
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II)-catalyzed chemoselective deacylative annulation of 1,3-diones with sulfoxonium ylides via C–C bond activation†
Si
Wen
,
Weiwei
Lv
,
Dan
Ba
,
Jing
Liu
and
Guolin
Cheng
 *
*
College of Materials Science & Engineering, Huaqiao University, Xiamen 361021, China. E-mail: glcheng@hqu.edu.cn
First published on 12th August 2019
Abstract
The first successful example of deacylative annulation of 1,3-diones with sulfoxonium ylides was achieved through Ru(II)-catalyzed C–C bond activation. The excellent chemoselectivity and broad substrate scope render this method a practical and versatile approach for the preparation of (hetero)aryl and alkenyl substituted furans, which are valuable units in many biologically active compounds and functional materials. A preliminary mechanistic study reveals that this process involves a deacylative α-ruthenation to generate key alkyl Ru(II) intermediates with the release of a benzoic acid fragment.
Introduction
Carbon–carbon bonds are the most extensive and basic chemical bonds in organic molecules. The selective cleavage of C–C bonds in a constructive manner enables the reconstitution of the molecular skeleton and introduction of new functional groups, thus attracting great attention.1 In the past decade, chemoselective C–C(CO) bond cleavage has been extensively studied by employing the strategies of chelation assistance,2 ring-strain release,3 and aromatization.4 However, the selective cleavage of unstrained C–C(CO) moieties without an auxiliary directing group still remains an unmet challenge.5,6 Recently, transition metal-catalyzed C–C(CO) bond functionalization of 1,3-diones has been achieved by Jiao,6a Bolm,6b Peng,6c and Wu.6d These methods are understood to proceed through oxidative α-functionalization of 1,3-diones, followed by retro-Claisen condensation to provide α-substituted ketones (Scheme 1a). However, Lei's work demonstrated an alternative reaction pathway for the C–C(CO) bond cleavage of 1,3-diones, in which deacylative α-cupration could occur to form alkyl Cu(III) complexes that subsequently underwent cross-coupling to give α-aryl ketones (Scheme 1b).7 Inspired by Lei's work, we questioned whether this open shell deacylative α-cupration mechanism might be translated to other transition metals, such as ruthenium, thereby allowing catalytic formation