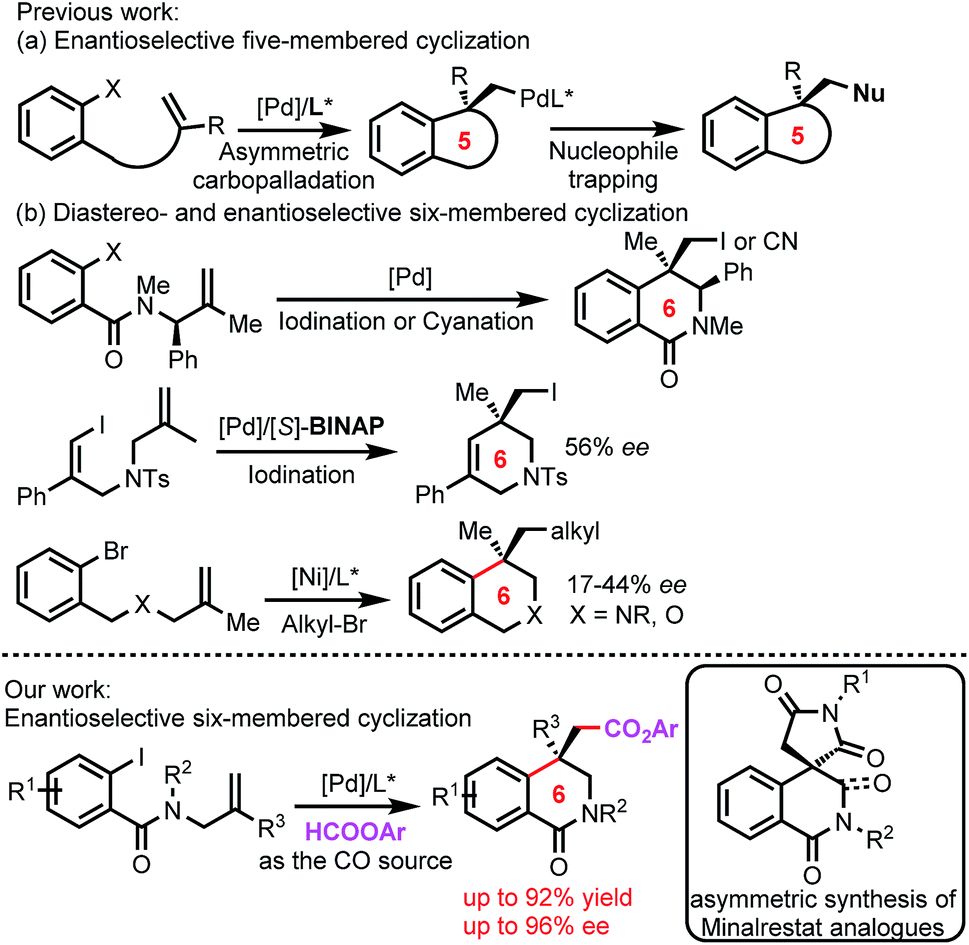 | ||
| Fig. 2 Stereoselective carbopalladation/trapping reactions. | ||
Orally active ARIs generally consist of two classes: the carboxylic acids and the cyclic imides. While Minalrestat and its analogues are the cyclic imide ARIs, 4-carboxyl isoquinolinones also showed activities for aldose reductase enzyme.4a Thus, transforming the alkylpalladium species in the carbopalladation/trapping reactions into a carboxyl or ester group should be ideal for the synthesis of isoquinolinone-based ARIs, because it can not only provide a carboxyl group directly but also the resulting carboxyl and ester groups can be converted to cyclic imides. Although carbonylation using CO as the trapping reagent has been reported in the domino intramolecular Heck reaction,16 like other reactions involving CO, the use of a toxic gas is undesirable. Formate esters are the desirable source of CO due to their commercial availability, low cost and low toxicity.21 However, they have not been used in the domino intramolecular Heck reaction. Herein, we report an enantioselective intramolecular carbonylative Heck reaction using formate esters as the CO source for the construction of six-membered isoquinolinone skeletons bearing an all-carbon quaternary stereocenter.
