
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi-catalyzed cross-electrophile coupling between vinyl/aryl and alkyl sulfonates: synthesis of cycloalkenes and modification of peptides†
Jicheng
Duan
,
Yun-Fei
Du
,
Xiaobo
Pang
and
Xing-Zhong
Shu
 *
*
State Key Laboratory of Applied Organic Chemistry (SKLAOC), College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou, 730000, China. E-mail: shuxingzh@lzu.edu.cn
First published on 5th August 2019
Abstract
We report here the coupling reactions between vinyl/aryl and alkyl C–O electrophiles that can be derived from chemical feedstocks and naturally occurring functional groups. This method provides an efficient approach to the synthesis of a wide range of functionalized, and/or secondary alkyl substituted cycloalkenes. These compounds are difficult to produce by conventional methods. The reaction proceeds with broad substrate scope, and tolerates various functional groups such as alcohol, aldehyde, ketone, ester, amide, alkene, alkyne, heterocycles, organotin and organosilicon compounds. The synthetic utility of this method has been demonstrated by providing facile access to important building blocks. We also demonstrated the possibility to apply this method for late-stage modification of peptides. A broad range of functionalized alkyl groups could be selectively introduced into tyrosine in peptides via C–C bond formation, which has been a challenge to the existing procedures.
Introduction
The design of a novel C–C bond forming reaction that relies on naturally occurring functional groups and/or chemical feedstocks as coupling fragments instead of organometallics is an ongoing effort in the synthetic community.1 One strategy that could be used to satisfy this demand would be the cross-electrophile reaction.2 This strategy has enabled construction of C–C bonds directly from electrophiles, demonstrating excellent functional group compatibility, and unique selectivity orthogonal to the classic cross-couplings.3 Over the past years, there have been great achievements in the development of reactions using organic halides (Scheme 1(1)).2 However, the reactions of C–O electrophiles have been less studied,4 and there are only isolated examples that have shown the cross-coupling between two C–O electrophiles.5 The development of such reactions would help expand the applicability of this method, and take advantage of the abundant C–O bonds in natural and artificial products. In this manuscript, we report the coupling reactions between vinyl/aryl and alkyl C–O electrophiles, which enable synthesis of cycloalkenes from ketones and alcohols (Scheme 1(2a)), and selective modification of tyrosine in peptides (Scheme 1(2b)).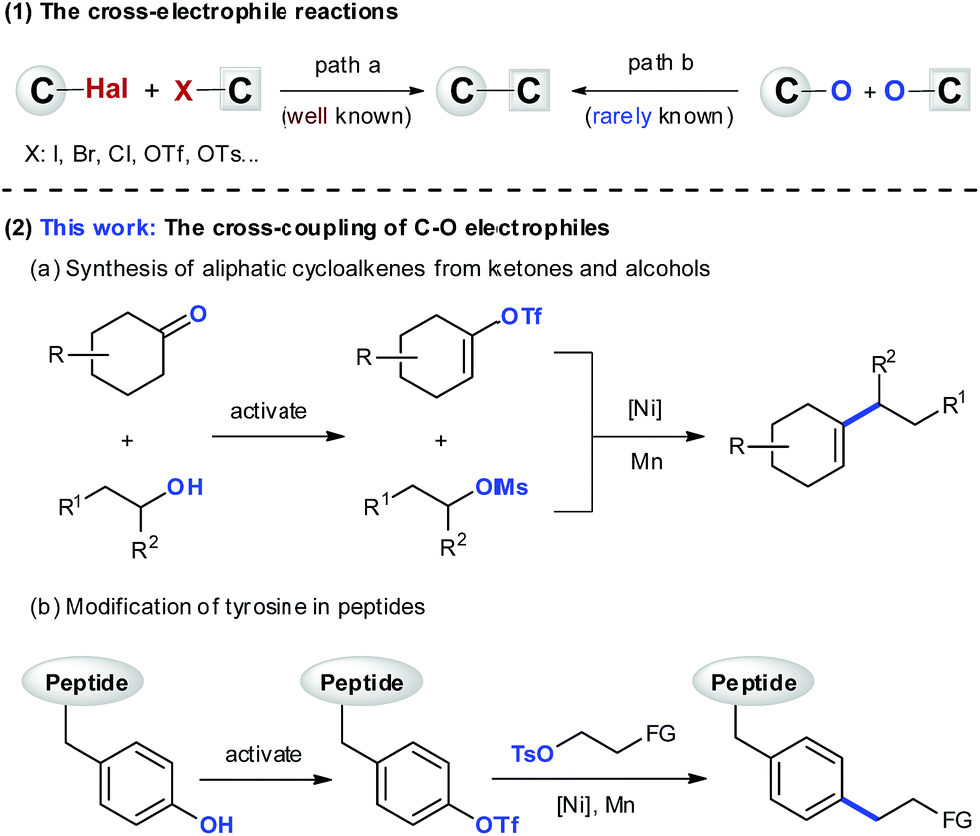 | ||
| Scheme 1 The cross-coupling of electrophiles. | ||
Considering the development of reactions between C–O electrophiles, we became interested in the synthesis of cycloalkenes. Cycloalkenes represent key structural elements in a variety of natural products, pharmaceuticals, and functional materials, and they constitute the foundation for numerous important reactions.6 Unlike the many synthetic approaches to acyclic alkenes, the synthesis of cycloalkenes is much less advanced and highly relies on chemistry developed decades ago.7 One of the most reliable methods is the metal-catalyzed cross-coupling reaction using well-defined vinyl species, which circumvents the regio- and stereoselectivity problems associated with conventional alkylation–elimination processes.8 To date, this method has received considerable attention,9 and numerous applications in the field of total synthesis of biologically relevant targets have been found.10 Despite the impressive advances, much remains to be improved. For instance, the synthesis of highly functionalized and/or secondary alkyl substituted cycloalkenes remains difficult,7,11 and the requirement for cyclic vinyl metals and alkyl metals is still less than ideal because of their limited availability, and high reactivity profiles.12
Encouraged by recent progress in reductive vinylation reactions using organic halides,13 we wondered about the possibility of constructing aliphatic cycloalkenes from ketones and alcohols through the coupling of their derivatives, vinyl triflates14 and alkyl mesylates15 (Scheme 1(2a)). Whereas the former are highly reactive towards oxidative addition to low valent metals (vinyl-OTf ≫ vinyl-Br > PhBr),16 unactivated alkyl mesylates remain one of the most challenging substrates in the cross-coupling arena.17 Such highly mismatched reactivity of two electrophiles means that control of selectivity in the formation of the cross-product would be difficult.18
