
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmon-promoted electrocatalytic water splitting on metal–semiconductor nanocomposites: the interfacial charge transfer and the real catalytic sites†‡
Lili
Du
,
Guodong
Shi
,
Yaran
Zhao
,
Xiang
Chen
,
Hongming
Sun
,
Fangming
Liu
,
Fangyi
Cheng
 and
Wei
Xie
*
and
Wei
Xie
*
Key Lab of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center, College of Chemistry, Nankai University, Weijin Rd. 94, Tianjin 300071, China. E-mail: wei.xie@nankai.edu.cn
First published on 29th August 2019
Abstract
Plasmonic metal nanoparticles (NPs) have emerged as promising visible light harvesters to facilitate solar-to-chemical energy conversion via the generation of hot electrons by non-radiative decay of plasmons. As one of the most promising renewable energy production methods for the future, electrocatalytic water splitting is an ideal chemical reaction in which plasmonic NPs can be utilized for direct solar-to-fuel conversion. Due to the rapid carrier recombination on plasmonic NPs, hybrid photocatalysts integrating metals and semiconductors are usually employed to separate the hot electrons and holes. However, an understanding of the catalytic mechanism, which is critical for rational design of plasmonic electrocatalysts, including the interfacial charge transfer pathway and real reactive sites, has been lacking. Herein, we report on the combination of plasmonic Au NPs and semiconductors (Ni and/or Co hydroxides) for plasmon-promoted electrocatalytic water splitting. By using surface-enhanced Raman spectroscopy (SERS), we find a strong spontaneous interfacial charge transfer between Au and NiCo layered double hydroxide (LDH), which facilitates both the oxygen and hydrogen evolution reactions. The real catalytic sites on the hybrid material are confirmed by selective blocking of the metal surface with a thiol molecular monolayer. It is found that the plasmon-promoted oxygen evolution occurs on the LDH semiconductor but surprisingly, the hydrogen evolution sites are mainly located on the Au NP surface. This work demonstrates the critical role of interfacial charge transfer in hot electron-driven water splitting and paves the way for rational design of high-performance plasmonic electrocatalysts.
Introduction
Plasmonic metal NPs are excellent visible light harvesters.1–3 Under resonant irradiation, their optical absorption and scattering are enhanced due to the localized surface plasmon resonance (LSPR), a collective oscillation of free electrons driven by electromagnetic waves.4–8 The surface plasmons undergo non-radiative decay and generate hot electron–hole pairs that are energy sources for chemical reactions.9–11 Specifically, the hot holes can act as electron trappers to capture external electrons and promote oxidation reactions.12 Meanwhile, the hot electrons can be utilized in reduction reactions taking place on the metal surface.13–16Electrocatalytic water splitting involving both oxidation (oxidation evolution reaction, OER) and reduction (hydrogen evolution reaction, HER) half-reactions is one of the most promising clean and renewable energy production methods for the future.17–20 It is therefore an ideal chemical reaction for the application of plasmonic NPs, in which solar energy can be directly converted to hydrogen fuel.21 Because of the rapid charge-carrier recombination and the intrinsically low electrocatalytic activity of plasmonic NPs, the plasmonic NPs were usually employed together with conventional semiconductor catalysts to separate the charge carriers.22,23 Au NP-decorated Ni(OH)2 nanosheets and Au nanorod-decorated MoS2 nanosheets represent the few examples of plasmonic metal–semiconductor electrocatalysts for the OER and HER, respectively.24,25 In oxygen production, the hot holes facilitate electron transfer from a conventional semiconductor catalyst to plasmonic metal NPs and thus enhance the water oxidation on the semiconductor surface. In contrast, the hot electrons transferred from the metal to the semiconductor were thought to be responsible for the plasmon-promoted hydrogen production. Generally, the semiconductors were believed to be the catalytically active sites for both oxidation and reduction of water. However, experimental evidence of the real catalytic site on the hybrid surfaces has been lacking. To design high performance plasmonic electrocatalysts for visible light-assisted water splitting, it is important to know how the hot carriers are transferred and where the catalytic sites of the reactions are.
Besides the hot carriers, plasmonic metal NPs also provide localized electromagnetic fields that enable surface-enhanced Raman spectroscopic (SERS) detection of adsorbed chemical species.26,27 In the past decade, SERS has been proved to be a powerful analytical technique for interfacial characterization.28 Because of the high sensitivity, surface selectivity and fingerprint specificity, SERS is very sensitive to the electronic properties of the metal surface and therefore promising for investigating the interfacial charge transfer to understand the mechanism of plasmon-promoted electrocatalysis.29,30
In this study, we prepared Au NP-decorated Co(OH)2, Ni(OH)2 and Ni–Co layered double hydroxide (Au/Co(OH)2, Au/Ni(OH)2 and Au/NiCo LDH) as plasmonic electrocatalysts for water splitting under visible light irradiation. The hydroxide semiconductors are conventional water splitting electrocatalysts, while the Au NPs with stable chemical properties and strong plasmonic activity act as visible light harvesters.31–33 Before the water splitting experiments, we employed SERS to study the interfacial charge transfer between the hydroxide semiconductors and the decorated Au NPs in a semiconductor–metal–molecule hybrid system, where 2,6-dimethylphenyl isocyanide (DMPIC) was used as a probe molecule. DMPIC interacts with the metal surface via orbital hybridization and the strength of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond is affected by the electron abundance of the SERS substrates (Au NPs).34 According to the SERS spectra, we can determine if the plasmonic Au NPs lose or gain electrons after immobilizing them on the different hydroxides.
C bond is affected by the electron abundance of the SERS substrates (Au NPs).34 According to the SERS spectra, we can determine if the plasmonic Au NPs lose or gain electrons after immobilizing them on the different hydroxides.