Results and discussion
We commenced our studies using benzamide 1a and phenyl formate 2a as the model substrates. After extensive survey of the conditions including the evaluation of various ligands, the desired carbonylated isoquinolinone 3aa was obtained in 42% yield and 81% ee by using (R)-SEGPHOS (L11) under the conditions as shown in Scheme 1.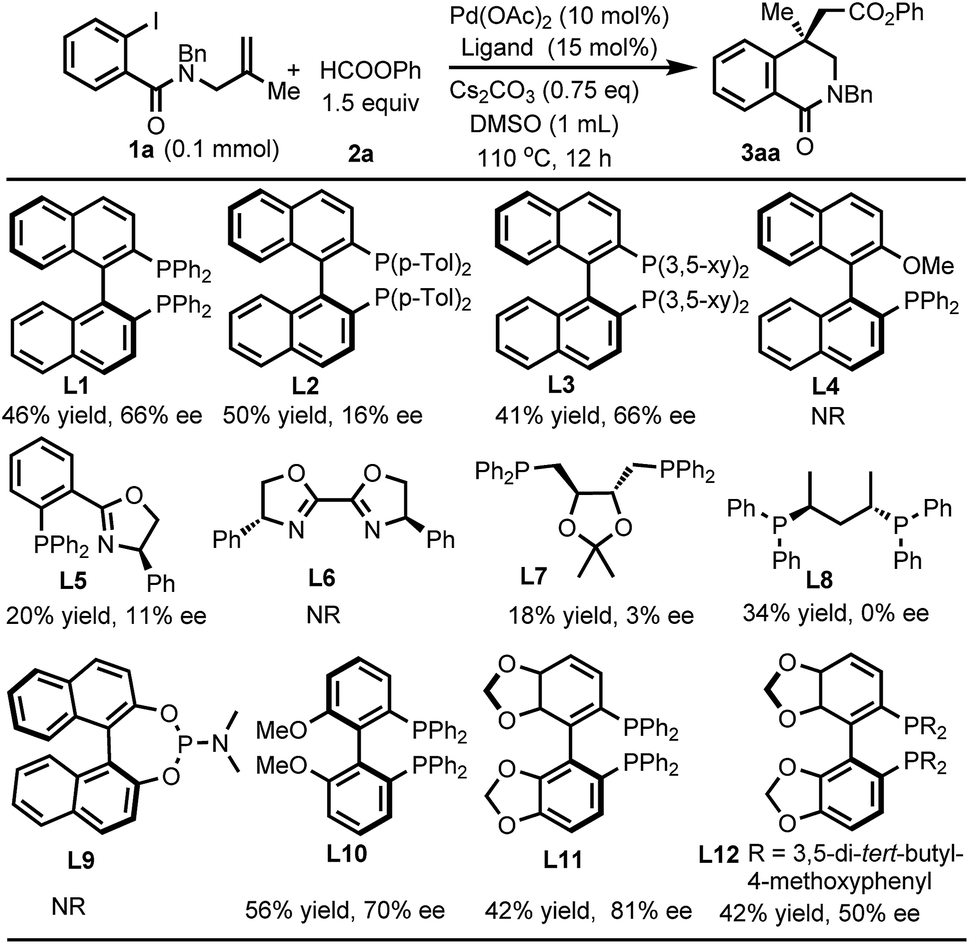 | ||
| Scheme 1 Ligands screened. | ||
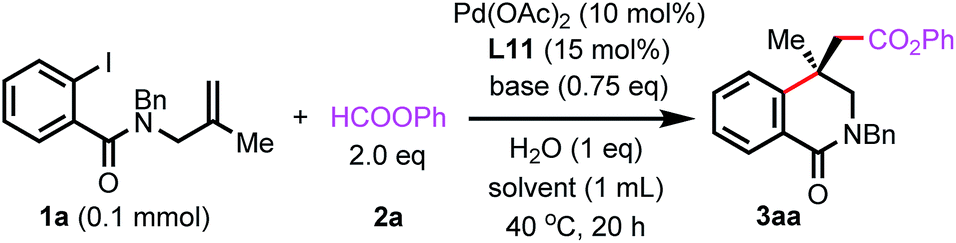
We further optimized the conditions using L11 to improve the yield and entantioselectivity. As shown in Table 1, replacing Cs2CO3 with other carbonates led to a dramatic increase in yields (entries 2–4). In the reaction using Cs2CO3, a side-product resulting from the capture of the alkylpalladium intermediate by a hydride was observed. The other carbonates suppressed the side reaction. By using Na2CO3 as the base, the ee value was enhanced to 93% when the reaction was carried out at 40 °C. However, the yield decreased to 50% (entry 5). The addition of 1 equivalent of H2O improved the yield to 75% (entry 6). Although the exact role of H2O remains to be investigated, it has been reported that H2O could accelerate the reduction of PdII to Pd0 by bidentate phosphine ligands.22 Reducing the amount of the ligand to 12 mol% failed to increase the yield, and the ee value remained unchanged (entry 7). The use of 1.5 equivalents of 2a gave a similar yield (entry 8). Gratefully, the yield was improved to 82% by increasing the quantity of Na2CO3 (entry 9) and further enhanced to 88% by running the reaction in 2 mL DMSO (entry 10). High enantioselectivity was also achieved when the reaction was carried out in NMP or DMF, albeit in a lower yield (entries 11 and 12). However, the reaction failed to form 3aa in other solvents. In these reactions, 1a just remained intact (entries 13–15). Although reducing catalyst and ligand loading led to lower yields, the ee values remained unchanged (entries 16 and 17).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Yield (%)a | ee (%)b |
| a Isolated yields. b Determined by HPLC analysis. c 110 °C. d Without 1 equiv. of H2O. e 12 mol% of L11. f 1.5 equiv. of 2a. g 1.5 equiv. of Na2CO3. h 2 mL solvent. i 5 mol% of Pd(OAc)2 and 6 mol% of L11. j 2 mol% of Pd(OAc)2 and 2.4 mol% of L11. | ||||
| 1 | Cs2CO3 | DMSO | 42c,d | 81 |
| 2 | K2CO3 | DMSO | 75c,d | 84 |
| 3 | Na2CO3 | DMSO | 78c,d | 86 |
| 4 | Li2CO3 | DMSO | 81c,d | 83 |
| 5 | Na2CO3 | DMSO | 50d | 93 |
| 6 | Na2CO3 | DMSO | 75 | 92 |
| 7 | Na2CO3 | DMSO | 75e | 92 |
| 8 | Na2CO3 | DMSO | 77e,f | 92 |
| 9 | Na2CO3 | DMSO | 82e,f,g | 92 |
| 10 | Na2CO3 | DMSO | 88e,f,g,h | 92 |
| 11 | Na2CO3 | NMP | 68e, | 93 |
| 12 | Na2CO3 | DMF | 56e,f,g,h | 87 |
| 13 | Na2CO3 | CH3CN | 0e,f,g,h | — |
| 14 | Na2CO3 | THF | 0e,f,g,h | — |