of alkyl Ru intermediates that can be captured by suitable coupling partners. In continuation of our research on C–C(CO) bond cleavage reactions,8 herein we disclose the first ruthenium-catalyzed deacylative annulation of 1,3-diones with sulfoxonium ylides (Scheme 1c). This method provides a practical and mild synthetic route to substituted furans,9 which are essential structural moieties in many biologically active compounds, natural products, and functional materials.10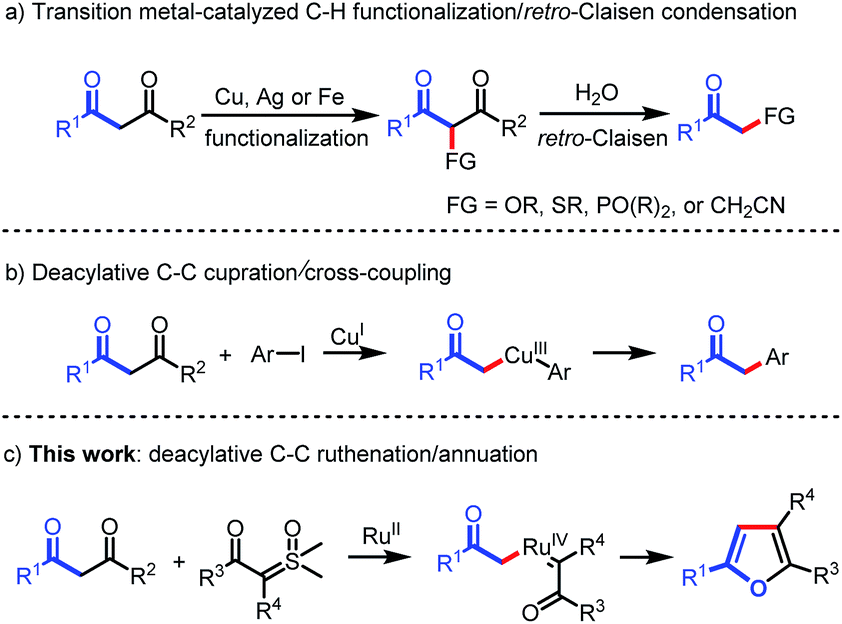 | ||
| Scheme 1 Transition metal-catalyzed C–C bond activation of 1,3-diones. | ||
On the other hand, sulfoxonium ylides are readily available and bench-stable carbene precursors, which have been extensively explored for the transition metal-catalyzed functionalization of C–H bonds.11 However, sulfoxonium ylide carbene-involved C–C bond functionalization has not yet been realized due to it being challenging to control the chemoselectivity from the same starting materials.12
Results and discussion
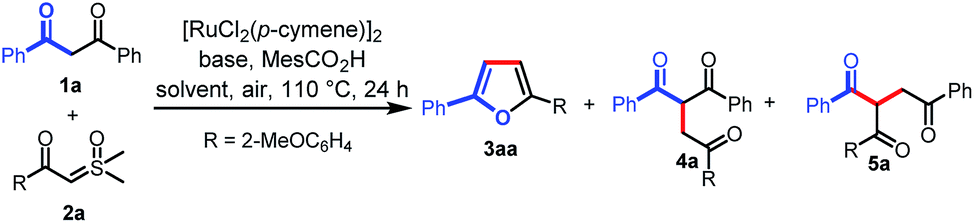
We initiated our investigation on the model reaction of 1,3-diphenylpropane-1,3-dione (1a) with sulfoxonium ylide (2a) to optimize various reaction parameters. The results are summarized in Table 1. With [RuCl2(p-cymene)]2 as the catalyst, MesCO2H as the additive, and Na3PO4 as the base, the desired reaction occurred in HFIP to afford the desired furan product (3aa) in 23% yield (entry 1). However, the C–H carbene insertion product (4a)12a,b and the C–C carbene insertion product (5a)12b,c were also obtained as an inseparable mixture in 45% combined yield with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 chemoselectivity. Investigations on various solvents indicated that toluene performed better than others, affording 3aa in a yield of 35% with excellent chemoselectivity (Table 1, entries 1–6). The use of a proper base was crucial for this reaction, and the exploration of different bases revealed that tBuOLi provided the best yield of 72% (entries 7–12). Only 10% yield of 3aa was obtained in the absence of a base (entry 13). The MesCO2H additive was proved to be necessary to ensure the generation of 3aa. In the absence of MesCO2H, trace 3aa was observed (entry 14). A comparative yield was observed when MesCO2Li was used instead of tBuOLi and MesCO2H (entry 15). The yield of 3aa could be improved