
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplate catalysis by manganese pincer complexes: oxa- and aza-Michael additions to unsaturated nitriles†
Shan
Tang
 and
David
Milstein
*
and
David
Milstein
*
Department of Organic Chemistry, The Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: david.milstein@weizmann.ac.il
First published on 7th August 2019
Abstract
Activation of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds by metal–ligand cooperation provides a new route for the functionalization of nitriles. Herein, we report the electrophilic activation of unsaturated nitriles by dearomatized manganese pincer complexes for the oxa- and aza-Michael addition reactions under very mild and neutral conditions. Derivatives of acrylonitrile and allyl cyanide furnished the corresponding β-addition products by reacting with alcohols and amines. Mechanistically, the catalysis is mostly ligand based. Reaction of the dearomatized PNN–Mn complex with 2-pentenenitrile or 3-pentenenitrile furnished an enamido–Mn complex. The equilibrium between an enamido complex and a ketimido complex, and reversible C–C bond formation with the ligand are proposed to play central roles in the catalysis.
N bonds by metal–ligand cooperation provides a new route for the functionalization of nitriles. Herein, we report the electrophilic activation of unsaturated nitriles by dearomatized manganese pincer complexes for the oxa- and aza-Michael addition reactions under very mild and neutral conditions. Derivatives of acrylonitrile and allyl cyanide furnished the corresponding β-addition products by reacting with alcohols and amines. Mechanistically, the catalysis is mostly ligand based. Reaction of the dearomatized PNN–Mn complex with 2-pentenenitrile or 3-pentenenitrile furnished an enamido–Mn complex. The equilibrium between an enamido complex and a ketimido complex, and reversible C–C bond formation with the ligand are proposed to play central roles in the catalysis.
Introduction
Nitriles are very important constituents in a diverse variety of pharmaceuticals and natural products. Moreover, nitriles serve as versatile synthetic intermediates since the cyano groups can be easily transformed into carbonyl compounds. The synthesis and transformations of nitriles have attracted much interest from organic chemists.1–4 Metal–ligand cooperation (MLC) of pincer metal complexes provides a unique way for the activation of unsaturated chemical bonds.5–11 In 2013, our group reported the activation of nitriles by a dearomatized pyridine-based PNP–Re complex through metal–ligand-cooperation.12 Stoichiometric reactions of nitriles with the dearomatized PNP–Re complex furnished either ketimido or enamido complexes with C–C and Re–N bond formation. Importantly, the formation of enamido complexes enabled the Michael addition of benzylic nitriles to α,β-unsaturated carbonyl compounds. Later