| 15 | Na2CO3 | Toluene | 0e,f,g,h | — |
| 16 | Na2CO3 | DMSO | 70f,g,h,i | 92 |
| 17 | Na2CO3 | DMSO | 40f,g,h,j | 92 |
Next, we investigated the substrate scope of the enantioselective intramolecular carbonylative Heck reaction. The compatibility of various functional groups on the benzene rings of the benzamides was first examined. As shown in Scheme 2, a range of electron-donating groups were compatible, and good or excellent ee values were obtained (3ba–3ea). In the reaction of 1e, the yield was low, and the major side-product was the compound resulting from the capture of the alkylpalladium species by a hydride. The yield was improved to 50% by running the reaction at 60 °C with a slight decrease of ee value. A slightly lower ee value and yield were observed for the benzamides bearing an electron-withdrawing group (3fa–3ha). Fluoro, chloro, and even bromo groups were well-tolerated, and the corresponding isoquinolinones were formed in high ee values (3ia–3ka). A fluoro group para to the amide group was compatible, and the corresponding product was obtained in 70% yield and 90% ee (3la). The substrates bearing a substituent at the ortho-position (3ma and 3na) or two substituents (3pa–3qa) were also suitable. Notably, heteroarene-derived amides also underwent the asymmetric reaction with high enantioselectivities (3ra and 3sa). It should be mentioned that the resulting structures are the core motifs in ubiquitous bioactive compounds, such as MAPKAP-K2 inhibitors23a and MCH receptor antagonists.23b
 | ||
| Scheme 2 Scope of benzamides with respect to substituents on the arene rings.[a] Isolated yields.[b] 60 °C. | ||
The reactions of benzamides bearing different substituents on the allyl groups were also investigated (Scheme 3). The ethyl, n-butyl, and benzyl groups were compatible (5aa–5ca), and even methoxy and p-methoxybenzyl groups were tolerated (5da and 5ea), which allows for further manipulation of the resulting products. In the presence of a bulky tert-butyl group, only a trace amount of desired product 5fa was formed. A phenyl or ester group suppressed the carbonylative Heck reaction (5ga and 5ha). In these reactions, the arylpalladium intermediates were directly carbonylated to form phenyl benzoates. For the reaction of the substrate bearing a benzyloxy group, carbopalladation occurred. However, the resulting alkylpalladium species underwent β-O-elimination before it reacted with CO (5ia). Furthermore, a range of functionalities on the amide groups were suitable, and the reactions were highly enantioselective (5ja–5na). However, the unprotected benzamide failed to form the desired product (5oa) and remained intact in the reaction. For phenyl-protected benzamide, only a trace amount of product 5pa was observed, and several unidentified side-products were formed.