to 78% when the reaction was run at an elevated temperature (entry 16). However, a lower yield was obtained when the reaction was carried out at 130 °C (entry 17). Excitedly, when the solvent volume was increased to 2 mL, the desired product (3aa) was obtained in 85% yield (entry 18), but when the solvent volume was further increased to 3 mL, the yield of 3aa was reduced to 66% (entry 19). Finally, in the absence of a catalyst, no desired product was observed (entry 20). Then, the optimized reaction conditions were identified as follows: 1a (0.1 mmol), 2a (2 equiv.), [RuCl2(p-cymene)]2 (5 mol%), MesCO2Li (1.5 equiv.) in toluene (2 mL) at 120 °C in air for 24 h (entry 18).
:6 chemoselectivity. Investigations on various solvents indicated that toluene performed better than others, affording 3aa in a yield of 35% with excellent chemoselectivity (Table 1, entries 1–6). The use of a proper base was crucial for this reaction, and the exploration of different bases revealed that tBuOLi provided the best yield of 72% (entries 7–12). Only 10% yield of 3aa was obtained in the absence of a base (entry 13). The MesCO2H additive was proved to be necessary to ensure the generation of 3aa. In the absence of MesCO2H, trace 3aa was observed (entry 14). A comparative yield was observed when MesCO2Li was used instead of tBuOLi and MesCO2H (entry 15). The yield of 3aa could be improved to 78% when the reaction was run at an elevated temperature (entry 16). However, a lower yield was obtained when the reaction was carried out at 130 °C (entry 17). Excitedly, when the solvent volume was increased to 2 mL, the desired product (3aa) was obtained in 85% yield (entry 18), but when the solvent volume was further increased to 3 mL, the yield of 3aa was reduced to 66% (entry 19). Finally, in the absence of a catalyst, no desired product was observed (entry 20). Then, the optimized reaction conditions were identified as follows: 1a (0.1 mmol), 2a (2 equiv.), [RuCl2(p-cymene)]2 (5 mol%), MesCO2Li (1.5 equiv.) in toluene (2 mL) at 120 °C in air for 24 h (entry 18).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Yieldb (%) | |
| 3aa |
4a + 5a (4a:5a) |
|||