on, we extended this reaction protocol to unactivated aliphatic nitriles in Michael addition reactions catalyzed by a dearomatized pyridine-based PNP–Mn complex.13 Cooperative activation of aliphatic nitriles to form nucleophilic species was the key to the success of this reaction. In both cases, the metal center was not involved in the bond forming process. Otten, de Vries and co-workers developed the oxa-Michael addition reaction of alcohols to unsaturated nitriles by using a dearomatized pyridine-based PNN–Ru pincer complex (Scheme 1a).14,15 Metal–ligand-cooperation was the key to the success of the transformation. Compared with the oxa-Michael addition, much lower activity was observed for the aza-Michael addition reactions of amines to unsaturated nitriles even at higher temperatures. Since β-amino nitriles are both pharmacologically and synthetically of great importance,16,17 it is desirable to discover metal catalysts with high activity under mild conditions for the aza-Michael addition reactions of unsaturated nitriles with amines.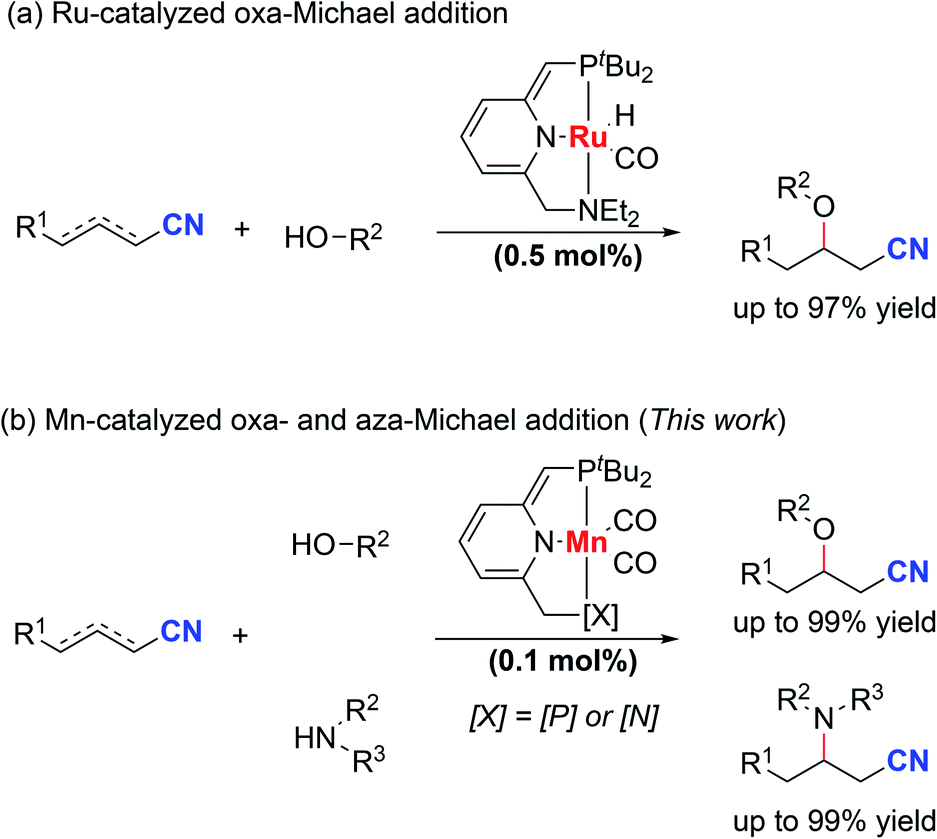 | ||
| Scheme 1 Activation of unsaturated nitriles through metal–ligand cooperation. | ||
In recent years, much effort has been made to replace catalysts based on noble metals with complexes of earth abundant, base transition metals.18–21 Base metals including copper and nickel were used in the hetero-Michael addition of alcohols to unsaturated nitriles.22–28 As manganese is the third most abundant transition metal in the Earth's crust, manganese complexes have received much attention recently as catalysts in organic transformations.29–44 Generally, higher catalyst loading was required and lower reactivity was observed compared with noble metal-based catalysts.45–47 These issues hinder the practical application of manganese catalysts. Herein, we report a manganese-catalyzed hetero-Michael addition reaction to unsaturated nitriles. Dearomatized pyridine-based PNP–Mn and PNN–Mn complexes can be used as catalysts in this transformation (Scheme 1b). Quite remarkably, the oxa- and even aza-Michael reactions of alcohols and amines proceed smoothly at room temperature using only 0.1 mol% catalyst loading and no added base. Quite unusually for a homogeneously catalyzed reaction, the aza-Michael reaction proceeds more favorably using a complex of an earth abundant metal (manganese) than with a ruthenium complex.
Results and discussion
Crotononitrile (1a) and isopropanol (2a) were chosen as model substrates in this research (Table 1). Using 0.5 mol% pyridine-based dearomatized PNP–Mn complex ([Mn]-1) as the catalyst in THF at room temperature, 44% oxa-Michael addition product was obtained using 2 mL (∼13 equiv.) of isopropanol (Table 1, entry 1) while only 26% yield was obtained with 2 equiv. of isopropanol (Table 1, entry 2). A higher yield was obtained in the absence of THF (Table 1, entry 