Finding out the real catalytic site on a hybrid surface is very challenging. The distinct Au and hydroxide surfaces offer the opportunity of selective coating of thiol molecules on the Au surface. After coating, although the semiconductor surface remains unchanged, the Au surface will not be available any more for the reactions. We incubated the Au/NiCo LDH hybrid material in a solution of thiophenol (TP) before the visible light-assisted electrocatalytic water splitting. Interestingly, a remarkably decreased catalytic activity of the Au/NiCo LDH hybrid material was detected in the HER, while no significant decrease was observed in the OER. It is clear that the main catalytic sites of the OER and HER are different. Based on the SERS study of the interfacial charge transfer between Au NPs and NiCo LDH, a reasonable mechanism of the plasmon-promoted electrocatalysis has been proposed. This new mechanism is different from the previously proposed ones: (1) the electron transfer in the plasmonic metal–semiconductor hybrid system is unidirectional, both in the OER and HER. (2) The HER takes place mainly on the plasmonic Au surface. The present study highlights the important role of the interfacial charge transfer between a plasmonic light harvester and a conventional electrocatalyst, and also identifies the real catalytic sites on the hybrid electrocatalyst for plasmon-promoted electrocatalytic water splitting.
Experimental
Materials
Chloroauric acid (HAuCl4, Sigma-Aldrich), sodium borohydride (NaBH4, Sigma-Aldrich), cobalt acetate (Co(Ac)2, 99%, Aladdin), nickel acetate (Ni(Ac)2, 99%, Aladdin), 2,6-dimethylphenyl isocyanide (98%, Energy-Chemical), methanol (Tianjin Kangkede Technology Co., Ltd.), potassium hydroxide (KOH, 97%, Aladdin), and Ni foam (Innochem) were used without further purification.Synthesis of NiCo LDH, Ni(OH)2 and Co(OH)2
NiCo LDH was prepared through a modified solvothermal method.35 Ni(Ac)2·4H2O (1 mmol) and Co(Ac)2·4H2O (0.5 mmol) were dissolved together in 15 mL methanol. After stirring for 30 min, the mixture was transferred into a 50 mL Teflon-lined autoclave with a piece of treated Ni foam inside. The autoclave was then sealed and kept in an oven at 180 °C for 12 hours and naturally cooled down to room temperature. The substrate was washed several times with deionized water/ethanol and dried in an oven at 60 °C for 2 hours. The catalyst loading on the Ni foam was determined to be ∼1.4 mg cm−2. A powdery sample was prepared using the same method, just without the presence of Ni foam. Ni(OH)2 and Co(OH)2 were prepared using similar methods.Synthesis of Au NPs
According to a previous study,36 Au nanoparticles were synthesized by NaBH4 (0.1 M) reduction of HAuCl4 (0.25 mM). The color of the colloid is wine red, indicating the formation of plasmonic Au nanoparticles. Then, 1 mL toluene was added to a Petri dish containing 5 mL colloid of Au NPs. Subsequently, 0.5 mL dimethylformamide (DMF) was added dropwise. After the evaporation of methylbenzene, a compact Au NP film was formed at the water/oil interface. The film was then transferred gently to a piece of Ni foam to form the Au NPs/Ni foam electrode.37Synthesis of Au/NiCo LDH, Au/Ni(OH)2, and Au/Co(OH)2
NiCo LDH was decorated with Au NPs via electrostatic interactions. NiCo LDH supported on Ni foam was immersed into a colloidal suspension of Au NPs and shaken slightly until the colloid became colorless. After complete immobilization of the Au NPs on the NiCo LDH, the Au/NiCo LDH nanocomposites were dried in air. For comparison, 5, 10 and 15 mL Au colloids were used and the Au mass percentage was determined to be 1.7, 3.3, and 5.1 wt%, respectively. Au/Ni(OH)2 and Au/Co(OH)2 were prepared using the same method (10 mL Au colloid).Preparation of DMPIC-functionalized Au NP, Au/NiCo LDH, Au/Ni(OH)2 and Au/Co(OH)2 substrates for SERS measurements
Powdery NiCo LDH, Ni(OH)2 and Co(OH)2 were dispersed in 1 mL Au colloid (60 nm). Subsequently, 10 μL of 10 mM ethanolic DMPIC solution was added. The mixture was incubated for 30 min to allow the formation of a molecular monolayer on the Au surface. The samples were then washed two times with ethanol and resuspended in 1 mL water. The prepared samples were directly used for SERS measurements.Characterization