| a Reaction conditions: except where otherwise noted, all of the reactions were performed with 1a (0.1 mmol), 2a (0.2 mmol), base (0.15 mmol), MesCO2H (0.15 mmol), and [RuCl2(p-cymene)]2 (5 mol%) in a solvent (1 mL) at 110 °C in air for 24 h. b The yields were determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard. c Without MesCO2H. d Reaction was carried out at 120 °C. e Reaction was carried out at 130 °C. f 2 mL of toluene was used. g Isolated yield. h 3 mL of toluene was used. i Without [RuCl2(p-cymene)]2. HFIP = 1,1,1,3,3,3-hexafluoro-2-propanol. DMF = N,N-dimethylformamide. DCE = 1,2-dichloroethane. MesCO2H = 2,4,6-trimethylbenzoic acid. | ||||
| 1 | HFIP | Na3PO4 | 23 | 45 (1:6) |
| 2 | i PrOH | Na3PO4 | 26 | 8 (1:3) |
| 3 | DMF | Na3PO4 | 30 | 20 (1:3) |
| 4 | CH3CN | Na3PO4 | 25 | 14 (1:2) |
| 5 | DCE | Na3PO4 | 15 | 24 (1:5) |
| 6 | Toluene | Na3PO4 | 35 | Trace |
| 7 | Toluene | Na2CO3 | 30 | Trace |
| 8 | Toluene | K2CO3 | 20 | Trace |
| 9 | Toluene | Cs2CO3 | 25 | Trace |
| 10 | Toluene | NaHCO3 | 24 | 0 |
| 11 | Toluene | KH2PO4 | 52 | 0 |
| 12 | Toluene | t BuOLi | 72 | Trace |
| 13 | Toluene | — | 10 | 0 |
| 14c | Toluene | t BuOLi | Trace | 0 |
| 15c | Toluene | MesCO2Li | 71 | Trace |
| 16c,d | Toluene | MesCO2Li | 78 | Trace |
| 17c,e | Toluene | MesCO2Li | 40 | Trace |
| 18c,d,f | Toluene | MesCO2Li | 85(82)g | Trace |
| 19c,d,h | Toluene | MesCO2Li | 66 | Trace |
| 20c,d,i | Toluene | MesCO2Li | 0 | 0 |
With the optimal conditions in hand, we turned our attention to the scope of 1,3-diones for this transformation (Scheme 2). It was found that the 1,3-diones bearing methyl, -methoxy, -halogen, and -CF3 groups could all be smoothly transformed to afford the substituted furan products in moderate to good yields (3aa–ao). The structure of 3ak was unambiguously verified by single-crystal X-ray diffraction.13 The reactivity of this transformation was significantly influenced by the steric hindrance. 1,3-diones with ortho-substituted phenyl rings (3al–ao) generally gave lower yields of desired products than those with meta- and para-substituents (3ab–af). The electronic properties of the phenyl rings in 1,3-diones were observed to affect the reaction efficiency obviously. The substrates with electron-withdrawing groups (3ad–af, 3ah, 3ai, and 3ak) gave higher yields than those with electron-donating groups (3ab, 3ac, and 3ag). However, a lower yield was observed when 1,3-dione with a bromo group was used (3aj). It is noteworthy that furan and thiophene rings were also tolerated, giving the desired products in moderate yields (3ap and 3aq), which could be expected to find wide applications in organic electronics.10b,14 Finally, this reaction was not applicable to pentane-2,4-dione (3ar).