3). Under these conditions, the major side reaction was the formation of homo-coupling products of crotononitrile (Table 1, entries 1–3). Since the homo-coupling of crotononitrile could be reduced at low catalyst loading, lowering the catalyst loading led to increased reaction selectivity (Table 1, entries 4–6). The best result was obtained by using 0.1 mol% [Mn]-1 as the catalyst (Table 1, entry 5). Other dearomatized pincer manganese complexes were also tested in this transformation. The complex [Mn]-2 demonstrated a slightly higher reaction efficiency and selectivity than complex [Mn]-1 (Table 1, entry 7). The bipyridine-based dearomatized PNP–Mn complex [Mn]-3 also demonstrated good reactivity in this transformation (Table 1, entry 8). A control experiment utilizing a strong base, KOtBu, as the catalyst gave a very low yield of the oxa-Michael addition product (Table 1, entry 9). Moreover, no reaction was detected in the absence of any catalyst (Table 1, entry 10).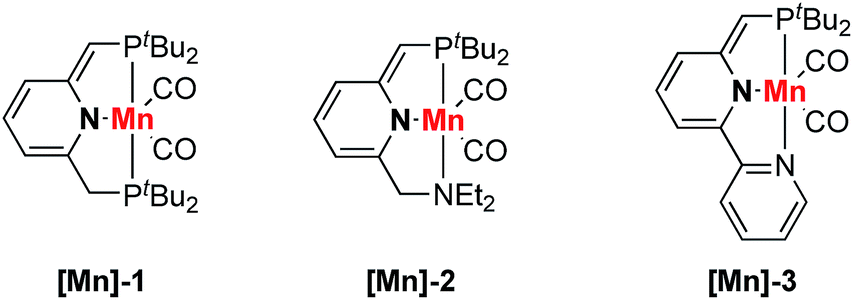
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield (%) |
| a Reaction conditions: crotononitrile (2.0 mmol), isopropanol (2.0 mL, ∼13 equiv.), catalyst, solvent (2.0 mL), r.t., 24 h. Yields determined by GC with mesitylene as the internal standard. n.r. = no reaction. b Isopropanol (0.31 mL, 2.0 equiv.) was used. c 1 M solution in THF. | |||
| 1 | 0.50 mol% [Mn]-1 | THF | 44 |
| 2 | 0.50 mol% [Mn]-1 | THF | 26b |
| 3 | 0.50 mol% [Mn]-1 | 67 | |
| 4 | 0.25 mol% [Mn]-1 | 82 | |
| 5 | 0.10 mol% [Mn]-1 | 93 | |
| 6 | 0.04 mol% [Mn]-1 | 30 | |
| 7 | 0.10 mol% [Mn]-2 | 96 | |
| 8 | 0.10 mol% [Mn]-3 | 94 | |
| 9 | 0.25 mol% KOtBuc | 17 | |
| 10 | None | n.r. | |
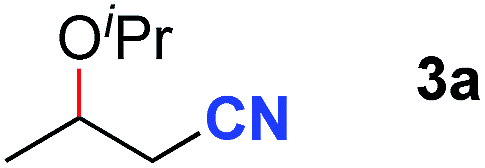
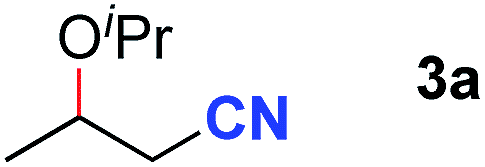
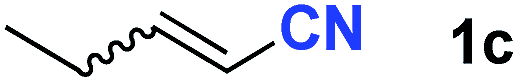
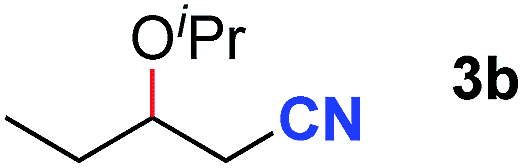
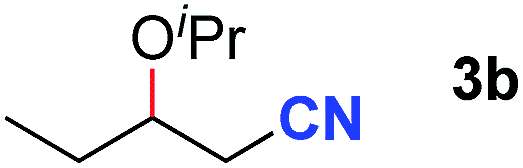
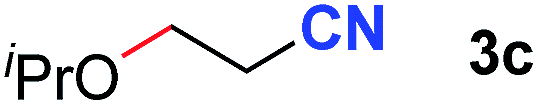
After achieving oxa-Michael addition by manganese catalysis, the scope of unsaturated nitriles was explored (Table 2). With [Mn]-2 as the catalyst, allyl cyanide furnished the same product as crotononitrile but with a slightly decreased yield (Table 2, entries 1 and 2). Similarly, both 2-pentenenitrile and 3-pentenenitrile afforded the β-alkoxylation product 3b (Table 2, entries 3 and 4). However, only a moderate yield was obtained in the case of 3-pentenenitrile due to the homo-coupling side-reaction of 1d (Table 2, entry 4). An excellent yield was obtained using acrylonitrile as the substrate (Table 2, entry 5). Using 0.5 mol% [Mn]-2 as the catalyst and a linear α,β-unsaturated nitrile bearing an ester group the desired oxa-Michael addition product was obtained in 71% yield (Table 2, entry 6). Notably, the ester group was tolerated under the standard, base-free conditions, while it could readily undergo the ester exchange reaction under basic conditions.