SEM images were obtained on a JEOL JSM-7500F electron microscope and high-resolution TEM was performed on a Tecnai G2 F20 S-TWIN at 200 kV. X-ray diffraction patterns were recorded on a Rigaku Mini Flex 600 powder diffractometer (Cu Kα radiation). The surface material valence states of the composite were analyzed using X-ray photoelectron spectroscopy (XPS) (Thermo Scientific ESCALAB 250Xi). The potential values of samples dispersed in water were determined on a Malvern Nano ZS90 Zetasizer. The extinction of the samples was measured using a UV-Vis spectrometer (PerkinElmer Lambda 750). The content of Au was determined by ICP-AES (PerkinElmer Optima 8300).SERS measurements
The SERS spectra were acquired with a Horiba LabRAM HR Evolution Raman microscope with an excitation wavelength of 632.8 nm for all measurements. And the measurements under different potentials were performed by using a homemade two-electrode configuration with a CHI-660E electrochemical workstation to control the applied potential.Photoelectrochemical (PEC) measurements
PEC measurements were carried out on an electrochemical workstation (CHI660E, Shanghai, CH. Instruments, China) using a typical three electrode system, and a solar simulator (NBeT Solar-150 Inc.) applying AM 1.5G irradiation was employed as the light source (100 mW cm−2). Colored sources were supported by filters with different passing wavelengths. A graphite rod and standard Hg/HgO electrode were used as the counter and reference electrodes, respectively, in an aqueous solution of 1 M KOH. The OER catalytic activity was investigated using linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1 in an O2-saturated solution of KOH, while the HER tests were carried out using LSV at a sweep rate of 1 mV s−1 in a H2-saturated solution. The water splitting electrolysis was performed in 1 M KOH using a two-electrode configuration. All tested potentials vs. the Hg/HgO electrode were converted to the RHE scale according to the Nernst equation ERHE = EHg/HgO + 0.0591 × pH + 0.098 V. All polarization curves were reported with iR compensation for ohmic losses: E = Em − iRs, where E is the iR-corrected potential, Em is the measured potential and Rs is the resistance of the solution. Electrochemical impedance spectra (EIS) were measured at an overpotential of 250 mV from 0.1 Hz to 100 KHz.Results and discussion
Characterization of nanocomposites
Fig. S1‡ shows the preparation process of Au/NiCo LDH. TEM images of the NiCo LDH prepared via a solvothermal method are shown in Fig. S2.‡ Au NPs with a size of ∼5.75 nm (Fig. S3‡) were prepared by NaBH4 reduction of HAuCl4. Because of the opposite interfacial charges (Fig. S4‡), the plasmonic Au NPs were successfully loaded on the hydroxides via electrostatic interactions, forming the Au/NiCo LDH nanocomposites. The SEM/TEM images and the energy dispersive X-ray spectroscopy (EDS) mapping images shown in Fig. 1 reveal that the Au NPs were uniformly interspersed throughout the surface of NiCo LDH nanosheets. Using similar methods, Au NP-decorated Co(OH)2 and Ni(OH)2 were successfully prepared and the characterization results are shown in Fig. S5–S7‡. | ||
| Fig. 1 Characterization of the Au/NiCo LDH. SEM (a), TEM (b), high-resolution TEM (c), and EDS elemental mapping images (d) of the Au/NiCo LDH nanocomposite. | ||
Detection of the interfacial charge transfer by using SERS
As a surface-sensitive molecular spectroscopic technique, SERS was used to investigate the interfacial charge transfer between the Au NPs and the conventional hydroxide electrocatalysts (semiconductor). The NC symmetric stretching of DMPIC located within the spectral range of 2150 to 2200 cm−1 is very sensitive to the electronic structure of the metal surface.38 In this work, we used DMPIC as a molecular probe and compared the NC stretching enhanced by Au NPs immobilized on the three different semiconductors: Co(OH)2, Ni(OH)2 and NiCo LDH. Fig. 2a illustrates the NC band shift due to the metal–molecule electron transfer. The DMPIC molecule interacts with the Au surface via the σ lone electron pair and unoccupied π* frontier orbitals of NC, which hybridize with the d