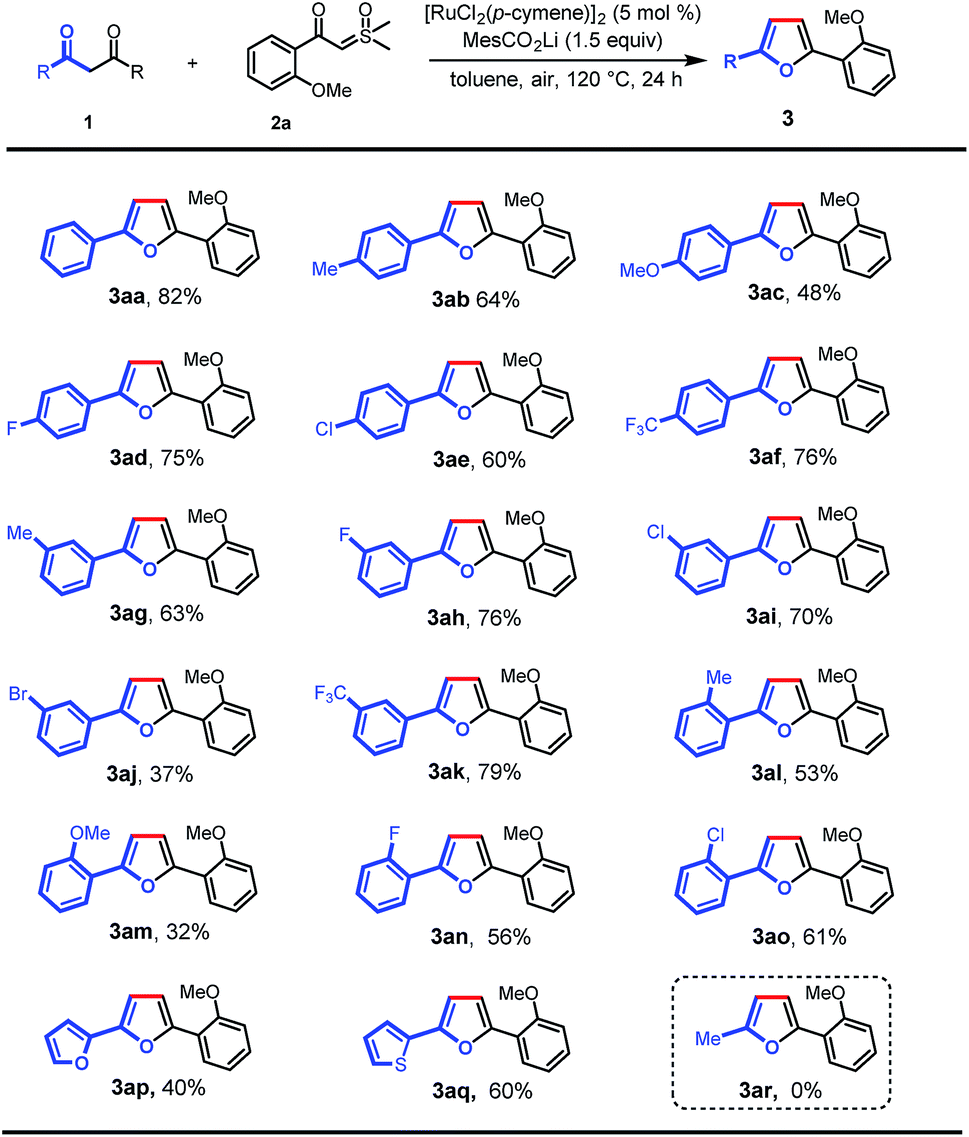 | ||
| Scheme 2 Scope of 1,3-diones. Reaction conditions: 1 (0.1 mmol), 2a (0.2 mmol), MesCO2Li (0.15 mmol), and [RuCl2(p-cymene)]2 (5 mol%) in toluene (2 mL) at 120 °C in air for 24 h. | ||
Next, we further investigated the reaction of 1,3-diphenylpropane-1,3-dione with a variety of aroyl sulfoxonium ylides under the optimal reaction conditions (Scheme 3). Various valuable functional groups were tolerated, such as methyl, phenyl, phenoxyl, methoxyl, halogen, and trifluoromethyl. The reactivity was not sensitive to the steric hindrance and electronic properties of the phenyl rings on the sulfoxonium ylides. Substrates with ortho-substituted phenyl rings, having electron-donating moieties (3ba–bc) and electron-withdrawing groups (3bd–bf), gave the desired products in moderate to good yields. The substrates with meta- and para-substituted phenyl rings could be smoothly converted into the desired products in moderate yields (3bg–bt). Then, a phenyl group was introduced at the para-position which formed the tetra(aryl ring)-containing product 3bu in 60% yield. In addition, 1-naphthalenyl (3bv), 2-naphthalenyl (3bw), 2-furyl (3bx), and 2-thienyl (3by) substrates were also tolerated, giving the corresponding products in the yields of 34%, 45%, 54%, and 55%, respectively. Importantly, triphenyl substituted furan (3bz) could also be obtained using α-phenyl sulfoxonium ylide as the substrate, albeit in low yield due to the increased bulkiness. Alkenoyl sulfoxonium ylide was also a capable substrate, giving 2-alkenyl substituted furan (3ca) in 27% yield. The aryl/alkenyl groups in conjugation with carbonyl are indispensable moieties for the successful formation of the corresponding furans, and the sulfoxonium ylides with alkanoyl groups failed to provide the desired products (3cb and 3cc).
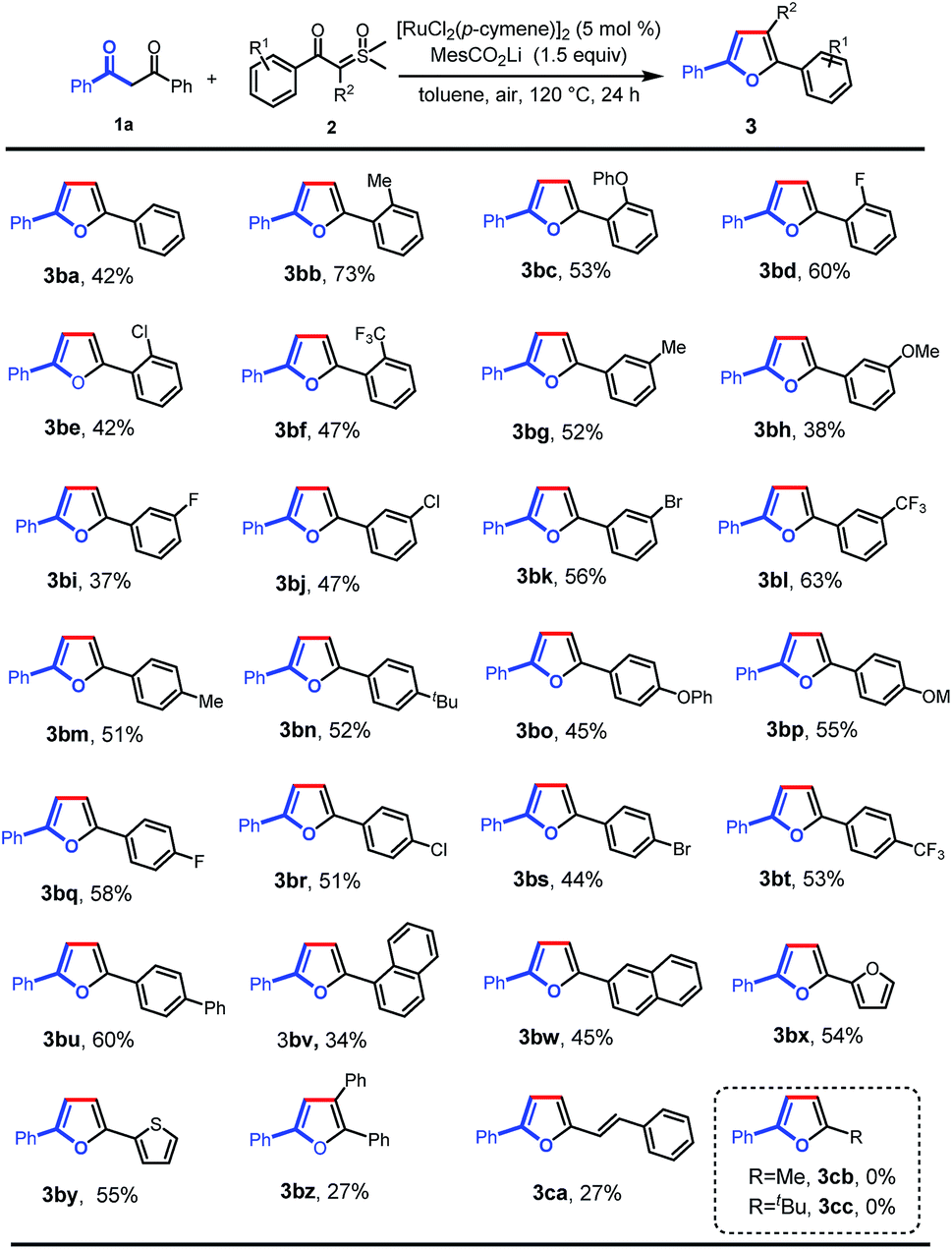 | ||
| Scheme 3 Scope of sulfoxonium ylides. Reaction conditions: 1a (0.1 mmol), 2 (0.2 mmol), MesCO2Li (0.15 mmol), and [RuCl2(p-cymene)]2 (5 mol%) in toluene (2 mL) at 120 °C in air for 24 h. | ||