|
|
|||
|---|---|---|---|
| Entry | Nitrile | Product | Yield (%) |
| a Reaction conditions: crotononitrile (2.0 mmol), isopropanol (2.0 mL), [Mn]-2 (0.0020 mmol), r.t., 24 h. The reaction yield was determined by 1H NMR with mesitylene as the internal standard. b Isolated yields. c 0.5 mol% [Mn]-2 was used. | |||
| 1 |

|
|
96 |
| 2 |

|
|
92 |
| 3 |
|
|
83b |
| 4 |

|
|
66 |
| 5 |

|
|
>99 |
| 6 |

|

|
71c |
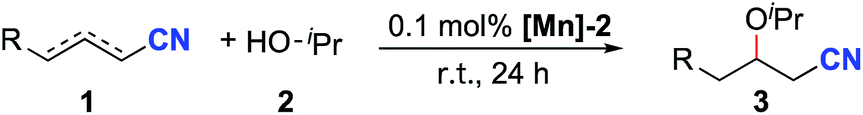
Next, the reactions of unsaturated nitriles with various alcohols and amines were conducted (Scheme 2). Linear aliphatic alcohols such as ethanol, methanol and n-butanol afforded high yields of the oxa-Michael addition products (Scheme 2, 3e–3g, 3p). The reaction with 2-methoxyethanol furnished the oxa-Michael addition product in 85% yield (Scheme 2, 3h). With a slightly increased catalyst loading, the unsaturated 2-methylallyl alcohol afforded the desired product in a high yield (Scheme 2, 3i). Tertiary alcohols and benzyl alcohol were unsuitable substrates under the standard reaction conditions; the latter case is possibly due to the competing addition of benzyl alcohol by MLC to [Mn-2].
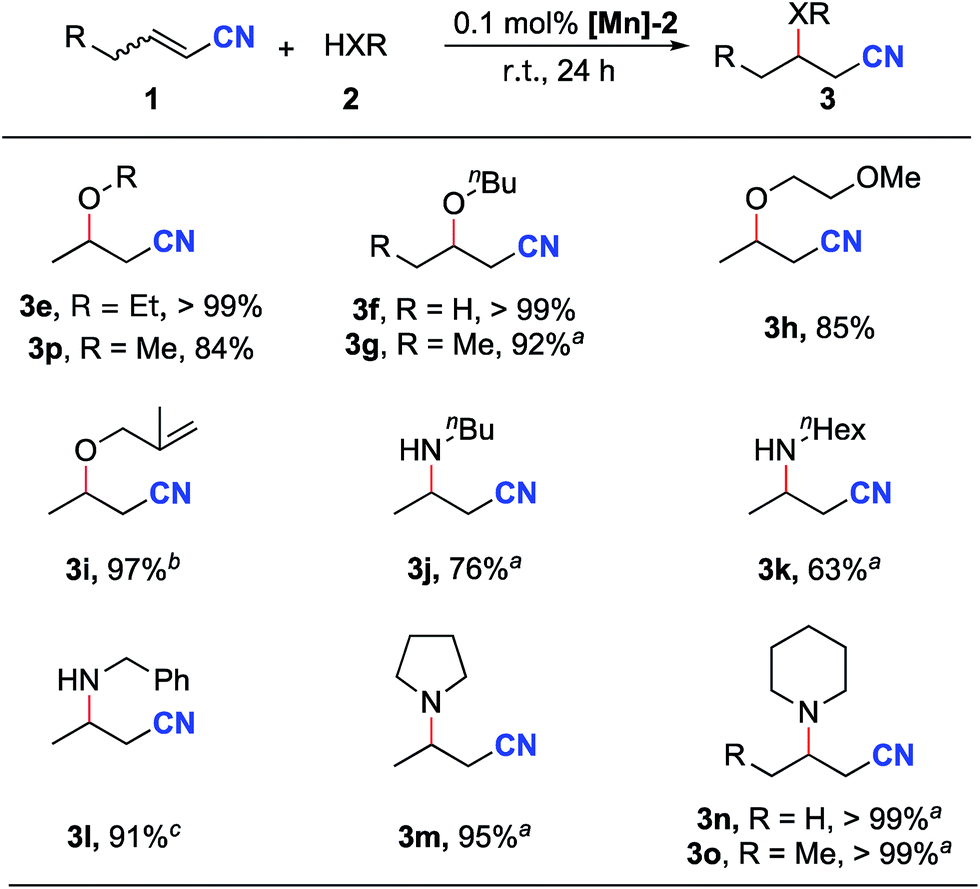 | ||
| Scheme 2 Scope of alcohols and amines. Reaction conditions: crotononitrile or 2-pentenenitrile (2.0 mmol), alcohol or amine (2.0 mL), [Mn]-2 (0.0020 mmol, 0.1 mol%), r.t., 24 h. Yields determined by 1H NMR with mesitylene as the internal standard. aIsolated yields. b0.5 mol% [Mn]-2 was used. cBenzylamine (1.0 mL) was used. | ||
Significantly, this reaction protocol was also applicable to amines for aza-Michael addition reactions. Moderate reaction yields were observed using linear aliphatic amines such as n-butylamine or n-hexylamine (Scheme 2, 3j and 3k). Benzylamine afforded the aza-Michael addition product in 91% yield (3l). Notably, secondary aliphatic amines demonstrated excellent reactivity (Scheme 2, 3m–3o). In the case of piperidine, the same aza-Michael addition product (3n) was obtained in quantitative yield when allyl cyanide was used instead of crotononitrile.