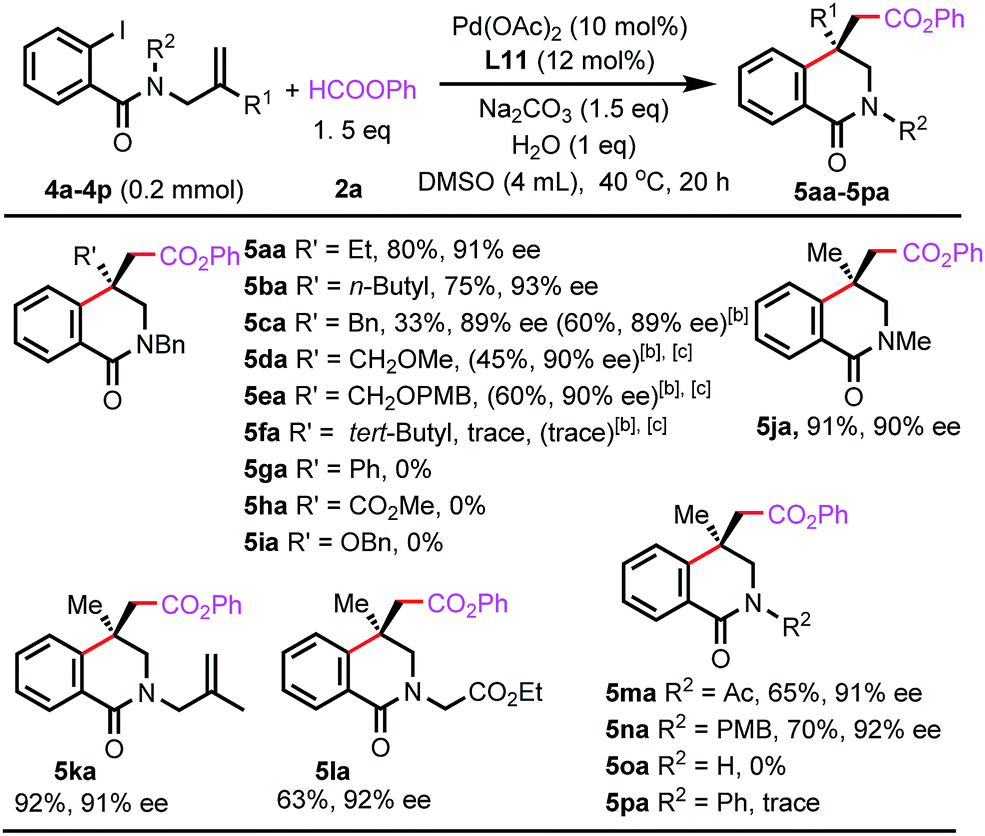 | ||
| Scheme 3 Scope of benzamides with respect to substituents on the allyl and amide groups.[a] Isolated yield.[b] 60 °C.[c] 1 equiv. of HCOOPh. | ||
The reaction scope with respect to aryl formates was also evaluated (Scheme 4). Aryl formates bearing an electron-donating methoxy group or electron-withdrawing ester group were efficient carbonylating reagents (3ab and 3ac). The chloro group was well-tolerated and naphthalen-2-yl formate was also suitable (3ad and 3ae). Reactions using alkyl formates were also investigated. The desired carbonylated isoquinolinone products were not observed for methyl or benzyl formate.