A gram-scale experiment of this deacylative annulation was demonstrated employing 1a and 2a as model substrates; the product 3aa was obtained in 61% yield, along with an isocoumarin byproduct (6) (Scheme 4a). The 5 mmol scale reaction of 1d and 2a could give the product 3ad in 66% yield (Scheme 4b). Ackermann recently reported Ru(II)/Ag(I)-catalyzed C–H activation/annulation of benzoic acids with sulfoxonium ylides for the synthesis of isocoumarins.11f Indeed, we notice that isocoumarin (6) could also be generated under the silver-free conditions in 25% yield from benzoic acid (7) and 2a (Scheme 4c). These results indicated that benzoic acid might be generated during the C–C bond cleavage process.
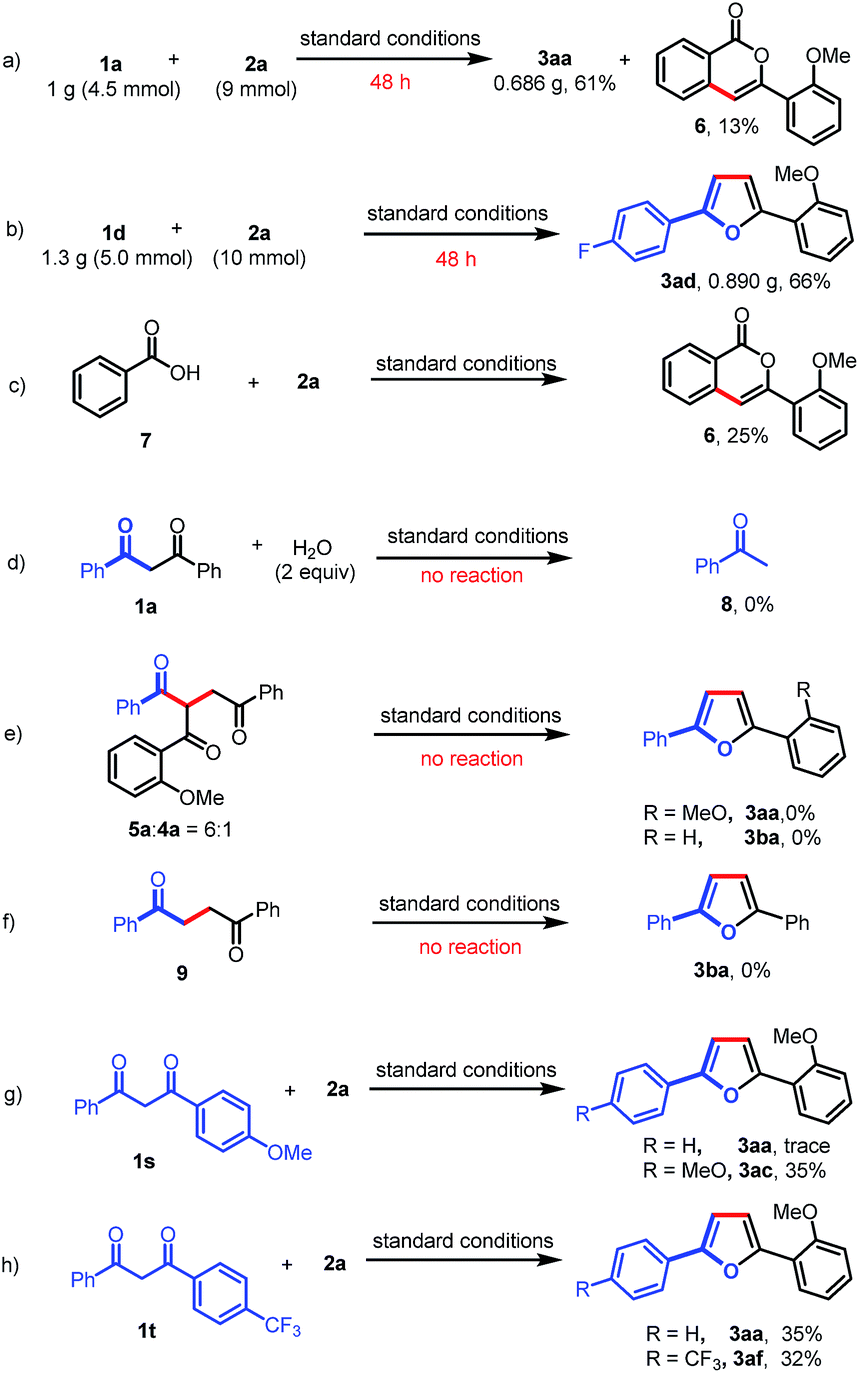 | ||
| Scheme 4 Gram-scale reactions and control experiments. | ||
To further understand the reaction mechanism, the reaction of 1,3-dione (1a) and H2O was investigated under the standard reaction conditions. No reaction was observed, which indicated that the Ru(II)-catalyzed C–C activation could not occur in the absence of a sulfoxonium ylide (Scheme 4d). Furthermore, a mixture of 5a and 4a (6:1) could not afford furan products (3aa or 3ba) under the standard reaction conditions (Scheme 4e). To rule out the possibility that Paal–Knorr furan synthesis is involved in our transformation, we also prepared 1,4-diphenylbutane-1,4-dione (9); however, no desired product (3ba) was obtained when this 1,4-dione was subjected to the standard reaction conditions (Scheme 4f). Finally, the intramolecular competitive reactions of unsymmetrical 1,3-diones (1s or 1t) with 2a indicated that the chemoselectivity of this reaction was affected by the electron density of aryl-groups, and the C–C bond cleavage tended to occur at the less electron-rich moieties (Scheme 4g and h). The electron-deficient carbonyls are more likely to be attacked by a nucleophile, such as H2O, which may induce the subsequent C–C bond cleavage.
On the basis of these results, we proposed that the reaction would proceed as shown in Scheme 5. The transformation begins with the generation of Ru complex A under basic conditions, which is subsequently captured by sulfoxonium ylide to form Ru carbene complex B. Then, C–C bond activation occurs in the presence of H2O, giving Ru complex C. Migratory insertion of C affords intermediate D. Finally, intramolecular annulation of D results in intermediate E, followed by β-O elimination to furnish the furan products (3) and regenerate the Ru(II) catalyst.