The scalability of both the oxa- and aza-Michael additions was evaluated by performing reactions on the 10 mmol scale. After reacting at room temperature for 48 h, the oxa-Michael addition reaction of 2-pentenenitrile with n-butanol (∼5.5 equiv.) afforded 1.34 g (86%) of the desired product 3g (Scheme 3a). Under similar reaction conditions, the aza-Michael addition of crotononitrile with n-butylamine (∼5.0 equiv.) furnished 0.87 g (62%) of the desired product 3j (Scheme 3b).
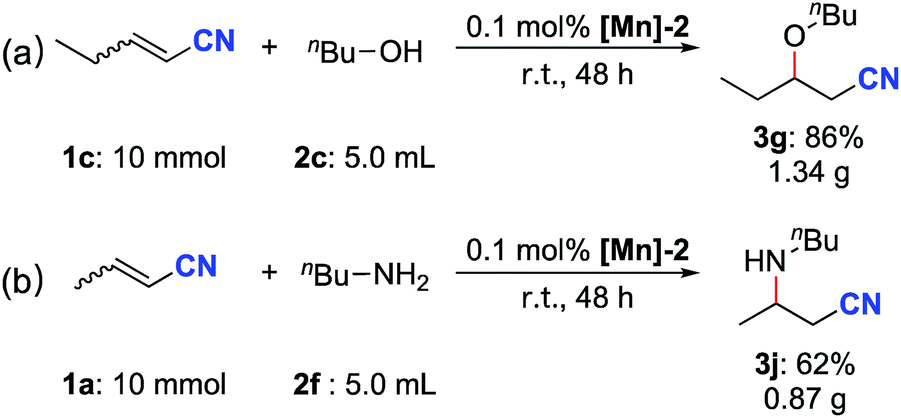 | ||
| Scheme 3 Gram scale reactions. | ||
In order to gain insights into the reaction mechanism, we explored the stoichiometric reactivity of the dearomatized complex [Mn]-2 with the substrates (see Fig. S1, ESI†). According to 31P{1H} NMR analysis, no reaction was detected between complex [Mn]-2 and isopropanol in THF at room temperature (Scheme 4a). In contrast, both nitriles 1c and 1d reacted with complex [Mn]-2 to furnish an enamido–Mn complex A (Scheme 4b and c). By layering a concentrated THF solution with pentane at −38 °C, complex A was isolated in 56% yield as orange crystals. Compared with the dearomatized complex [Mn]-2, the 31P{1H} NMR spectrum of complex A exhibited a clear down-field chemical shift to 140.5 ppm. In the 1H NMR spectrum, three different olefinic CH signals were detected and an NH resonance appeared at 3.99 ppm as a singlet, indicating the formation of an enamido–Mn complex. The proton at the benzylic position which underwent C–C coupling was observed at 3.64 ppm as a characteristic singlet. The lack of correlation between this benzylic proton and the phosphine indicated that the nitrile reacted with the amine arm, likely following a tautomerization step in the reaction of [Mn]-2 to form complex A.9,10,14
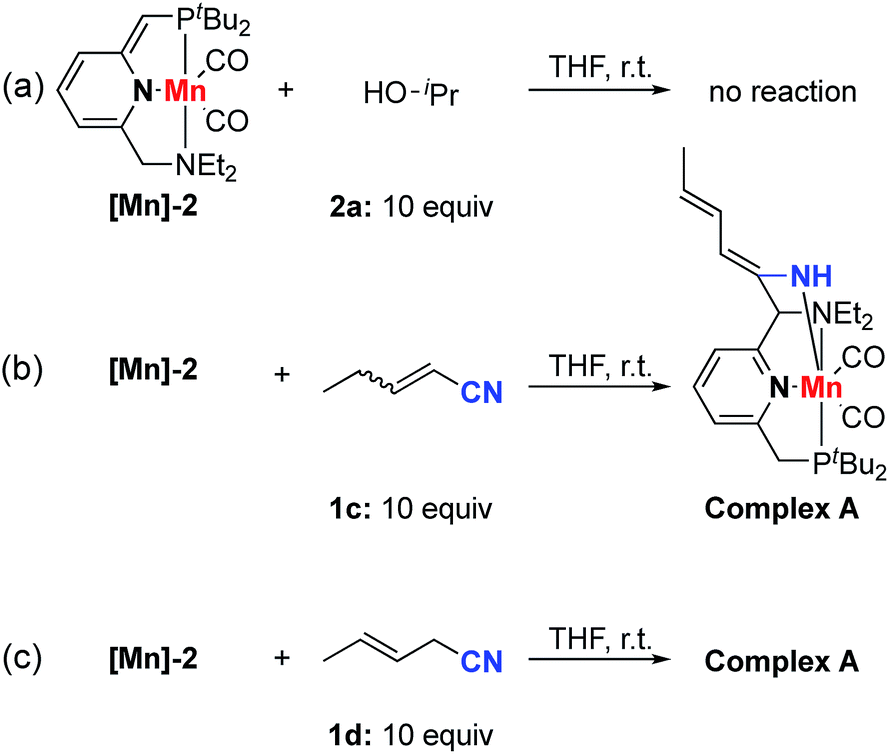 | ||
| Scheme 4 Mechanistic reactions. | ||
To explore the participation of complex A in the catalytic reactions, oxa- and aza-Michael addition reactions were explored by using it as the catalyst. The reaction between nitrile 1c and isopropanol resulted in the generation of the oxa-Michael addition product 3b in 61% yield after 48 h (Scheme 5a). In addition, complex A also catalyzed the aza-Michael addition reaction between 1c and piperidine, affording the desired product 3o in 79% yield (Scheme 5b). These results support the involvement of complex A in the nitrile activation process.