Results and discussion
With these challenges in hand, we started our investigation by focusing on the reaction of triflate 1a with 2a and screening a range of conditions (Table 1, also see Table S1 in the ESI†). As expected, our initial studies revealed that, in most cases, vinyl triflate was converted into the protonated and dimerized byproducts, while alkyl mesylate remained intact. Further studies established that performing the reaction with NiI2 (10 mol%), tpy (15 mol%), and Mn (3.0 equiv.) in DMA at 100 °C gave the best result, affording 3a with 81% isolated yield (entry 1). The use of NiCl2 or NiBr2 gave either trace amounts or a low yield of the desired product, with most of the 2a recovered, indicating the importance of the catalytic amount of iodide for the activation of alkyl mesylate (entries 2 and 3).19 This is also consistent with the observation that the use of NaI (0.5 equiv.) improved the reactivity of 2a under NiBr2 catalysis, affording 3a with 63% yield along with a large amount of alkyl–alkyl dimers (entry 3 vs. 4). Most of the 2a was converted into the alkyl–alkyl dimer when NaI (0.5 equiv.) was used under the standard conditions (entry 1 vs. 5). The bipyridine ligands L1–L3 and bathophenanthroline L4, which proved effective in the vinylation of alkyl halides13c,h and arylation of alkyl tosylates,4f,5 gave 3a with low yields (entries 6–9). Further studies revealed that the tridentate ligand afforded the best result (entries 1 and 11). The use of Zn instead of Mn gave a trace amount of 3a (entry 12). The use of TDAE instead of Mn afforded 3a in 8% yield, suggesting that Mn might act as a reductant in this process (entry 13). No desired product was observed in the absence of either Ni or Mn (entry 14).|
|
||
|---|---|---|
| Entry | Change of conditions | 3a (%) |
| a 1a (0.2 mmol) was used and reacted for 12 h; the yields were determined by GC analysis with dodecane as the internal standard. b Isolated yield. c TDAE: tetrakis(dimethylamino)ethylene. | ||
| 1 | None | 87 (81)b |
| 2 | NiCl2 instead of NiI2 | Trace |
| 3 | NiBr2 instead of NiI2 | 15 |
| 4 | NiBr2 with NaI (0.5 equiv.) | 63 |
| 5 | NiI2 with NaI (0.5 equiv.) | 38 |
| 6 | L1 instead of tpy | 26 |
| 7 | L2 instead of tpy | 34 |
| 8 | L3 instead of tpy | 28 |
| 9 | L4 (BPhen) instead of tpy | 37 |
| 10 | L5 instead of tpy | 21 |
| 11 | L6 instead of tpy | 77 |
| 12 | Zn instead of Mn | 4 |
| 13 | TDAEc instead of Mn | 8 |
| 14 | No Ni or Mn | 0 |
|
||
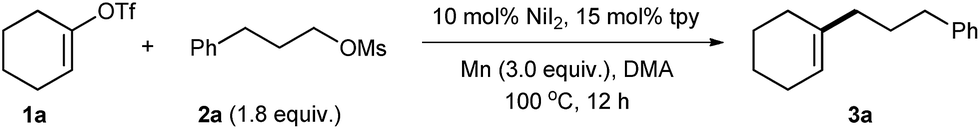
With the optimized reaction conditions in hand, we then studied the scope of the reaction with respect to vinyl triflates (Table 2). Cyclic vinyl triflates ranging from five- to eight-membered rings coupled with 2a to give target products with moderate to good yields (3a–d). Nonaromatic heterocycles are prevalent in pharmaceuticals, but their introduction through C–C bonds represents a challenge.20 Under our conditions, various heterocycles such as 3-piperideine (3e), 3,6-dihydro-2H-pyran (3f), and 3,6-dihydro-2H-thiopyran (3g) were efficiently incorporated into the alkyl chain. Vinyl triflate with a tBu substituent on the ring performed well (3h). Unlike vinyl halides, 3- and 6-substituted vinyl triflates were readily available from unsymmetrical ketones and converted into products 3i and 3j smoothly. A moderate yield of 3k was obtained when indenyl triflate was used. Incorporation of functionalized vinyl groups is important to increase the diversity of molecules. In this respect, the reactions of vinyl triflates derived from dicarbonyl compounds gave 3l, 3m, and 3n with moderate yields. The last two compounds are key building blocks in natural product synthesis.21 Reactions of acyclic vinyl electrophiles were also effective when more reactive alkyl tosylates were employed, affording the desired products with useful yields (3o–u).4f,i,5 The presence of ester and alcohol groups was tolerated (3p–r). While the reaction of terminal vinyl triflate 1s (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 1:9) afforded 3s with a ratio of E:Z is 2:1, only the E-product was obtained when internal substrate 1t (E:Z = 3:1) was used (3t). On the other hand, the reaction of Z-1u afforded 3u in 60% yield with a ratio of E:Z = 1:4, whereas only a trace of 3u was observed when E-1u was used. Presently, the reaction of fully substituted vinyl triflates has not been successful.