orbitals of the metal to form σ–d donation and d–π* back donation.39 Electron transfer from the support material to the metal decreases the σ–d interaction but enhances the d–π* back-donation; this weakens the NC bond strength and leads to a red-shift of the SERS band.40 To verify the relation between the band shift and the interfacial charge transfer, we constructed a two-electrode system and the DMPIC modified Au NPs were immobilized on the working electrode. As shown in Fig. 2b, under an external voltage (−0.4 to 0 V), the SERS band of NC red-shifts. In contrast, a blue-shift of the band is observed on the electron-deficient Au at positive potentials (0 to +0.4 V). Fig. 2c shows the corresponding SERS spectra in the spectral range from 2100 to 2250 cm−1. The NC stretching on the Co(OH)2, Ni(OH)2 and NiCo LDH exhibits a red-shift of 8, 12 and 14 cm−1, respectively, compared with the SERS band at 2172 cm−1 on Au NPs in colloidal suspension (Fig. 2c, red curve). According to the band position, the electron transfer to Au NPs from Co(OH)2, Ni(OH)2 and NiCo LDH corresponds to an external potential of −0.19, −0.29 and −0.34 V, respectively.
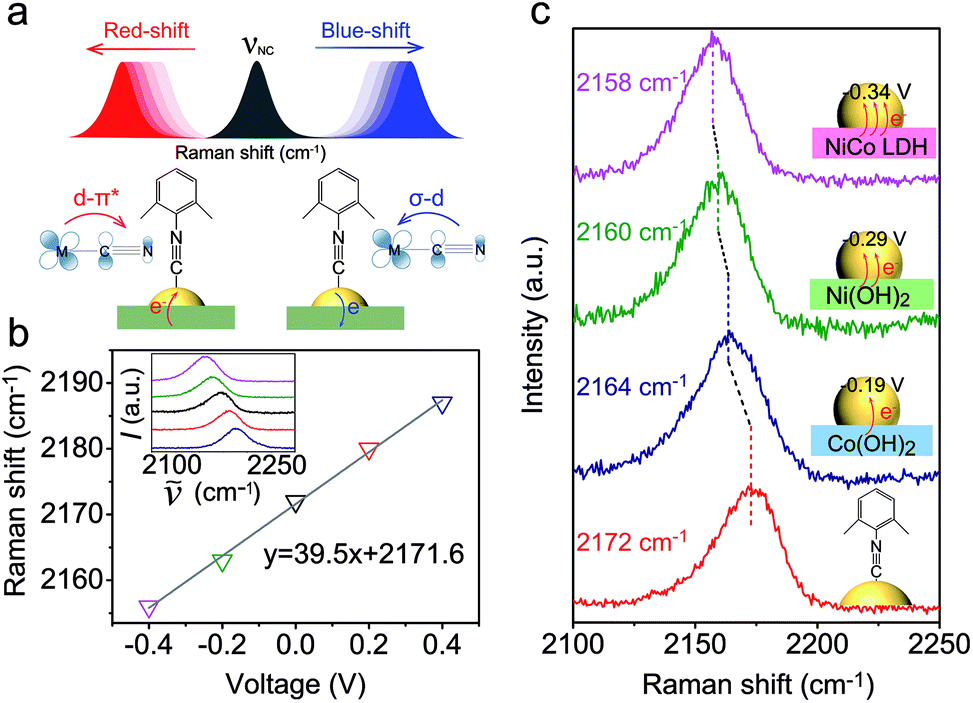 | ||
| Fig. 2 SERS detection of interfacial charge transfer between Au NPs and semiconductors. (a) Schematic illustration of the interfacial charge transfer in the semiconductor–metal–molecule (DMPIC) system and the corresponding SERS band shift. (b) Raman shift of the symmetric stretching mode of NC as a function of applied voltage. The inset is the original SERS spectra showing the corresponding bands. (c) SERS spectra of DMPIC (from the bottom to the top) enhanced by Au NPs in colloidal suspension, Au/Co(OH)2, Au/Ni(OH)2, and Au/NiCo LDH, respectively. | ||
NiCo LDH is a better electron donor for Au NPs compared with Co(OH)2 and Ni(OH)2. This spontaneous electron transfer is further confirmed by XPS analysis. Compared with the pristine NiCo LDH, the binding energy of Ni and Co in the Au/NiCo LDH is positively shifted by 0.3 and 0.4 eV, respectively, suggesting the strong tendency of electron transfer from NiCo LDH to Au NPs (Fig. S8‡).25
Charge transfer promoted electrocatalytic activity
The electrocatalytic performances of Au/NiCo LDH, Au/Ni(OH)2 and Au/Co(OH)2 nanocomposites were evaluated. As shown in Fig. S9,‡ the Au/NiCo LDH exhibits the best performance in the OER among the three Au NP-decorated semiconductors. Compared with the corresponding materials without Au NPs, Au/NiCo LDH also shows the largest improvement of catalytic activity upon the Au NP decoration (Fig. 3a), which can be attributed to the enhanced oxidative reactivity of NiCo LDH after donating electrons to the metal. The same trend was observed in the HER (Fig. 3b): the electron-abundant Au may contribute additional reductive sites for the hydrogen evolution (vide infra). Taken together, the spontaneous charge transfer between Au and hydroxides promotes the intrinsic electrocatalytic activity of the materials in both the OER and HER. Because of the highest charge transfer tendency and catalytic activity among the three nanocomposites, we chose Au/NiCo LDH as the plasmonic electrocatalyst for water splitting under visible light irradiation.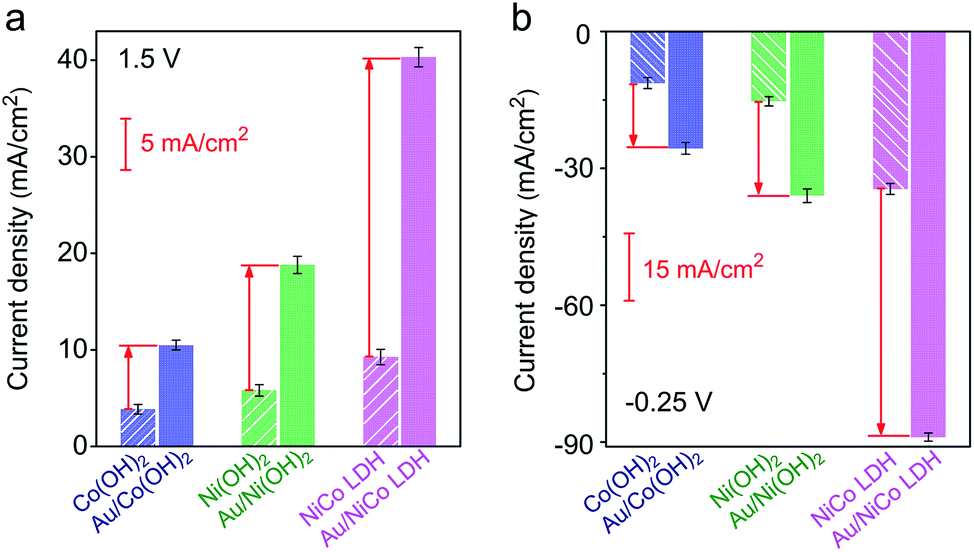 | ||
| Fig. 3 Electrochemical performance before and after decoration of Au NPs. Current densities of Co(OH)2, Ni(OH)2 and NiCo LDH with (solid column) and without (striped column) Au NPs toward the OER (a) and HER (b). | ||
Photoelectrocatalytic performance of Au/NiCo LDH for water splitting
Upon resonant electromagnetic excitation, the plasmonic hot electrons generated by Au NPs were expected to further improve the electrocatalytic performance. The experiment was performed using a typical three-electrode system to investigate the electrocatalytic water splitting under visible light irradiation. As shown in Fig. 4a and b, to reach a current density of 10 mA cm−2, the Au/NiCo LDH requires an overpotential of 203 and 119 mV for the OER and HER, respectively (details are shown in Fig. S10‡). Both the Tafel slopes of 42.3 mV dec−1 in the OER and 57.5 mV dec−1 in the HER are much lower than the corresponding values obtained from control experiments by using NiCo LDH or Au NPs. Fig. S11‡ shows the electrochemical impedance spectra (EIS) and the corresponding equivalent circuit model. It can be obviously seen that the Au/NiCo LDH possesses the lowest charge-transfer resistance under irradiation, suggesting that the plasmonic excitation of Au leads to higher charge transport efficiency in the electrode.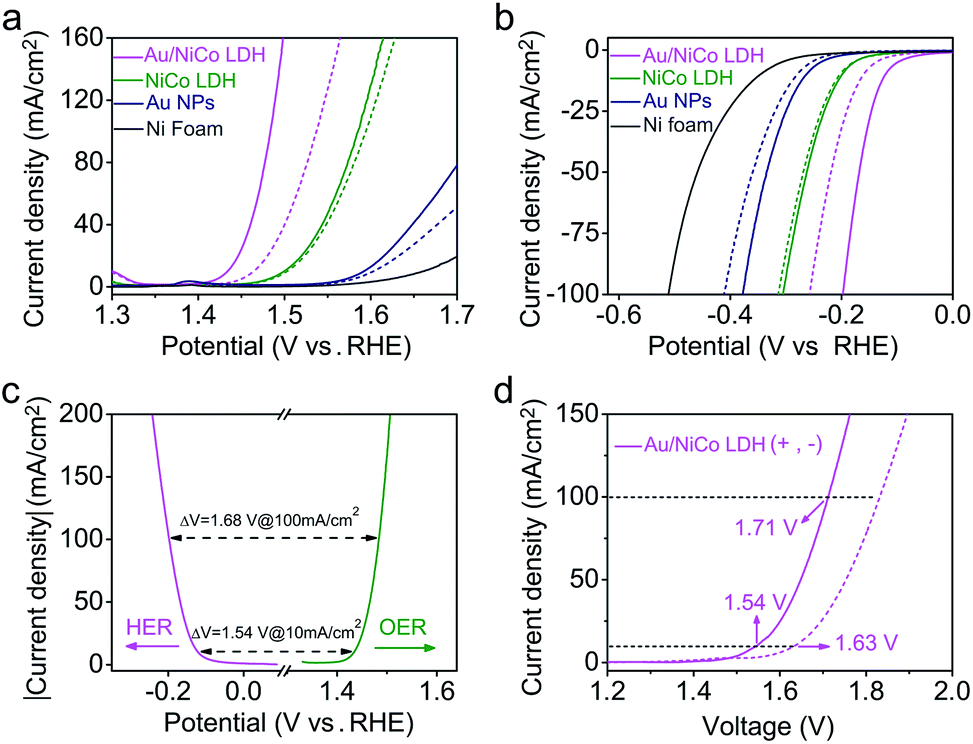 | ||