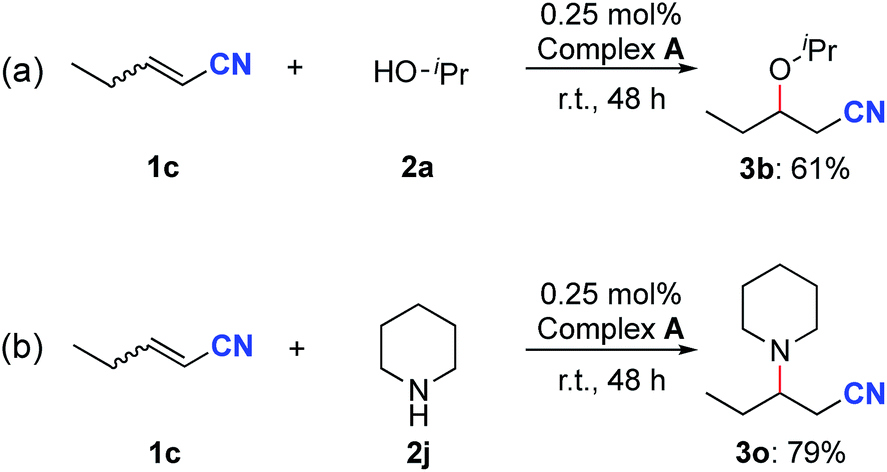 | ||
| Scheme 5 Catalytic reactivity of complex A. | ||
Next, a competition experiment between n-butanol and n-butylamine in the reaction with crotononitrile was carried out (Scheme 6). The aza-Michael addition product 3j was obtained in 91% yield while only a trace amount of the oxa-Michael addition product 3f was detected. This selectivity is completely opposite to that observed by Otten, de Vries and co-workers by using a dearomatized pyridine-based PNN–Ru pincer complex as the catalyst.14 The higher nucleophilicity of n-butylamine compared with n-butanol is likely to be the key factor for the observed selectivity in our case.
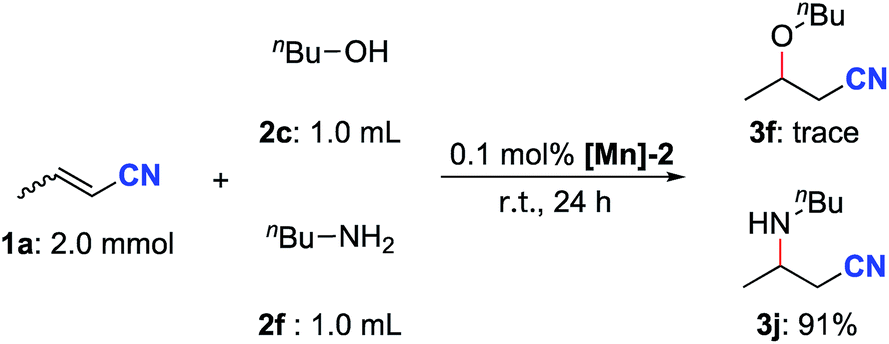 | ||
| Scheme 6 Competing reaction between n-butanol and n-butylamine in the reaction with crotononitrile. | ||
According to the experimental results and previous reports,13,14 a plausible mechanism is proposed for the reaction between 2-pentenenitrile or 3-pentenenitrile and isopropanol (Scheme 7). First, complex [Mn]-2 tautomerizes to form complex [Mn]-2*. Reaction of [Mn]-2* with the unconjugated unsaturated nitrile 1d gives an enamido complex, A, while the reaction of [Mn]-2* with conjugated unsaturated nitrile 1c furnishes a ketimido complex, B. The enamido complex A and the ketimido complex B are in equilibrium with each other. Isopropanol undergoes direct nucleophilic addition to the ketimido complex B to form a new enamido complex, C. Finally, complex C tautomerizes to form the imine complex D, which undergoes C–C cleavage, releasing the product 3b, thus regenerating the dearomatized complex [Mn]-2*. The homo-coupling of nitriles 1c or 1d is caused by the nucleophilic addition of complex A to another molecule of unsaturated nitrile.