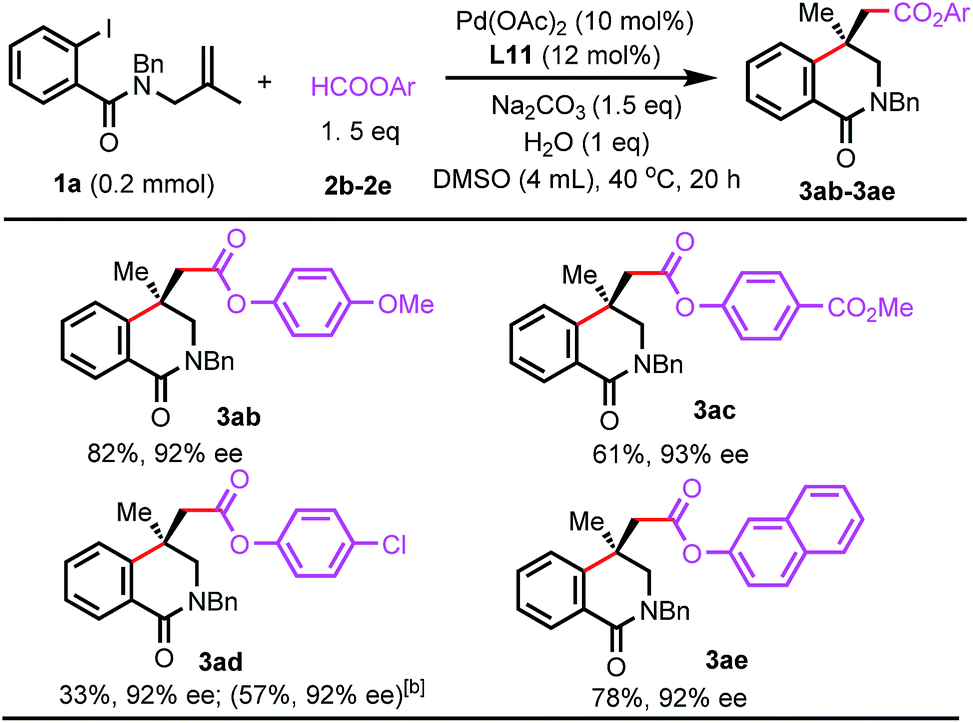 | ||
| Scheme 4 Scope of formate esters.[a] Isolated yield.[b] 60 °C. | ||
Next, we tried to synthesize Minalrestat with our enantioselective carbonylative Heck reaction. The quaternary carbon stereocenter in Minalrestat tends to racemize. However, the introduction of an N′-alkyl group could prevent the racemization, and it did not change the oral AR inhibitory potency of the N′-unsubstituted parent compounds. Furthermore, the analogues without the 6-fluoro group racemize more slowly and still have potent activity.4a Therefore, we chose Minalrestat analogue 10 as the target molecule (Scheme 5). On the other hand, a modular synthetic approach is highly desirable for drug discovery, because it would allow easy access to a number of analogues. To this end, we designed benzamide 4q as the substrate. 4q contains two PMB groups that can be readily removed. Gratefully, 4q underwent the carbonylative Heck reaction under the standard conditions to afford product 5qa in 93% ee. The two PMB groups could be removed using trifluoroacetic acid simultaneously, and the subsequent lactonization yielded 6. Lactone 6 was transformed into amide 7 with EtNH2. The free hydroxy group of 7 was oxidized with PCC, which led to the formation of succinimide 8. The α-methylene group of 8 was then oxidized to the carbonyl group, yielding the isoquinolinedione skeleton in 91% yield. The free amino group can be derivatized with various substituents for bioactivity studies. The 4-bromo-2-fluorobenzyl moiety could enhance the oral potency of aldose reductase inhibitors effectively,24 so it was introduced to the amide group of 9. The ee value of the final product 10 was 91%, which indicates that its quaternary carbon stereocenter did not racemize during the entire reaction process. Actually, the ee value of 10 remained unchanged even after one week.
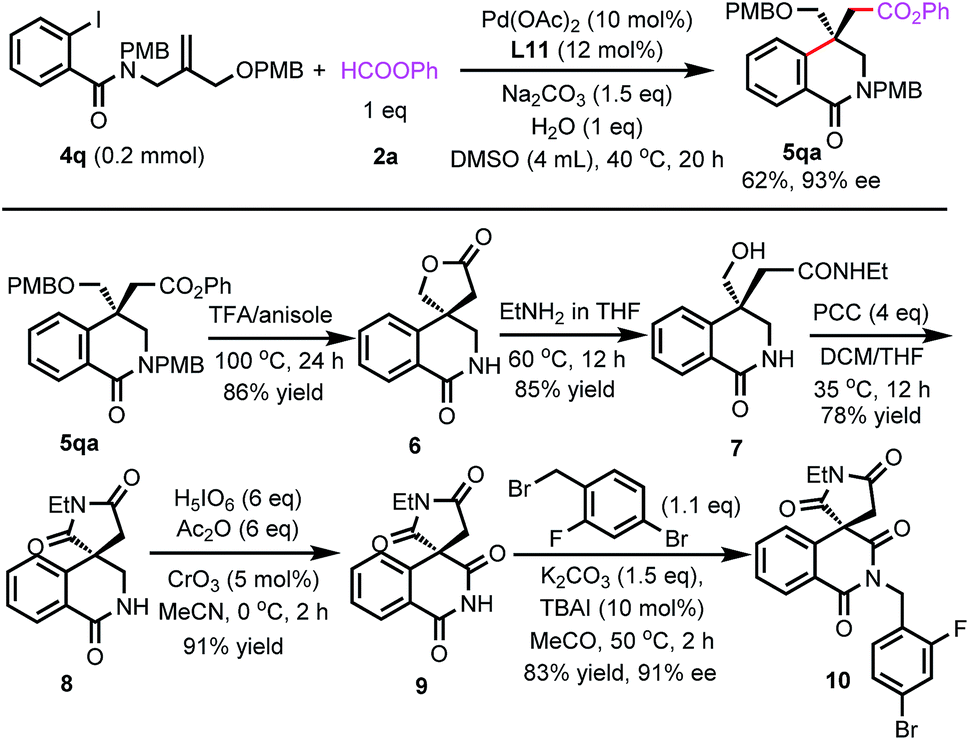 | ||
| Scheme 5 Synthesis of the Minalrestat analogue. | ||