| Fig. 4 PEC performance of the prepared catalysts. (a) OER and (b) HER polarization curves of Au/NiCo LDH, NiCo LDH, Au NPs, and Ni foam measured in 1.0 M KOH electrolyte with (solid curves) and without (dashed curves) visible light irradiation. (c) Polarization curves of Au/NiCo LDH for the HER (pink curve) and OER (green curve) and the voltage difference between the HER and OER at current densities of 10 and 100 mA cm−2. (d) Polarization curves for overall water splitting with (solid curve) and without (dashed curve) irradiation on Au/NiCo LDH. | ||
It would be interesting to see the impact of the amount of loaded Au NPs on the visible light-enhanced electrocatalytic performance. We compared the activity of Au/NiCo LDH with 1.7, 3.3 and 5.1 wt% Au content (Fig. S12‡). The medium loading amount of Au NPs (3.3 wt%) exhibits the best activity in both OER and HER experiments. The decrease in activity with more Au NPs is probably due to the aggregation of the NPs when the loading density is too high, which results in the reduced plasmonic activity of the Au/NiCo LDH. Thus, the nanocomposite with 3.3 wt% Au NPs was used in all further experiments. In order to investigate the influence of light irradiation on the counter electrode, we performed control experiments in which only the working electrode or counter electrode was under irradiation. As shown in Fig. S13,‡ there is no difference whether the counter electrode is illuminated or not. Besides, the high stability of this Au/NiCo LDH was confirmed by chronopotentiometry curves and high-resolution XPS results (see Fig. S14‡). Other electrochemical properties of the measured electrodes are summarized in Table S1.‡
Next, the Au/NiCo LDH was used on both the anode and cathode to perform overall water splitting. An applied potential of 1.54 V is required to reach a current density of 10 mA cm−2 under visible light irradiation, which is much lower than the value of 1.63 V in the dark. Finally, the gaseous products from the overall water splitting on Au/NiCo LDH electrodes were determined using gas chromatography. As illustrated in Fig. S15a,‡ 0.552 mmol of O2 and 1.107 mmol of H2 are produced after 3 h of irradiation, corresponding to faradaic efficiencies of 98.63% (photoanode) and 98.92% (photocathode), respectively (calculation details are shown in ESI S2‡). The long-term water splitting stability under visible light irradiation was evaluated by a chronopotentiometry method at current densities of 10 and 20 mA cm−2. As shown in Fig. S15b,‡ the voltage of the electrolyzer remains stable during the test. Moreover, the scanning electron microscopic (SEM)/TEM images of Au/NiCo LDH after the stability test in Fig. S16‡ reveal that the structure of Au/NiCo LDH is well preserved, which indicates that the catalyst has an excellent stability under the real electrocatalytic water splitting reaction conditions. Tables S2–S4‡ compare the performance of Au/NiCo LDH with that of other materials that are among the recently reported best catalysts for the OER, HER and overall water splitting.
Verification of the SPR effect
Since the plasmonic activity of Au NPs is wavelength-dependent, it is necessary to evaluate the catalytic performance using different light colors. Fig. S17a‡ shows the OER performance of Au/NiCo LDH under 420, 500, 550, 600, and 700 nm irradiation, respectively. We can see that the Au/NiCo LDH exhibits the best activity at 550 nm irradiation. Besides, we measured the photocurrent of Au/NiCo LDH to calculate the incident photon-to-current efficiency (IPCE) at single wavelengths (Fig. S17b‡). As shown in Fig. 5a, it's clear that the Au/NiCo LDH exhibits the highest IPCE at ∼550 nm, which is also close to the LSPR extinction peak of the Au NPs (Fig. 5a, red curve), further confirming the effect of SPR of Au NPs (details of IPCE are shown in S3†).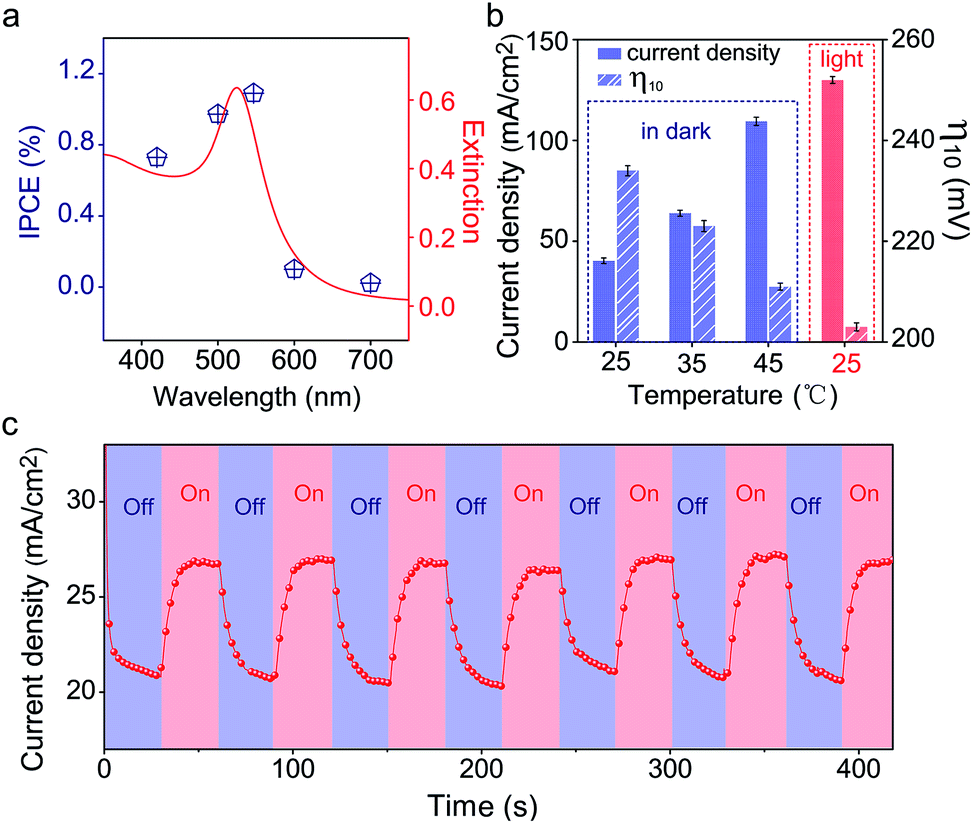 | ||