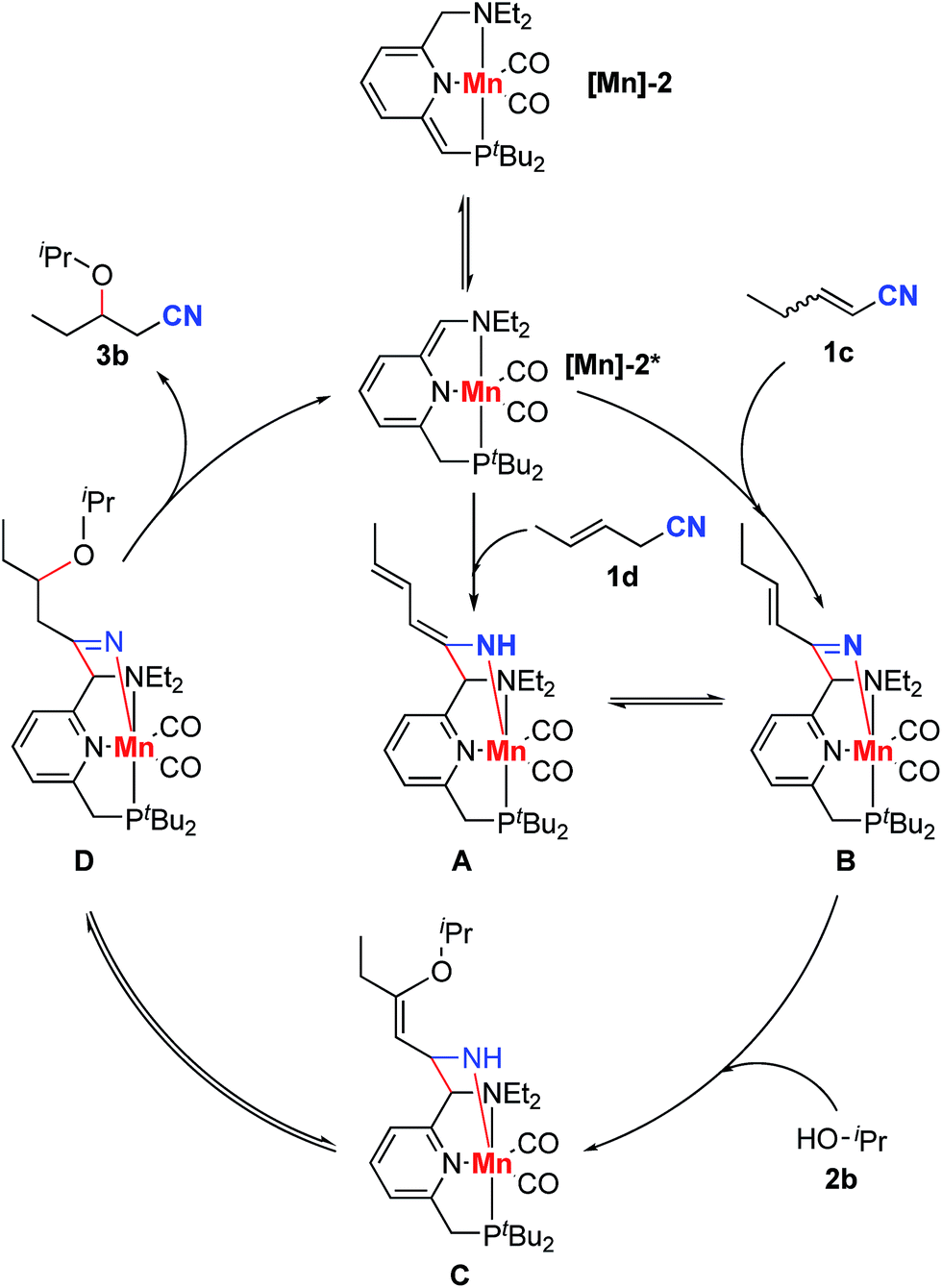 | ||
| Scheme 7 Proposed reaction mechanism. | ||
Conclusions
In summary, we have disclosed the manganese-catalyzed oxa- and aza-Michael addition to unsaturated nitriles at room temperature under base-free reaction conditions. Both conjugated unsaturated nitriles and allyl cyanides are suitable substrates in this manganese-catalyzed transformation. The reaction proceeded well with various alcohols and amines. Notably, the aza-Michael reaction is favored over the oxa-Michael reaction, unlike the previously reported ruthenium catalyzed reaction.14 The success of this transformation relies on metal–ligand cooperation, involving the activation of the CN group by reversible C–C and Mn–N bond formation with the dearomatized pincer complex. Ketimido–Mn complexes are proposed to be the active intermediates for the C–O or C–N bond formation. Notably, the metal center does not directly participate in the bond formation process; the pincer manganese complex acts as a template in the catalytic cycle.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council (ERC AdG 692775). D. M. holds the Israel Matz Professorial Chair of Organic Chemistry. S. T. is thankful to the Israel Planning and Budgeting Committee (PBC) for a postdoctoral fellowship.Notes and references
- K. Friedrich and K. Wallenfels, in The Cyano Group, ed. Z. Rappoport, Wiley, New York, 1970, pp. 67–122 Search PubMed.