:Z = 1:9) afforded 3s with a ratio of E:Z is 2:1, only the E-product was obtained when internal substrate 1t (E:Z = 3:1) was used (3t). On the other hand, the reaction of Z-1u afforded 3u in 60% yield with a ratio of E:Z = 1:4, whereas only a trace of 3u was observed when E-1u was used. Presently, the reaction of fully substituted vinyl triflates has not been successful.
|
a Isolated yields.
b 6-Methylhept-5-en-2-yl mesylate (2b) was used, NaI (0.5 equiv.), 80 °C.
c Bn-N(Boc)(CH2)4-OTs (2c) was used, NaI (0.5 equiv.).
d Ph(CH2)3-OTs (2a′) was used, NaI (0.5 equiv.), 60 °C.
e NMR yield.
f Vinyl triflate 1s (E:Z = 1:9, 3 equiv.) was used.
g Vinyl triflate 1t (E:Z = 3:1, 3 equiv.) was used.
h Vinyl triflate Z-1u (only Z isomer) was used.
|
|---|
|
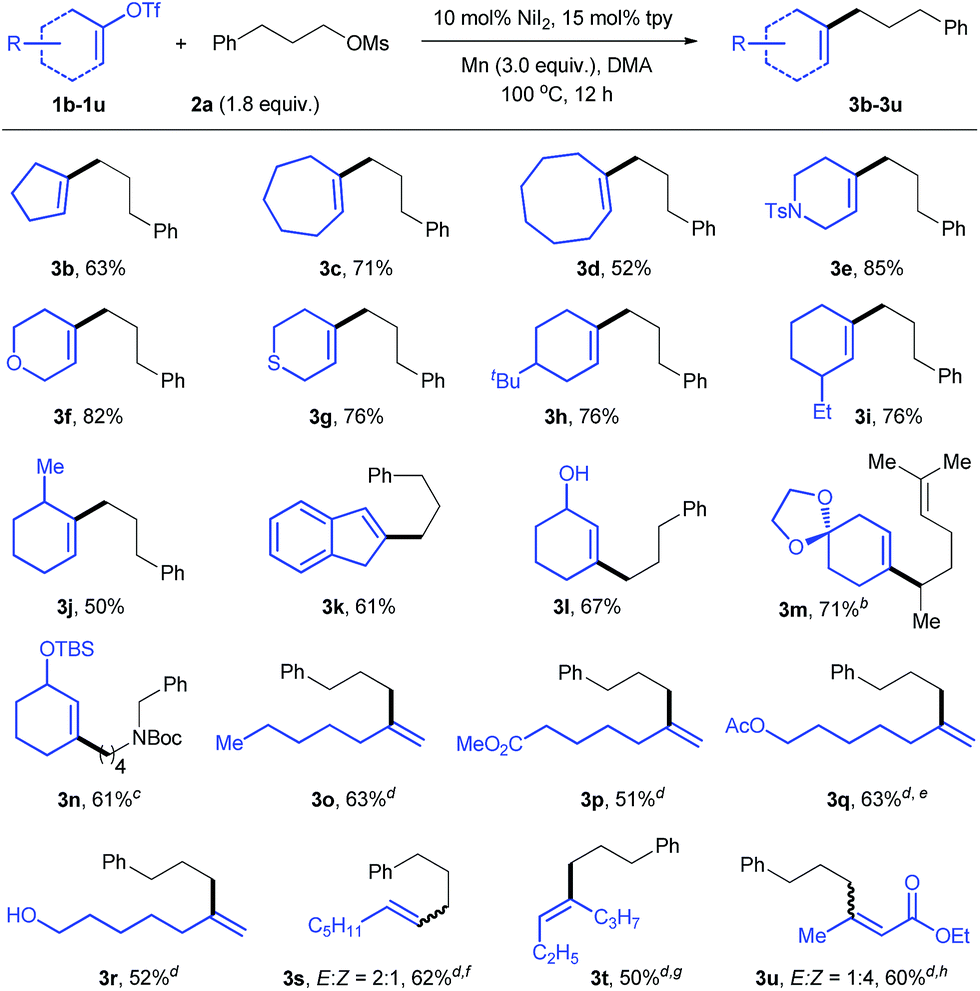
A wide range of sulfonated alcohols coupled efficiently with vinyl triflate 1e under the standard conditions (Table 3). The reaction of the β-substituted alkyl electrophile afforded product 3v with good yield. A simple long-chain alkyl substrate was converted into 3w smoothly. The reactions were highly chemoselective for the vinylation of alkyl mesylate, leaving a gamut of functionalities such as alcohol (3x), silyl ether (3y), alkyl fluoride (3z), alkyne (3aa), ester (3ab), amide (3ac), and aldehyde (3ad) intact. Both organosilicon (3ae) and organotin (3af) were also tolerated.

Incorporation of secondary alkyl groups at the alkene position remains a significant challenge. Under our conditions, a number of secondary alkyl mesylates coupled with vinyl triflate efficiently (3ag–an). The use of NaI (0.5 equiv.) was necessary to improve the reaction efficiency. Terminal alkene (3aj) and ester groups (3an) were compatible, and substrates derived from cyclic alcohols were also successfully employed (3al–an). The reaction could be scaled up to the gram scale and produced 3aj with 72% yield.
The abundance of alcohols in nature prompted us to investigate vinylation of substrates derived from biochemicals. Mesylates derived from ethanol (3ao) and oleyl alcohol (3ap) were found to be suitable for this transformation, affording the desired products with high yields. The reaction of a substrate derived from tetrahydrofurfuryl alcohol (THFA) gave 3aq with 71% yield. When the 1,4-pentanediol derivative was used, the reaction was highly selective for vinylation of the secondary C–OMs bond over the primary C–OTBS bond (3ar).