| Fig. 5 Verification of the SPR effect. (a) IPCE values of Au/NiCo LDH at different wavelengths (blue points) and the extinction spectrum of Au NPs (red curve). (b) The current densities (solid column) at 1.5 V and the overpotentials at 10 mA cm−2 (striped column) of Au/NiCo LDH under different reaction (OER) temperatures with or without light irradiation. (c) Chronoamperometric i–t curve of the Au/NiCo LDH under chopped irradiation. | ||
However, the non-radiative decay of plasmons not only generates hot electron–hole pairs but also produces heat via coupling with phonons.41 Thus it remains unclear whether the hot carriers or the photothermal effect dominates the plasmon-promoted electrocatalytic water splitting. Control experiments in the dark at different temperatures (Fig. S17c‡) indicate that the electrocatalytic activity of Au/NiCo LDH is not significantly enhanced at elevated temperatures of 35 and 45 °C. Fig. 5b compares the current densities and overpotentials for the OER at different temperatures with or without light irradiation. It can be observed that the Au/NiCo LDH under irradiation (25 °C) possesses the largest current density and lowest overpotential. To further verify whether the plasmon effect proceeds via charge separation or thermal enhancement, we isolated the Au NPs with an ultrathin SiO2 shell to prevent the hot electrons from moving to the NiCo LDH.42,43 As expected, bare Au decorated NiCo LDH exhibits a much higher PEC activity than Au@SiO2 decorated NiCo LDH (Fig. S18‡). Thus, it is the interfacial electron transfer rather than the heat that makes the dominant contribution to the enhanced catalytic activity.44 The i–t curve with irradiation switched on and off is shown in Fig. 5c; it can be observed clearly that switching off the light restricts the oxygen evolution.
In order to confirm the real catalytic surface on Au/NiCo LDH during the plasmon-promoted electrocatalytic water splitting, a thiol compound, thiophenol (TP), was used to coat the Au surface through strong covalent Au–S bonds (Fig. 6); thus the Au surface of the Au/NiCo LDH is selectively blocked for catalysis.45 As evidenced in Fig. S19,‡ after incubation with TP, Au NPs exhibit a significantly decreased activity but NiCo LDH shows no obviously change, in both the HER and OER. The overpotentials of the different catalysts for the HER and OER are presented in Fig. 6a and c, respectively. In the HER, the remarkably increased overpotential value of Au/NiCo LDH after the TP coating suggests that the plasmon-promoted water reduction mainly occurs on the Au surface (vide supra). In contrast, the overpotential value does not change significantly in the OER, indicating that NiCo LDH is the main catalytic material for oxygen evolution.
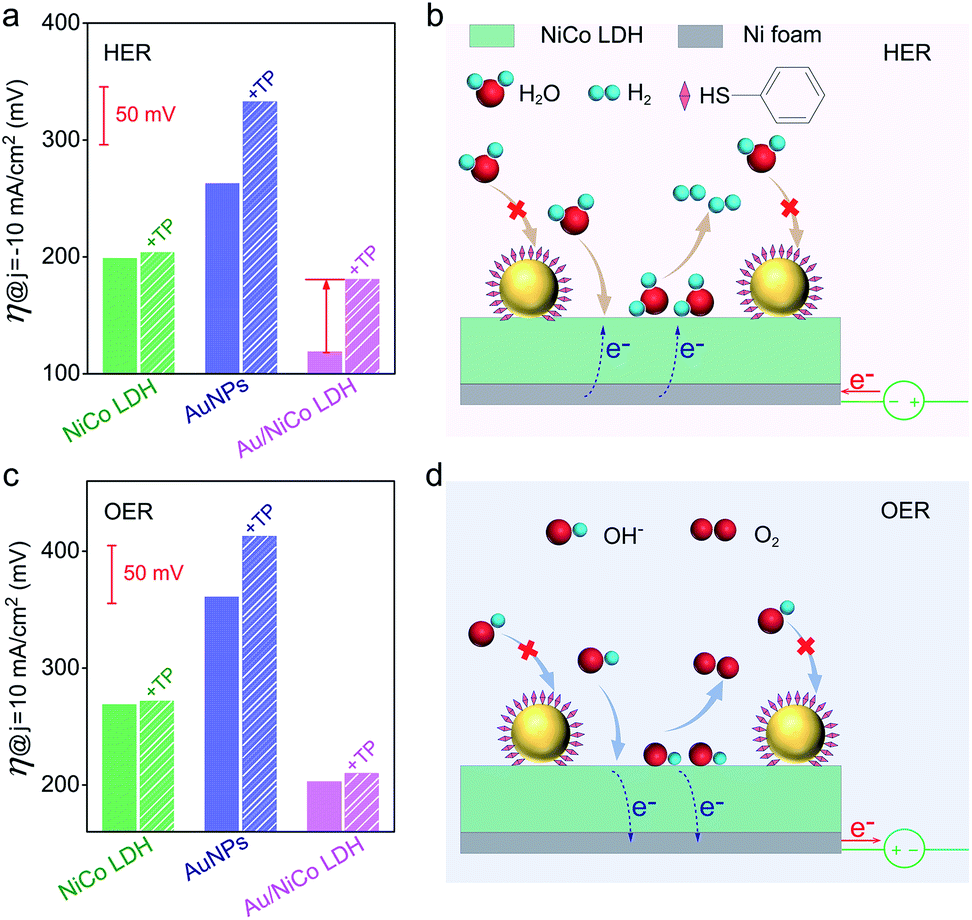 | ||
| Fig. 6 Determination of real catalytic surfaces. (a) Overpotentials of NiCo LDH, Au NPs and Au/NiCo LDH with (striped column) and without (solid column) TP monolayer coating on the Au NPs, under light irradiation toward the HER. (b) Schematic illustration of water reduction on the Au@TP/NiCo LDH. (c) Overpotentials of NiCo LDH, Au NPs and Au/NiCo LDH with (striped column) and without (solid column) TP coating under light irradiation toward the OER. (d) Schematic illustration of OH− oxidation on the Au@TP/NiCo LDH. | ||
Based on the above results, a proposed mechanism of the plasmon-promoted electrocatalysis is illustrated in Fig. 7. The electron transfer from NiCo LDH to Au enhances the intrinsic electrocatalytic activity of the nanocomposite. For NiCo LDH, the conduction band (CB) minimum and the valence band (VB) maximum are estimated to be −4.21 and −5.77 eV (Fig. S20‡), respectively.46 Under resonant light irradiation, hot electrons are generated on the Au NPs via non-radiative decay of the surface plasmons.47 On the cathode side (Fig. 7, left), hot electrons on the Au surface of Au/NiCo LDH are utilized more easily since the plasmon-generated holes are compensated for by both the electrons from the NiCo LDH and foreign electrons from the anode. As demonstrated in Fig. 6a, the main catalytic surface for the