Although the introduction of an N′-alkyl group can prevent the racemization of Minalrestat and its analogues, the acidity of the succinimides is very critical to the intrinsic activity of the parent molecules. The reason for the N′-alkylated analogues having equal oral AR inhibitory potency should be that the alkyl groups could be removed through biotransformation.4a The regenerated (N–H) compounds can still racemize, which results in the reduction of potency or might even cause unexpected side-effects. Therefore, it is highly desirable to develop new structures as ARIs that do not racemize. The racemization of Minalrestat is caused primarily by the presence of the 3-carbonyl group. Our asymmetric carbonylative Heck reaction provides a facile method for the synthesis of isoquinolinones. Therefore, we synthesized a decarbonylated analogue of the isoquinolinediones. As shown in Scheme 6, compound 6 was first allowed to react with 4-bromo-2-fluorobenzyl bromide to give 11. The subsequent aminolysis with NH3 and oxidation with PCC afforded isoquinolinone 13. The ee value of 13 was 95%, which indicates the high stability of the quaternary carbon stereocenter.
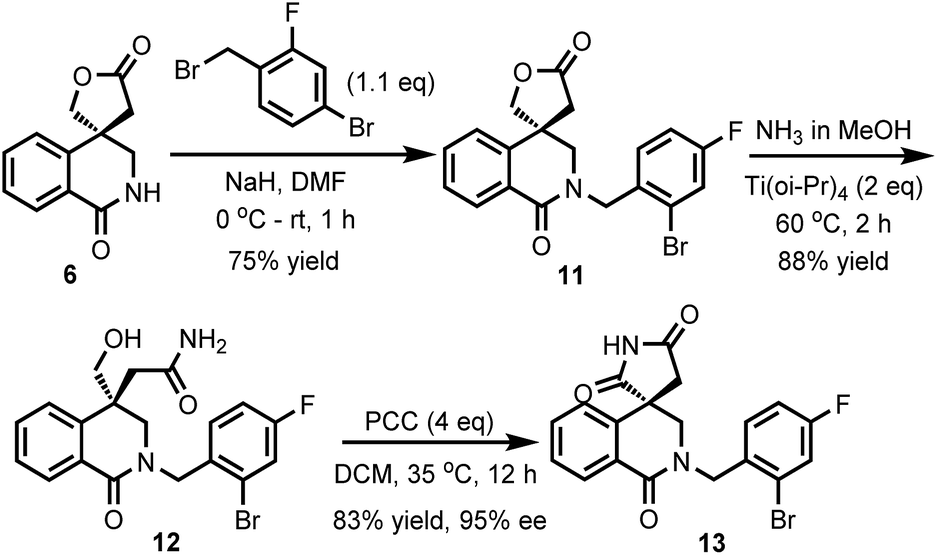 | ||
| Scheme 6 The synthesis of the non-racemizable derivative of Minalrestat. | ||
The isoquinolinone products can be readily transformed into other quaternary stereocarbon-containing compounds (Scheme 7). For example, the amide group could be reduced to give 14. It should be noted that the resulting 4,4-disubstituted 1,2,3,4-tetrahydroisoquinoline represents an essential skeleton widely found in bioactive compounds such as crinanes7d and Secoplicamine.7c Furthermore, the ester group could be hydrolyzed to give compound 15. Notably, 4-carboxyl isoquinolinediones also showed activities for aldose reductase enzyme.4b Finally, 15 was transformed to amide 16. The structure of 16 was characterized by X-ray crystallography.