Enyne 5 exemplifies the inefficiency of previous approaches to prepare the functionalized aliphatic cycloalkene (Scheme 2a). This compound is an important building block in the synthesis of a number of HCV NS5B inhibitors, and has been prepared in nine steps from nitrile 4.22 With reductive cross-coupling, a simpler starting material, 1a, can be used and the desired product 5 was accessed in a single step with 78% yield.
 | ||
| Scheme 2 Synthetic application and late-stage modification of biologically active molecules. aConditions as for Table 2, but NaI (0.5 equiv.), 40 °C was used, reaction for 24 h. | ||
The late-stage modification of complex molecules represents a promising approach to alter the pharmacological profiles of natural products. To further demonstrate the utility of our method, we then investigated the vinylation reactions of several biologically active compounds (Scheme 2b). Substrates derived from lithocholic acid and epiandrosterone reacted with 1e efficiently, affording the vinylated products 8 and 10 with 90 and 94% yields, respectively.
The control experiments shown in Table 1 indicate the importance of the catalytic amount of iodide for the activation of alkyl mesylate (entries 1–5). The observation by GC-MS analysis of tiny amounts of alkyl iodide during the reaction suggests that alkyl mesylate might be activated through mesylate-iodide exchange in situ.19 This prompted us to study the reaction of mesylate 2a with NaI (1.0 equiv.), and it was found that 2a was fully converted into iodide 11 at 100 °C after 12 h (Scheme 3a). While alkyl iodide 11 was too reactive to give good selectivity for the cross-product, the yield of 3a was significantly improved by slow addition of 11. These results indicate that catalytic mesylate/iodide exchange in situ is important for the success of this reaction.
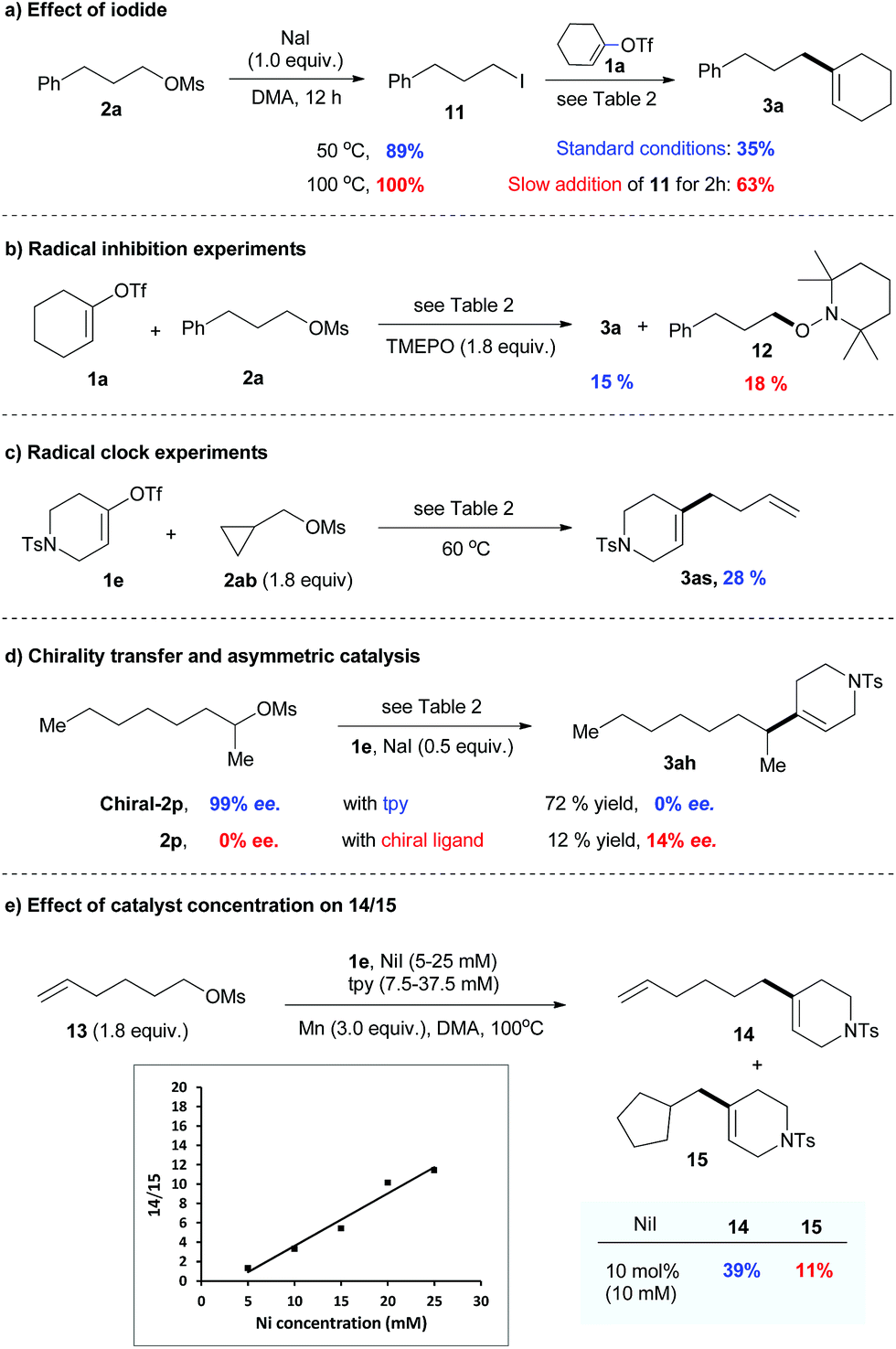 | ||
| Scheme 3 Mechanistic studies. | ||
The reaction of 1a with 2a was significantly inhibited in the presence of a radical scavenger such as TEMPO (Scheme 3b). The radical trapping product 12 was obtained with 18% yield. The reaction of 1e with cyclopropyl substrate 2ab resulted in a ring opening product 3as with 28% yield (Scheme 3c). The use of the optically pure alkyl mesylate chiral-2p gave 3ah with no enantiomeric excess (ee) under the standard conditions (Scheme 3d). Furthermore, the reaction of racemic 2p afforded 3ah with 14% ee when the chiral ligand was used. These results suggest a potential radical mechanism.