plasmon-enhanced HER is Au rather than NiCo LDH. Under resonant irradiation, the Au/NiCo LDH exhibits a Tafel slope of 57.5 mV dec−1 (Fig. S10c‡), indicating the Volmer–Heyrovsky path (Scheme S1‡) in which the hot electrons participate in the reduction of water and promote the evolution of hydrogen.48,49 On the anode side (Fig. 7, right), the hot electrons are further transferred to the cathode upon the applied potential.12 Since Au is not a good OER catalyst compared with NiCo LDH (as shown in Fig. 4a), electrochemical water oxidation on the Au surface of Au/NiCo LDH by the plasmon-generated holes is subordinate to that on the NiCo LDH surface. Meanwhile, beside the spontaneous charge transfer from NiCo LDH to Au, the plasmon-generated holes are effective electron trappers to capture electrons from NiCo LDH, remarkably enhancing the electron transfer from NiCo LDH to Au NPs for the formation of oxidative NiCo LDH. Then, the oxidative NiCo LDH species provide active sites to extract electrons from OH−, facilitating the formation and subsequent deprotonation of the key intermediates and thus promoting the OER catalysis (Scheme S1‡).50,51
 | ||
| Fig. 7 Proposed reaction mechanism. Schematic illustration of the plasmon-promoted electrocatalytic water splitting on Au/NiCo LDH under visible light irradiation. | ||
Conclusions
In summary, we presented a mechanistic study of plasmon-promoted electrocatalytic water splitting. Via integration of Au NPs and conventional semiconductor electrocatalysts including Co(OH)2, Ni(OH)2 and NiCo LDH, the catalytic performance was significantly enhanced under visible light irradiation. To investigate the interfacial charge transfer direction in the metal–semiconductor nanocomposites, SERS was employed to measure the molecular vibrational band shifts caused by the electronic property changes on the Au surface. We found that there is a simultaneous electron transfer from the semiconductor to the metal. As expected, Au/NiCo LDH with the highest electron transfer tendency among the three exhibited the best electrocatalytic performance, in both reactions with and without visible light irradiation. It has been confirmed by wavelength-dependent experiments that the LSPR effect is responsible for the enhanced activity under irradiation. To disentangle the contributions from hot carriers and heat, we performed temperature-dependent and ultrathin shell-isolation controls which demonstrated the dominant role of hot carriers. Finally, the real catalytic sites of the plasmonic electrocatalysis were determined by selective coating of the Au surface on the nanocomposite with a thiol monolayer. After coating, the catalytic activity for water reduction decreased remarkably while no obvious change was found in the oxidation half reaction, indicating that the HER and OER mainly took place on Au and semiconductor (NiCo LDH) surfaces, respectively. This mechanistic study highlights the important synergistic effect between the light harvester (i.e. the plasmonic NPs) and the conventional electrocatalyst for visible light-enhanced water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21775074, 51601098 & 21861132016), the Natural Science Foundation of Tianjin (17JCQNJC05400), and the National Key R&D Program (2017YFA0206702 & 2016YFB0901502).Notes and references
- S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS.
- B. Foerster, A. Joplin, K. Kaefer, S. Celiksoy, S. Link and C. Sonnichsen, ACS Nano, 2017, 11, 2886–2893 CrossRef CAS.
- C. Hu, T. Peng, X. Hu, Y. Nie, X. Zhou, J. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- Y. Kim, J. G. Smith and P. K. Jain, Nat. Chem., 2018, 10, 763–769 CrossRef CAS.
- J. Xu, W.-M. Yang, S.-J. Huang, H. Yin, H. Zhang, P. Radjenovic, Z.-L. Yang, Z.-Q. Tian and J.-F. Li, Nano Energy, 2018, 49, 363–371 CrossRef CAS.
- H. Jia, A. Du, H. Zhang, J. Yang, R. Jiang, J. Wang and C. Y. Zhang, J. Am. Chem. Soc., 2019, 141, 5083–5086 CrossRef CAS.
- H. Zhang, T. Wang, J. Wang, H. Liu, T. D. Dao, M. Li, G. Liu, X. Meng, K. Chang, L. Shi, T. Nagao and J. Ye, Adv. Mater., 2016, 28, 3703–3710 CrossRef CAS.
- J. Yang, Y. Guo, R. Jiang, F. Qin, H. Zhang, W. Lu, J. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS.