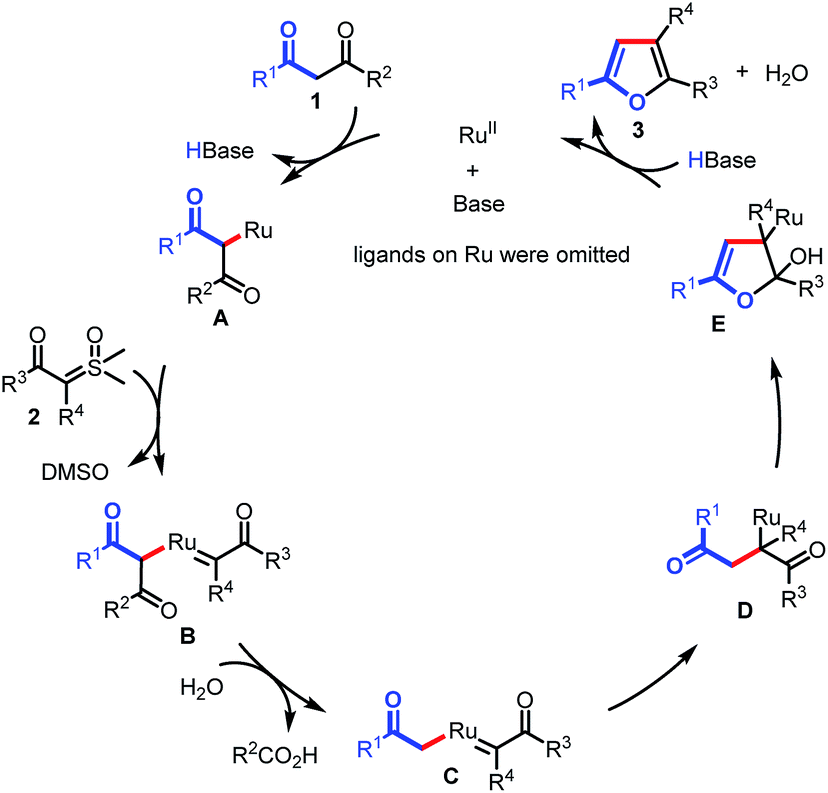 | ||
| Scheme 5 Plausible catalytic cycle. | ||
Conclusions
In conclusion, we have demonstrated the first example of Ru(II)-catalyzed chemoselective deacylative annulation of 1,3-diones with sulfoxonium ylides. A series of substituted furans have been synthesized in reasonable yields by this novel method. This protocol which uses unstrained C–C(CO) bonds as nucleophiles in transition metal-catalyzed cross coupling should be expected to find wide applications. More work to better understand the mechanistic information on this strategy is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF of China (21672075), the Program for New Century Excellent Talents in Fujian Province University, and the Instrumental Analysis Center of Huaqiao University.Notes and references
- For selected reviews, see: (a) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (b) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (c) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (d) M. A. Drahl, M. Manpadi and L. J. Williams, Angew. Chem., Int. Ed., 2013, 52, 11222 CrossRef CAS PubMed; (e) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (f) T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137 CrossRef CAS PubMed; (g) P.-h. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340 CrossRef CAS PubMed; (h) P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615 RSC. For selected examples, see: (i) M. Gozin, A. Weisman, Y. Bendavid and D. Milstein, Nature, 1993, 364, 699 CrossRef CAS; (j) J. Zhu, J. Wang and G. Dong, Nat. Chem., 2019, 11, 45 CrossRef CAS PubMed.
- (a) A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412 CrossRef CAS PubMed; (b) J. Wang, W. Chen, S. Zuo, L. Liu, X. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 12334 CrossRef CAS PubMed; (c) Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690 CrossRef CAS PubMed; (d) Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546 CrossRef CAS PubMed; (e) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef CAS PubMed; (f) Y. Xia, J. Wang and G. Dong, J. Am. Chem. Soc., 2018, 140, 5347 CrossRef CAS PubMed; (g) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586 CrossRef CAS PubMed.
- (a) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed; (b) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404 CrossRef CAS PubMed; (c) L. Deng, M. Chen and G. Dong, J. Am. Chem. Soc., 2018, 140, 9652 CrossRef CAS PubMed.
- Y. Xu, X. Qi, P. Zheng, C. C. Berti, P. Liu and G. Dong, Nature, 2019, 567, 373 CrossRef CAS PubMed.
- For examples on transition metal-catalyzed oxidative cleavage of unstrained C–C(CO) bonds, see: (a) L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303 CrossRef CAS PubMed; (b) X. Huang, X. Li, M. Zou, S. Song, C. Tang, Y. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS PubMed; (c) A. Maji, S. Rana, Akanksha and D. Maiti, Angew. Chem., Int. Ed., 2014, 53, 2428 CrossRef CAS PubMed; (d) C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528 CrossRef CAS PubMed; (e) N. Vodnala, R. Gujjarappa, C. K. Hazra, D. Kaldhi, A. K. Kabi, U. Beifuss and C. C. Malakar, Adv. Synth. Catal., 2019, 361, 135 CrossRef CAS.
- (a) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257 CrossRef CAS PubMed; (b) L.-H. Zou, D. L. Priebbenow, L. Wang, J. Mottweiler and C. Bolm, Adv. Synth. Catal., 2013, 355, 2558 CrossRef CAS; (c) L. Li, W. Huang, L. Chen, J. Dong, X. Ma and Y. Peng, Angew. Chem., Int. Ed., 2017, 56, 10539 CrossRef CAS PubMed; (d) Q. Wu, Y. Li, C. Wang, J. Zhang, M. Huang, J. K. Kim and Y. Wu, Org. Chem. Front., 2018, 5, 2496 RSC.