To verify a potential radical chain mechanism,23 we then studied the effect of catalyst concentration on the products of reaction 13 with 1e (Scheme 3e). Given that the C-centered radical is more easily trapped by the catalyst before cyclization under high catalyst concentration, we expected that, if a radical chain mechanism was followed, the ratio of 14/15 would increase with increasing catalyst concentration. Our results, presented in Scheme 3e, were consistent with this expectation, indicating that a radical chain mechanism may be involved.
Based on the above results and on reports presented by other investigators, we tentatively proposed a catalytic cycle for the coupling between vinyl and alkyl C–O electrophiles as shown in Scheme 4. The oxidative addition of vinyl triflate to Ni(0) would give vinyl-Ni(II) (A), which may be trapped by the alkyl radical and undergo reductive elimination to afford the desired product. The alkyl radical may be generated by reaction of alkyl-I with [NiI].23a Alkyl-I can be catalytically formed through mesylate/iodide exchange,19 and iodide will be regenerated during the cycle.
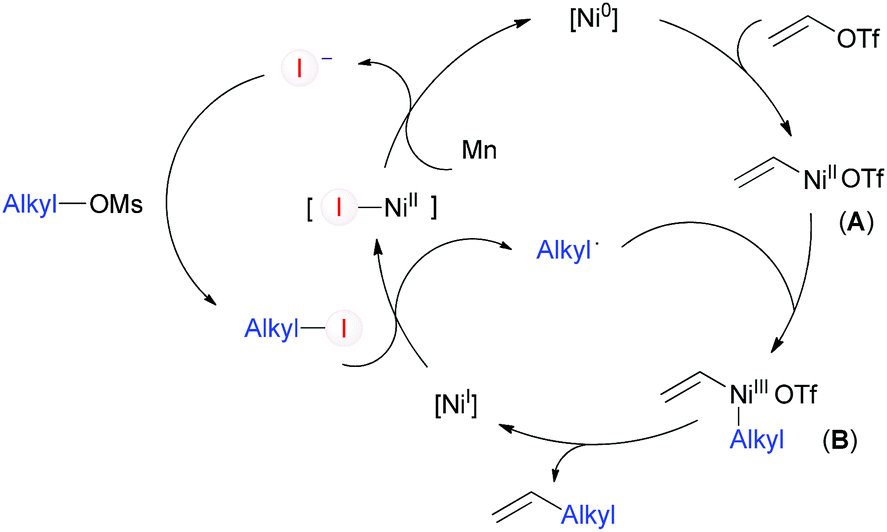 | ||
| Scheme 4 Proposed mechanism. | ||
Phenols are common and important structural motifs in natural products like tyrosine. To further illustrate the potential synthetic application of the cross-coupling between C–O electrophiles, we then studied the modification of tyrosine-containing peptides via reductive alkylation of the sulfonated phenol group.24 Late-stage modification of peptides has emerged as a significant task of current interest.25 The construction of covalent bonds to peptides provides a means of accessing non-natural peptides, which are of key relevance for numerous applied research areas such as asymmetric synthesis, drug delivery, proteomics and diagnosis.26 Tyrosine is a natural amino acid found in almost all proteins. While various cross-coupling methods have been developed for diversification of tyrosine in peptides,27 the introduction of an alkyl group via C–C bond formation remains largely unexplored. Moreover, unlike the conventional cross-coupling method that has been widely applied in this area,27,28 the potential of the cross-electrophile reaction for peptide modification is still waiting to be disclosed.29
With these considerations in mind, we started our investigation by studying the reaction of tyrosine 16a with tosylate 2a′. However, under the standard conditions for reductive alkylation of vinyl triflates, no desired product 17a was observed (Scheme S1a†). The use of Hosoya's conditions13h that are effective for reductive alkylation of aryl-OTf with alkyl iodide gave a trace of 17a (Scheme S1b†). Further optimization of the reaction conditions revealed that the use of NiBr2/BPhen (5/7.5 mol%) as a catalyst, LiBr/KBr (1.0/1.0 equiv.)30 and styrene (1.0 equiv.) as additives, and DMSO/CH3CN as a solvent afforded 17a in 83% yield (see Tables S3 and S4 in the ESI† for details). Although the role of styrene is presently unclear, the use of styrene (1.0 equiv.) has totally inhibited the protonation of 16a. The reaction of dipeptide 16b with alkyl tosylate 2a′ afforded the desired product in 74% yield (Table 4, 17b). No racemization at the stereogenic centers of the product was observed.31 A wide range of amino acids, including phenylalanine (17b), methionine (17c), glutamate (17d), tryptophan (17e), glycine (17f), valine (17g), leucine (17g), and proline (17h), embedded in the peptides are compatible with the reductive conditions. Tyrosines in tripeptides (17g, 17h) and tetrapeptide (17i) were alkylated selectively with moderate to good yields.