- S. Mubeen, J. Lee, N. Singh, S. Kramer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS.
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS.
- J. Xu, P. Gu, D. J. S. Birch and Y. Chen, Adv. Funct. Mater., 2018, 28, 1801573 CrossRef.
- S. Atta, A. M. Pennington, F. E. Celik and L. Fabris, Chem, 2018, 4, 2140–2153 CAS.
- R. Shi, Y. Cao, Y. Bao, Y. Zhao, G. I. N. Waterhouse, Z. Fang, L. Z. Wu, C. H. Tung, Y. Yin and T. Zhang, Adv. Mater., 2017, 29, 1700803 CrossRef.
- B. S. Hoener, S. R. Kirchner, T. S. Heiderscheit, S. S. E. Collins, W.-S. Chang, S. Link and C. F. Landes, Chem, 2018, 4, 1560–1585 CAS.
- J. Lee, S. Mubeen, X. Ji, G. D. Stucky and M. Moskovits, Nano Lett., 2012, 12, 5014–5019 CrossRef CAS.
- L. Yu, J. F. Yang, B. Y. Guan, Y. Lu and X. W. D. Lou, Angew. Chem., Int. Ed., 2018, 57, 172–176 CrossRef CAS.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS.
- Y. Chao, P. Zhou, N. Li, J. Lai, Y. Yang, Y. Zhang, Y. Tang, W. Yang, Y. Du, D. Su, Y. Tan and S. Guo, Adv. Mater., 2019, 31, e1807226 CrossRef.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, Adv. Funct. Mater., 2014, 24, 4698–4705 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS.
- N. Zhang, C. Han, X. Fu and Y.-J. Xu, Chem, 2018, 4, 1832–1861 CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS.
- Y. Shi, J. Wang, C. Wang, T. T. Zhai, W. J. Bao, J. J. Xu, X. H. Xia and H. Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef CAS.
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng, T. Kako and J. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef CAS.
- W. Xie, R. Grzeschik and S. Schlücker, Angew. Chem., Int. Ed., 2016, 55, 13729–13733 CrossRef CAS.
- Y. L. Li, Y. F. Hu, F. X. Shi, H. X. Li, W. Xie and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 9049–9053 CrossRef CAS.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- J. Ye, F. Wen, H. Sobhani, J. B. Lassiter, P. Van Dorpe, P. Nordlander and N. J. Halas, Nano Lett., 2012, 12, 1660–1667 CrossRef CAS.
- H. Zhang, X. G. Zhang, J. Wei, C. Wang, S. Chen, H. L. Sun, Y. H. Wang, B. H. Chen, Z. L. Yang, D. Y. Wu, J. F. Li and Z. Q. Tian, J. Am. Chem. Soc., 2017, 139, 10339–10346 CrossRef CAS.
- H. Zhang, X. Li, A. Hähnel, V. Naumann, C. Lin, S. Azimi, S. L. Schweizer, A. W. Maijenburg and R. B. Wehrspohn, Adv. Funct. Mater., 2018, 28, 1706847 CrossRef.
- Z. Zheng, W. Xie, M. Li, Y. H. Ng, D.-W. Wang, Y. Dai, B. Huang and R. Amal, Nano Energy, 2017, 41, 233–242 CrossRef CAS.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS.
- R. Gómez, J. Solla-Gullón, J. M. Pérez and A. Aldaz, J. Raman Spectrosc., 2005, 36, 613–622 CrossRef.
- D. Zha, Y. Fu, L. Zhang, J. Zhu and X. Wang, J. Power Sources, 2018, 378, 31–39 CrossRef CAS.
- B. D. Busbee, S. O. Obare and C. J. Murphy, Adv. Mater., 2003, 15, 414–416 CrossRef CAS.
- Y. J. Li, W. J. Huang and S. G. Sun, Angew. Chem., Int. Ed., 2006, 45, 2537–2539 CrossRef CAS.
- H. S. Su, X. G. Zhang, J. J. Sun, X. Jin, D. Y. Wu, X. B. Lian, J. H. Zhong and B. Ren, Angew. Chem., Int. Ed., 2018, 57, 13177–13181 CrossRef CAS.
- Y.-H. Wang, M.-M. Liang, Y.-J. Zhang, S. Chen, P. Radjenovic, H. Zhang, Z.-L. Yang, X.-S. Zhou, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2018, 57, 11257–11261 CrossRef CAS.
- S. Hu, B. J. Liu, J. M. Feng, C. Zong, K. Q. Lin, X. Wang, D. Y. Wu and B. Ren, J. Am. Chem. Soc., 2018, 140, 13680–13686 CrossRef CAS.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS.
- C. Li, P. Wang, Y. Tian, X. Xu, H. Hou, M. Wang, G. Qi and Y. Jin, ACS Catal., 2017, 7, 5391–5398 CrossRef CAS.
- C. Li, D. Cahen, P. Wang, H. Li, J. Zhang and Y. Jin, iScience, 2018, 8, 213–221 CrossRef CAS.
- L. Mascaretti, A. Dutta, S. Kment, V. M. Shalaev, A. Boltasseva, R. Zboril and A. Naldoni, Adv. Mater., 2019, e1805513 CrossRef.
- W. Xie, B. Walkenfort and S. Schlücker, J. Am. Chem. Soc., 2013, 135, 1657–1660 CrossRef CAS.
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef.
- H. X. Zhang, Y. Li, M. Y. Li, H. Zhang and J. Zhang, Nanoscale, 2018, 10, 2236–2241 RSC.
- Q. Q. Chen, C. C. Hou, C. J. Wang, X. Yang, R. Shi and Y. Chen, Chem. Commun., 2018, 54, 6400–6403 RSC.
- L. Yu, I. K. Mishra, Y. Xie, H. Zhou, J. Sun, J. Zhou, Y. Ni, D. Luo, F. Yu, Y. Yu, S. Chen and Z. Ren, Nano Energy, 2018, 53, 492–500 CrossRef CAS.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS.
Footnotes |
| † Dedicated to the 100th anniversary of Nankai University. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03360b |
| This journal is © The Royal Society of Chemistry 2019 |