- M. North, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Elsevier Science, Oxford, 1995, pp. 611–640 Search PubMed.
- J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563–570 CrossRef CAS PubMed.
- S. Chakraborty, Y. J. Patel, J. A. Krause and H. Guan, Angew. Chem., Int. Ed., 2013, 52, 7523–7526 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325–10328 CrossRef CAS PubMed.
- M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2012, 18, 9194–9197 CrossRef CAS PubMed.
- M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2014, 5, 2043–2051 RSC.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 RSC.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef CAS.
- G. A. Filonenko, D. Smykowski, B. M. Szyja, G. Li, J. Szczygieł, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 1145–1154 CrossRef CAS.
- M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed.
- A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed.
- S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 CrossRef CAS PubMed.
- L. E. Eijsink, S. C. P. Perdriau, J. G. de Vries and E. Otten, Dalton Trans., 2016, 45, 16033–16039 RSC.
- L. Yin, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 9610–9611 CrossRef CAS PubMed.
- J. Zhao, X. Liu, W. Luo, M. Xie, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 3473–3477 CrossRef CAS PubMed.
- M. Beller, Chem. Rev., 2019, 119, 2089 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- C. Munro-Leighton, E. D. Blue and T. B. Gunnoe, J. Am. Chem. Soc., 2006, 128, 1446–1447 CrossRef CAS PubMed.
- C. Munro-Leighton, S. A. Delp, E. D. Blue and T. B. Gunnoe, Organometallics, 2007, 26, 1483–1493 CrossRef CAS.
- F. Wang, H. Yang, H. Fu and Z. Pei, Chem. Commun., 2013, 49, 517–519 RSC.
- S. Uesugi, Z. Li, R. Yazaki and T. Ohshima, Angew. Chem., Int. Ed., 2014, 53, 1611–1615 CrossRef CAS PubMed.
- D. M. Spasyuk and D. Zargarian, Inorg. Chem., 2010, 49, 6203–6213 CrossRef CAS PubMed.
- A. B. Salah, C. Offenstein and D. Zargarian, Organometallics, 2011, 30, 5352–5364 CrossRef CAS.
- X. Lefèvre, G. Durieux, S. Lesturgez and D. Zargarian, J. Mol. Catal. A: Chem., 2011, 335, 1–7 CrossRef.
- N. V. Kulkarni and W. D. Jones, in Pincer Compounds, ed. D. Morales-Morales, Elsevier, 2018, pp. 491–518 Search PubMed.
- J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 3912–3929 CrossRef CAS.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS.
- B. Maji and M. K. Barman, Synthesis, 2017, 49, 3377–3393 CrossRef CAS.
- M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef CAS.
- F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- N. Gorgas and K. Kirchner, Acc. Chem. Res., 2018, 51, 1558–1569 CrossRef CAS PubMed.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2016, 138, 15543–15546 CrossRef CAS PubMed.
- S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed.
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS.
- F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 7261–7265 CrossRef CAS PubMed.
- M. Mastalir, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8812–8815 CrossRef CAS PubMed.
- A. Kaithal, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS PubMed.
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS PubMed.
- Y. Wang, Z. Shao, K. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2018, 57, 15143–15147 CrossRef CAS PubMed.
- A. Kumar, N. A. Espinosa-Jalapa, G. Leitus, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 14992–14996 CrossRef CAS PubMed.
- N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed.
- S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229–4233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03269j |
| This journal is © The Royal Society of Chemistry 2019 |