| a Peptide 16 (0.1 mmol) was used, isolated yields. |
|---|
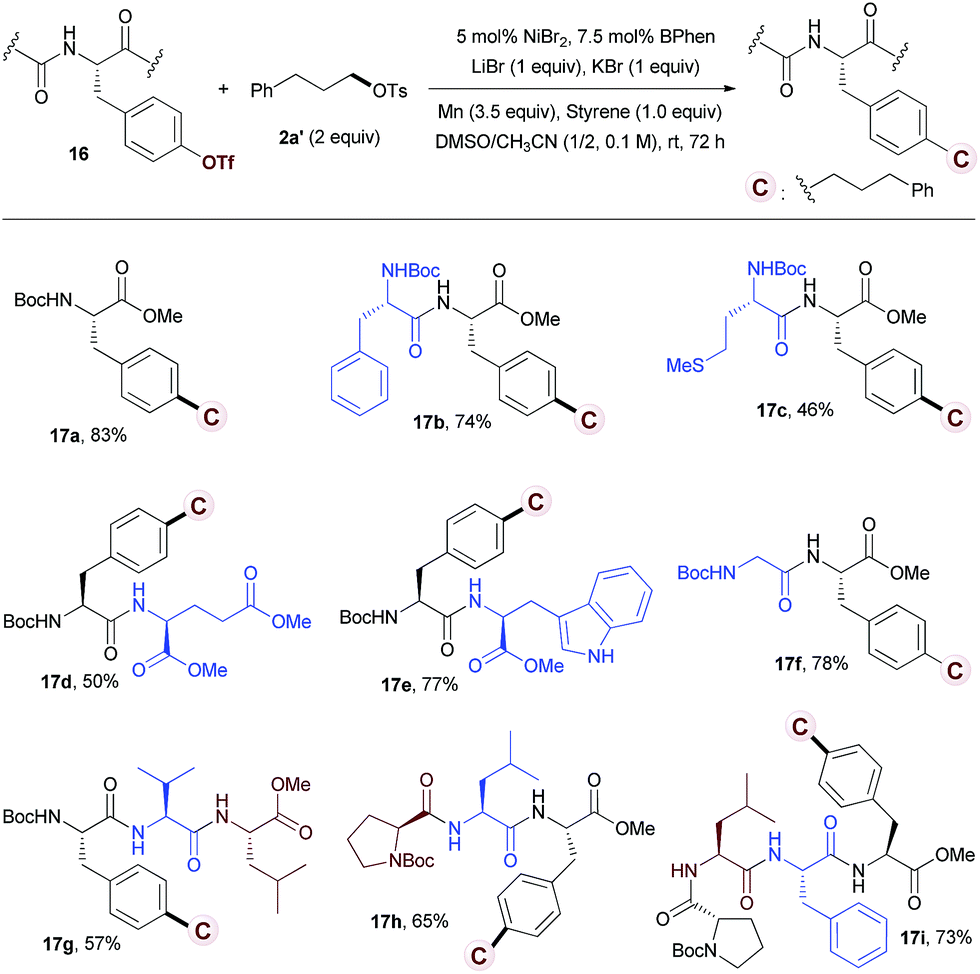
|
We then studied the scope of the reaction with respect to alkyl tosylates (Table 5). The reactions of 16b with non-functionalized alkyl tosylates afforded the products with high yields (17j, 17k). The introduction of functional groups into the peptides can provide the opportunity for further diversification. Alkyl tosylates that bear functional groups such as the methoxy group (17l), THP- or benzoyl-protected alcohol (17m, 17o), ester (17n), amides (17p, 17q), and alkene (17r) coupled with 16b efficiently. The use of aryl substituted alkyl electrophiles resulted in the products with moderate to high yields (17s–u). Unfortunately, in the current stage, attempts to introduce secondary alkyl groups have been unsuccessful.
| a Peptide 16b (0.1 mmol) was used, isolated yields. |
|---|
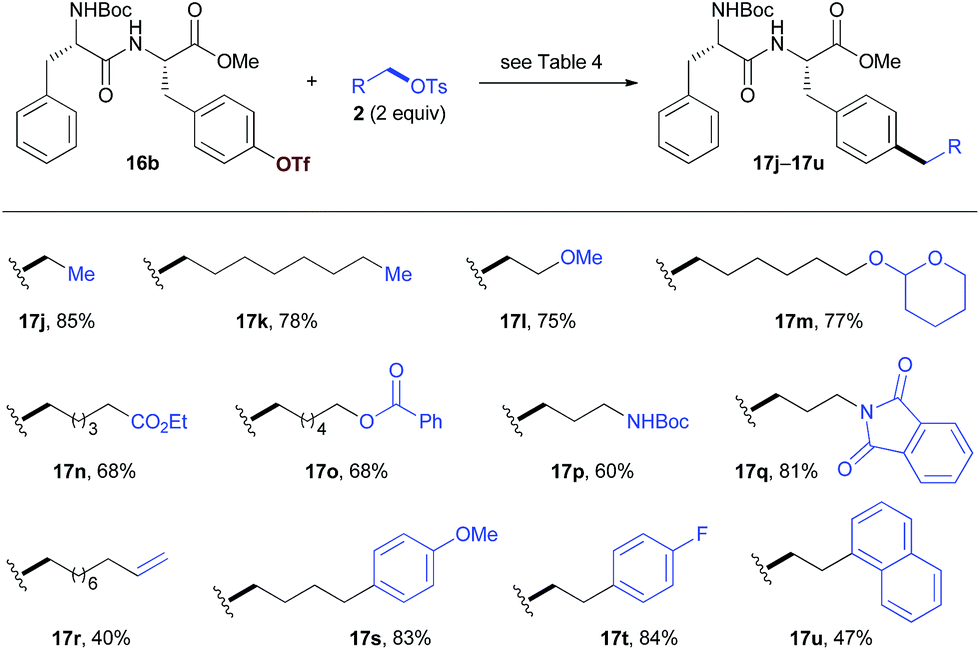
|
Conclusions
In summary, we have demonstrated a coupling reaction between vinyl/aryl and alkyl C–O electrophiles. This reaction enables facile access to a wide range of important, highly functionalized, and secondary alkyl substituted cycloalkenes that are difficult to obtain by conventional methods. This work established a framework for constructing aliphatic cycloalkenes from ketones and alcohols. It also provides an efficient approach for diversification of tyrosine in peptides. It thus expands the number of synthetic options that can be included in retrosynthetic analysis and allows facile modification of biochemicals. The synthetic application of this method, expanding the scope of cross-coupling between C–O electrophiles, and the asymmetric functionalization of C–O bonds are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (21772072 and 21502078) and the 1000 Talents Plan Program for their financial support. We also thank Prof. Xue-Yuan Liu (Lanzhou University) for helpful discussion.Notes and references
- Selected references: (a) G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342 CrossRef CAS PubMed; (b) X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 13862 CrossRef CAS PubMed; (c) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016 CrossRef CAS PubMed.
- Selected reviews: (a) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 682 CrossRef PubMed; (b) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242 CrossRef CAS PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (d) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC; (e) E. L. Lucas and E. R. Jarvo, Nat. Rev. Chem., 2017, 1, 65 CrossRef; (f) E. Richmond and J. Moran, Synthesis, 2018, 50, 499 CrossRef CAS.