- C. He, S. Guo, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273 CrossRef CAS PubMed.
- (a) G. Cheng, X. Zeng, J. Shen, X. Wang and X. Cui, Angew. Chem., Int. Ed., 2013, 52, 13265 CrossRef CAS PubMed; (b) X. Yang, G. Cheng, J. Shen, C. Kuai and X. Cui, Org. Chem. Front., 2015, 2, 366 RSC; (c) G. Cheng, W. Lv, C. Kuai, S. Wen and S. Xiao, Chem. Commun., 2018, 54, 1726 RSC; (d) G. Cheng, W. Lv and L. Xue, Green Chem., 2018, 20, 4414 RSC; (e) B. Ge, W. Lv, J. Yu, S. Xiao and G. Cheng, Org. Chem. Front., 2018, 5, 3103 RSC.
- For selected examples on furan synthesis, see: (a) R. C. D. Brown, Angew. Chem., Int. Ed., 2005, 44, 850 CrossRef CAS PubMed; (b) S. F. Kirsch, Org. Biomol. Chem., 2006, 4, 2076 RSC; (c) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed; (d) M. Zhang, H.-F. Jiang, H. Neumann, M. Beller and P. H. Dixneuf, Angew. Chem., Int. Ed., 2009, 48, 1681 CrossRef CAS PubMed; (e) S. Kramer and T. Skrydstrup, Angew. Chem., Int. Ed., 2012, 51, 4681 CrossRef CAS PubMed; (f) H. Lee, Y. Yi and C.-H. Jun, Adv. Synth. Catal., 2015, 357, 3485 CrossRef CAS; (g) L. Feng, H. Yan, C. Yang, D. Chen and W. Xia, J. Org. Chem., 2016, 81, 7008 CrossRef CAS PubMed; (h) T. Naveen, A. Deb and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 1111 CrossRef CAS PubMed; (i) F. Verma, P. K. Singh, S. R. Bhardiya, M. Singh, A. Rai and V. K. Rai, New J. Chem., 2017, 41, 4937 RSC; (j) A. R. White, R. A. Kozlowski, S.-C. Tsai and C. D. Vanderwal, Angew. Chem., Int. Ed., 2017, 56, 10525 CrossRef CAS PubMed; (k) C. Arroniz, G. Chaubet and E. A. Anderson, ACS Catal., 2018, 8, 8290 CrossRef CAS; (l) X. Wang, A. Lerchen, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 1712 CrossRef CAS PubMed; (m) S. Agasti, T. Pal, T. K. Achar, S. Maiti, D. Pal, S. Mandal, K. Daud, G. K. Lahiri and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 11039 CrossRef CAS PubMed.
- (a) A. Boto and L. Alvarez, Furan and Its Derivatives, Heterocycles in Natural Product Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 97–152 Search PubMed; (b) H. Tsuji and E. Nakamura, Acc. Chem. Res., 2017, 50, 396 CrossRef CAS PubMed.
- (a) L.-Q. Lu, T.-R. Li, Q. Wang and W.-J. Xiao, Chem. Soc. Rev., 2017, 46, 4135 RSC; (b) M. Barday, C. Janot, N. R. Halcovitch, J. Muir and C. Aissa, Angew. Chem., Int. Ed., 2017, 56, 13117 CrossRef CAS PubMed; (c) J. Vaitla, A. Bayer and K. H. Hopmann, Angew. Chem., Int. Ed., 2017, 56, 4277 CrossRef CAS PubMed; (d) Y. Xu, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 19, 5256 CrossRef CAS PubMed; (e) K. S. Halskov, M. R. Witten, G. L. Hoang, B. Q. Mercado and J. A. Ellman, Org. Lett., 2018, 20, 2464 CrossRef CAS PubMed; (f) S. Ji, K. Yan, B. Li and B. Wang, Org. Lett., 2018, 20, 5981 CrossRef CAS PubMed; (g) Y.-F. Liang, L. Yang, T. Rogge and L. Ackermann, Chem.–Eur. J., 2018, 24, 16548 CrossRef CAS PubMed; (h) X. Wu, H. Xiong, S. Sun and J. Cheng, Org. Lett., 2018, 20, 1396 CrossRef CAS PubMed.
- (a) Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817 CrossRef CAS PubMed; (b) Z. Liu, P. Sivaguru, G. Zanoni, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2018, 57, 8927 CrossRef CAS PubMed; (c) Z. Liu, X. Zhang, M. Virelli, G. Zanoni, E. A. Anderson and X. Bi, iScience, 2018, 8, 54 CrossRef CAS PubMed.
- CCDC 1918495 (3ak) contains the supplementary crystallographic data for this paper.†.
- O. Gidron, N. Varsano, L. J. W. Shimon, G. Leitus and M. Bendikov, Chem. Commun., 2013, 49, 6256 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1918495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03245b |
| This journal is © The Royal Society of Chemistry 2019 |