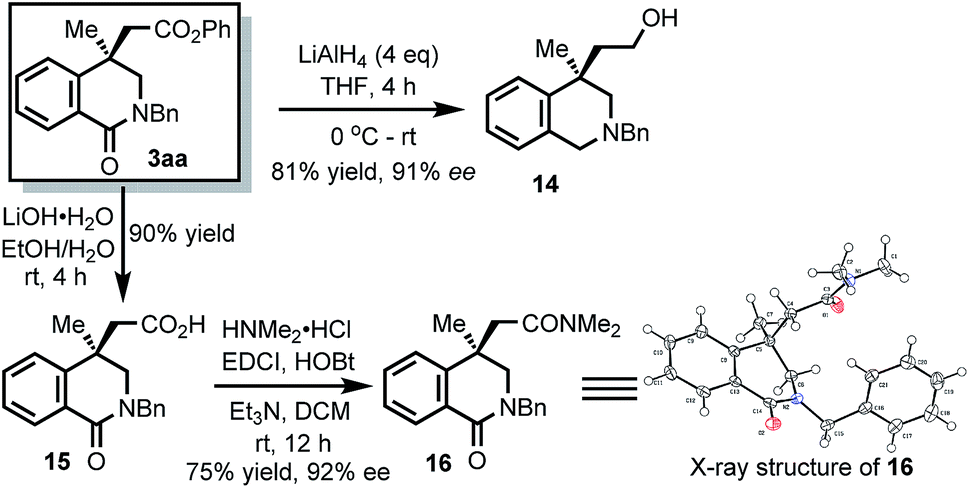 | ||
| Scheme 7 Transformation of the isoquinolinone product. | ||
Base on the previous report,16a a plausible mechanism for the Pd-catalyzed Heck carbonylation is depicted in Scheme 8. The oxidative addition of aryl iodide 1a to a Pd0 species that is generated in situ forms PdII intermediate I. I undergoes intramolecular carbopalladation to afford σ-alkyl PdII complex II. Phenyl formate 2b generates CO and phenol with Na2CO3 as the base.25 The migratory insertion of CO into the σ-alkyl–Pd bond gives acylpalladium complex III. The nucleophilic attack of the phenol and subsequent reductive elimination give the final product 3aa with regeneration of Pd0 catalyst.
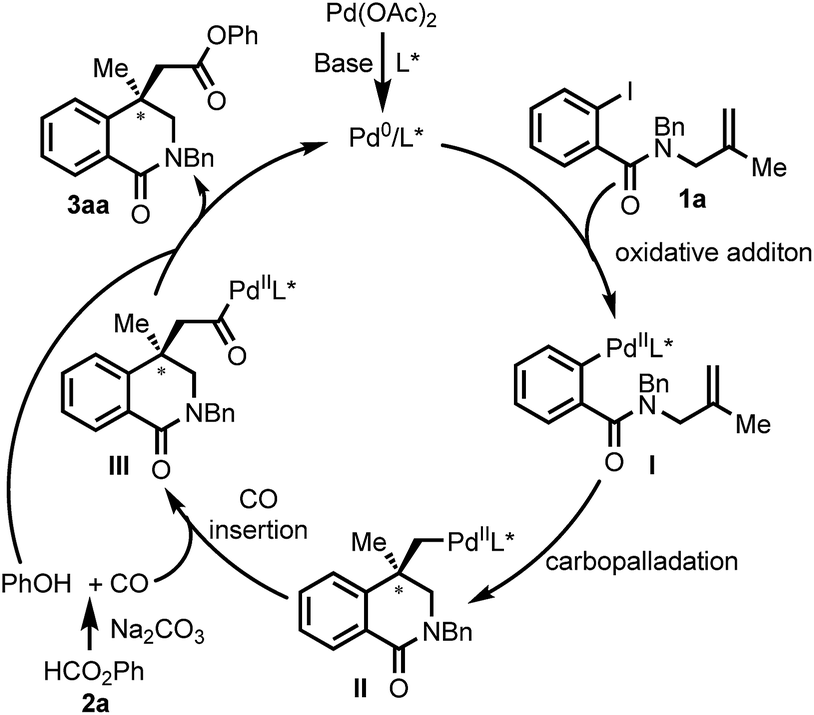 | ||
| Scheme 8 Plausible mechanism. | ||
Conclusions
In summary, we have developed a Pd-catalyzed enantioselective intramolecular carbonylative Heck reaction by using formate esters as the source of CO. A range of N-allyl benzamides were transformed into isoquinolinones bearing an all-carbon quaternary center in good yields and high enantioselectivities in the presence of (R)-SEGPHOS as the ligand. This reaction represents one of the rare examples of asymmetric six-membered cyclization reactions through a domino intramolecular Heck-nucleophilic capture sequence. The resulting isoquinolinone represents the core skeleton in ubiquitous bioactive compounds, and therefore the reaction should have great potential to be applied in organic synthesis. The reaction has been successfully applied to the first asymmetric construction of the isoquinolinedione skeleton in Minalrestat and allows easy access to a range of Minalrestat analogues for developing new aldose reductase inhibitors. Synthesizing other Minalrestat analogues and testing their bioactivities are currently underway in our laboratory.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by the National Natural Science Foundation of China (No. 21672162).Notes and references
- A. Pau, B. Asproni, G. Murineddu, G. Boatto, G. E. Grella, D. Rakowitz, L. Costantino and G. A. Pinna, Med. Chem., 2006, 2, 39 CrossRef CAS
.