- Metal-Catalyzed Cross-coupling Reactions, ed. F. Diederich and P. G. Stang, Wiley-VCH, New York, 1998 Search PubMed.
- Selected recent references: (a) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed; (b) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645 CrossRef CAS PubMed; (c) K. M. Arendt and A. G. Doyle, Angew. Chem., Int. Ed., 2015, 54, 9876 CrossRef CAS PubMed; (d) E. J. Tollefson, L. W. Erickson and E. R. Jarvo, J. Am. Chem. Soc., 2015, 137, 9760 CrossRef CAS PubMed; (e) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC; (f) G. A. Molander, K. M. Traister and B. T. O'Neill, J. Org. Chem., 2015, 80, 2907 CrossRef CAS PubMed; (g) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016 CrossRef CAS PubMed; (h) Z.-C. Cao and Z.-J. Shi, J. Am. Chem. Soc., 2017, 139, 6546 CrossRef CAS PubMed; (i) K. Komeyama, R. Ohata, S. Kiguchi and I. Osaka, Chem. Commun., 2017, 53, 6401 RSC; (j) T. Lin, J. Mi, L. Song, J. Gan, P. Luo, J. Mao and P. J. Walsh, Org. Lett., 2018, 20, 1191 CrossRef CAS PubMed; (k) X.-G. Jia, P. Guo, J.-C. Duan and X.-Z. Shu, Chem. Sci., 2018, 9, 640 RSC; (l) X.-B. Yan, C.-L. Li, W.-J. Jin, P. Guo and X.-Z. Shu, Chem. Sci., 2018, 9, 4529 RSC.
- Isolated examples of the coupling of Ar-OTs and MeOTs, see: J. Wang, J. Zhao and H. Gong, Chem. Commun., 2017, 53, 10180 RSC.
- (a) Comprehensive Organic Transformations, ed. R. C. Larock, Wiley, New York, 2nd edn, 1999 Search PubMed; (b) E. Piers and R. W. Friesen, Can. J. Chem., 1992, 70, 1204 CrossRef CAS; (c) Q. Jin, J. W. Lee, H. Jang, J. E. Choi, D. Lee, J. T. Hong, Y. Kim, M. K. Lee and B. Y. Hwang, J. Nat. Prod., 2016, 79, 1548 CrossRef CAS PubMed; (d) C. Gény, A. A. Samra, P. Retailleau, B. I. Iorga, H. Nedev, K. Awang, F. Roussi, M. Litaudon and V. Dumontet, J. Nat. Prod., 2017, 80, 3179 CrossRef PubMed.
- (a) Alkenes, ed. D. Meijere, Thieme Publisher, Stuttgart, Germany, 2009, vol. 47 Search PubMed; (b) Preparation of Alkenes, ed. J. M. J. Williams, Oxford, Oxford University Press, 1996 Search PubMed.
- Selected reviews: (a) K. Tamao, Coupling Reactions Between sp3 and sp2 Carbon Centers, ch. 2.2, 1991, vol. 3 Search PubMed; (b) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674 CrossRef CAS PubMed; (c) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (d) T. Iwasaki and N. Kambe, Top. Curr. Chem., 2016, 374, 66 CrossRef PubMed.
- Selected references: (a) J. E. McMurry and W. J. Scott, Tetrahedron Lett., 1980, 21, 4313 CrossRef CAS; (b) W. J. Scott, G. T. Crisp and J. K. Stille, J. Am. Chem. Soc., 1984, 106, 4630 CrossRef CAS; (c) I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155 CrossRef PubMed; (d) B.-J. Li, L. Xu, Z.-H. Wu, B.-T. Guan, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2009, 131, 14656 CrossRef CAS PubMed; (e) A. Krasovskiy and B. H. Lipshutz, Org. Lett., 2011, 13, 3822 CrossRef CAS PubMed.
- Selected reviews: (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (b) S. Chassaing, S. Specklin, J.-M. Weibel and P. Pale, Curr. Org. Synth., 2012, 9, 806 CrossRef CAS. Selected references: (c) S. Chiba, M. Kitamura and K. Narasaka, J. Am. Chem. Soc., 2006, 128, 6931 CrossRef CAS PubMed; (d) M. G. Tabor and R. A. Shenvi, Org. Lett., 2015, 17, 5776 CrossRef CAS PubMed; (e) B. Wang, H. Qin, F. Zhang and Y. Jia, Tetrahedron Lett., 2014, 55, 1561 CrossRef CAS.
- The oxidative addition of unactivated secondary alkyl electrophiles to a metal center is very difficult, see: (a) I. D. Hills, M. R. Netherton and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 5749 CrossRef CAS PubMed. Further, secondary metal species are relatively unstable, resulting in increased side reactions such as β-hydride elimination and isomerization. For related references, see: (b) S. Dupuy, K.-F. Zhang, A.-S. Goutierre and O. Baudoin, Angew. Chem., Int. Ed., 2016, 55, 14793 CrossRef CAS PubMed.
- Unlike many approaches to acyclic vinyl metals, cyclic vinyl metals are typically produced from cyclic vinyl halides by using strongly basic lithiation conditions. Cyclic vinyl halides themselves are difficult to access.