- M. Brownlee, Diabetes, 2005, 54, 1615 CrossRef CAS PubMed
- R. Maccari and R. Ottaná, J. Med. Chem., 2015, 58, 2047 CrossRef CAS PubMed
-
(a) M. S. Malamas, T. C. Hohman and J. Millen, J. Med. Chem., 1994, 37, 2043 CrossRef CAS PubMed
- The asymmetric reaction for the synthesis of Ranirestat is limited to pyrrole-based substrates: T. Mashiko, K. Hara, D. Tanaka, Y. Fujiwara, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 11342 CrossRef CAS PubMed
-
(a) G. R. Pettit, S. Ducki, S. A. Eastham and N. Melody, J. Nat. Prod., 2009, 72, 1279 CrossRef CAS PubMed
-
(a) I. Ninomiya, O. Yamamoto and T. Naito, J. Chem. Soc., Chem. Commun., 1976, 437 RSC
-
(a) M. Cushman and J.-K. Chen, J. Org. Chem., 1987, 52, 8 Search PubMed
-
(a) Y. Sato, M. Sodeoka and M. Shibasaki, J. Org. Chem., 1989, 54, 4738 CrossRef CAS
- Selected reviews:
(a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed
- A. Pinto, Y. Jia, L. Neuville and J. Zhu, Chem.–Eur. J., 2007, 13, 961 CrossRef CAS PubMed
-
(a) H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187 CrossRef CAS PubMed
-
(a) W. Kong, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 16028 CrossRef CAS PubMed
- W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 9714 CrossRef CAS PubMed
- R. Liu, Y. Wang, Y. Li, B. Huang, R. Liang and Y.-X. Jia, Angew. Chem., Int. Ed., 2017, 56, 7475 CrossRef CAS PubMed
-
(a) R. C. Carmona, O. D. Köster and C. R. D. Correia, Angew. Chem., Int. Ed., 2018, 57, 12067 CrossRef CAS PubMed
-
(a) P. Diaz, F. Gendre, L. Stella and B. Charpentier, Tetrahedron, 1998, 54, 4579 CrossRef CAS
-
(a) H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed
- For two asymmetric six-membered cyclization examples with low enantioselectivities, see: ref. 12a and 18e. Although an asymmetric reaction of vinyl iodides to form six-membered tetrahydropyridines has been reported, a mechanism involving the vinylborylation of alkenes is preferred.
(a) Z. Jiang, L. Hou, C. Ni, J. Chen, D. Wang and X. Tong, Chem. Commun., 2017, 53, 4270 RSC
- For selected racemic six-membered cyclization reactions, see:
(a) R. Grigg, S. Sukirthalingam and V. Sridharan, Tetrahedron Lett., 1991, 32, 2545 CrossRef CAS
- D. Formenti, F. Ferretti and F. Ragaini, ChemCatChem, 2018, 10, 148 CrossRef CAS
- F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 21, 2177 CrossRef
-
(a) J.-P. Wu, J. Wang, A. Abeywardane, D. Andersen, M. Emmanuel, E. Gautschi, D. R. Goldberg, M. A. Kashem, S. Lukas, W. Mao, L. Martin, T. Morwick, N. Moss, C. Pargellis, U. R. Patel, L. Patnaude, G. W. Peet, D. Skow, R. J. Snow, Y. Ward, B. Werneburg and A. White, Bioorg. Med. Chem. Lett., 2007, 17, 4664 CrossRef CAS PubMed
-
D. Stribling and D. R. Brittan, Structure Activity Relationships of Aldose Reductase Inhibitors and Their Possible Use in Diabetic Complications, in Innovative Approaches in Drug Research, ed. A. F. Hams, Elsevier Science Publishers B. V., Amsterdam, 1986, pp. 297–313 Search PubMed
- T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2011, 14, 3100 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation. CCDC 1921698. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03406d |
| This journal is © The Royal Society of Chemistry 2019 |