- (a) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146 CrossRef CAS PubMed; (b) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365 CrossRef CAS PubMed; (c) K. A. Johnson, S. Biswas and D. J. Weix, Chem.–Eur. J., 2016, 22, 7399 CrossRef CAS PubMed; (d) J. Liu, Q. Ren, X. Zhang and H. Gong, Angew. Chem., Int. Ed., 2016, 55, 15544 CrossRef CAS PubMed; (e) Y. Cai, A. D. Benischke, P. Knochel and C. Gosmini, Chem.–Eur. J., 2017, 23, 250 CrossRef CAS PubMed; (f) X. Lu, Y. Wang, B. Zhang, J.-J. Pi, X.-X. Wang, T.-J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632 CrossRef CAS PubMed; (g) J. Gu, C. Qiu, W. Lu, Q. Qian, K. Lin and H. Gong, Synthesis, 2017, 49, 1867 CrossRef CAS; (h) Y. Sumida, T. Sumida and T. Hosoya, Synthesis, 2017, 49, 3590 CrossRef CAS; (i) J. L. Hofstra, A. H. Cherney, C. M. Ordner and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 139 CrossRef CAS PubMed; (j) A. M. Olivares and D. J. Weix, J. Am. Chem. Soc., 2018, 140, 2446 CrossRef CAS PubMed.
- Vinyl triflates are readily available from carbonyl compounds and alkynes, see: (a) P. J. Stang, M. Hanack and L. R. Subramanian, Synthesis, 1982, 85 CrossRef CAS; (b) H. Ritter, Synthesis, 1994, 735 Search PubMed; (c) M. G. Suero, E. D. Bayle, B. S. L. Collins and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 5332 CrossRef CAS PubMed.
- We choose mesylate as a leaving group because it is the smallest sulfonate. The effect of various leaving groups on this reaction, see Table S2 in the ESI.†.
- A. Jutand and S. Négri, Organometallics, 2003, 22, 4229 CrossRef CAS.
- Unactivated alkyl mesylates have been rarely employed in Ni catalysis. An elegant study on homo-coupling, see: (a) M. R. Prinsell, D. A. Everson and D. J. Weix, Chem. Commun., 2010, 46, 5743 RSC. For relative reactivities of alkyl halide and sulfonate with transition metals, see: (b) R. G. Pearson and P. E. Figdore, J. Am. Chem. Soc., 1980, 102, 1541 CrossRef CAS; (c) J. Terao, H. Todo, S. A. Begum, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2007, 46, 2086 CrossRef CAS PubMed. A reductive coupling of alkyl mesylates by Cu catalysis, see: (d) J.-H. Liu, C.-T. Yang, X.-Y. Lu, Z.-Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem.–Eur. J., 2014, 20, 15334 CrossRef CAS PubMed.
- There are only isolated examples demonstrating the coupling of vinyl triflates with alkyl halides, see ref. 13c, h, i. However, under those conditions, no or trace of product 3a was observed from the reaction of 1a and 2a.
- An elegant study on iodide co-catalyzed cross-coupling: (a) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2014, 136, 48 CrossRef CAS PubMed. For previous studies proposed involving a sulfonate/halide exchange, see: (b) H.-Q. Do, E. R. R. Chandrashekar and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 16288 CrossRef CAS PubMed; (c) Z. Liang, W. Xue, K. Lin and H. Gong, Org. Lett., 2014, 16, 5620 CrossRef CAS PubMed.
- (a) M. Ferles and J. Pliml, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky and A. J. Boulton, Academic Press, New York, 1970, vol. 12, pp. 43–101 Search PubMed; (b) E. Vedejs and G. A. Kraff, Tetrahedron, 1982, 38, 2857 CrossRef CAS.
- (a) W. A. Kinney, G. D. Crouse and L. A. Paquette, J. Org. Chem., 1983, 48, 4986 CrossRef CAS; (b) D. Tanner and P. Somfai, Tetrahedron, 1986, 42, 5657 CrossRef CAS.
- S. R. LaPlante, M. Bös, C. Brochu, C. Chabot, R. Coulombe, J. R. Gillard, A. Jakalian, M. Poirier, J. Rancourt, T. Stammers, B. Thavonekham, P. L. Beaulieu, G. Kukolj and Y. S. Tsantrizos, J. Med. Chem., 2014, 57, 1845 CrossRef CAS PubMed.
- (a) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef CAS PubMed; (b) J. Breitenfeld, J. Ruiz, M. D. Wodrich and X. Hu, J. Am. Chem. Soc., 2013, 135, 12004 CrossRef CAS PubMed.
- Selected references on cross-electrophile coupling between aryl and alkyl electrophiles: (a) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920 CrossRef CAS PubMed; (b) S. Wang, Q. Qian and H. Gong, Org. Lett., 2012, 14, 3352 CrossRef CAS PubMed; (c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442 CrossRef CAS PubMed; (d) G. A. Molander and K. M. Traister, J. Org. Chem., 2014, 79, 5771 CrossRef CAS PubMed; (e) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562 CrossRef CAS PubMed; (f) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed . Also see: ref. 4e, f, i, 17d and 19c.
- (a) M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511 RSC; (b) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed; (c) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS PubMed.
- (a) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed; (b) Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91 RSC; (c) B. Albada and N. Metzler-Nolte, Chem. Rev., 2016, 116, 11797 CrossRef CAS PubMed.
- T. Willemse, W. Schepens, H. W. T. van Vlijmen, B. U. W. Maes and S. Ballet, Catalysts, 2017, 7, 74 CrossRef.
- Selected references: (a) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef CAS PubMed; (b) E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687 CrossRef CAS PubMed; (c) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (d) A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576 CrossRef CAS PubMed; (e) J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346 CrossRef CAS PubMed.
- Z.-X. Tian, J.-B. Qiao, G.-L. Xu, X. Pang, L. Qi, W.-Y. Ma, Z. Zhao, J. Duan, Y.-F. Du, P. Su, X.-Y. Liu and X.-Z. Shu, J. Am. Chem. Soc., 2019, 141, 7637 CrossRef CAS PubMed.
- Hosoya has recently revealed that the addition of LiBr can accelerate the oxidative addition of aryl-OTf to the Ni(0), and stabilize the aryl-Ni(II) intermediate. See: Y. Sumida, T. Sumida, D. Hashizume and T. Hosoya, Org. Lett., 2016, 18, 5600 CrossRef CAS PubMed.
- This was determined after analysis of the diastereomers.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03347e |
| This journal is © The Royal Society of Chemistry 2019 |