
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular basis of methylation and chain-length programming in a fungal iterative highly reducing polyketide synthase†
Xiao-Long
Yang‡
 ab,
Steffen
Friedrich
ab,
Sen
Yin
ab,
Oliver
Piech
ab,
Katherine
Williams
abc,
Thomas J.
Simpson
c and
Russell J.
Cox
*ab
ab,
Steffen
Friedrich
ab,
Sen
Yin
ab,
Oliver
Piech
ab,
Katherine
Williams
abc,
Thomas J.
Simpson
c and
Russell J.
Cox
*ab
aInstitute for Organic Chemistry, Leibniz University of Hannover, Schneiderberg 1B, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
bBMWZ, Leibniz University of Hannover, Schneiderberg 38, 30167, Hannover, Germany
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
First published on 30th July 2019
Abstract
Exchange of 32 different sub-fragments of the C-methyltransferase (C-MeT), pseudo-ketoreductase (ΨKR) and ketoreductase (KR) catalytic domains of the tenellin iterative Type I polyketide synthase non ribosomal peptide synthetase (PKS-NRPS) TENS by homologous fragments from the desmethylbassianin (DMBS) and militarinone (MILS) PKS-NRPS led to the creation of chimeric synthetases in which programming fidelity was altered, resulting in the production of mixtures of products with different methylation patterns and chain lengths. Swap of KR domain subfragments with the homologous fragments from the KR of the heptaketide militarinone synthetase resulted in the synthesis of penta, hexa and heptaketides. The results of these and previous experiments are rationalised by considering the existence of competition for acyl-carrier protein (ACP) bound substrates between different catalytic domains of the PKS. In particular, competition between the C-MeT and ketoreductase domains (KR) can account for methylation programming, and competition between the KR and the off-loading NRPS accounts for chain-length selectivity.
Introduction
All known fungal polyketide synthases (PKS) are multi-functional proteins which control starter unit selection and the programming of chain length and methylation.1 Since these proteins are almost universally iterative, they have the ability to catalyse different sets of reactions during each extension cycle using the same manifest of catalytic components. In the case of highly reducing fungal PKS (hr-PKS), the programming is particularly complex because specific C-methylation, reduction, dehydration and enoyl reduction reactions can occur during each cycle. While some aspects of programming in non-reducing Type I and II PKS (nr-PKS)2 are understood – particularly the roles of starter unit acyl transferase (SAT)3 domains and product template (PT)4 domains, and chain-length selection in Type II PKS,5 the features controlling programming in Type I hr-PKS remain cryptic. For example, during the biosynthesis of the pretenellin A 16 polyketide (Scheme 1), methylation occurs at the diketide and triketide stages, but not at the tetra- and pentaketide stages. This selectivity is enacted by a single catalytic C-methyltransferase (C-MeT) domain. The determinant of chain-length in these systems is not known.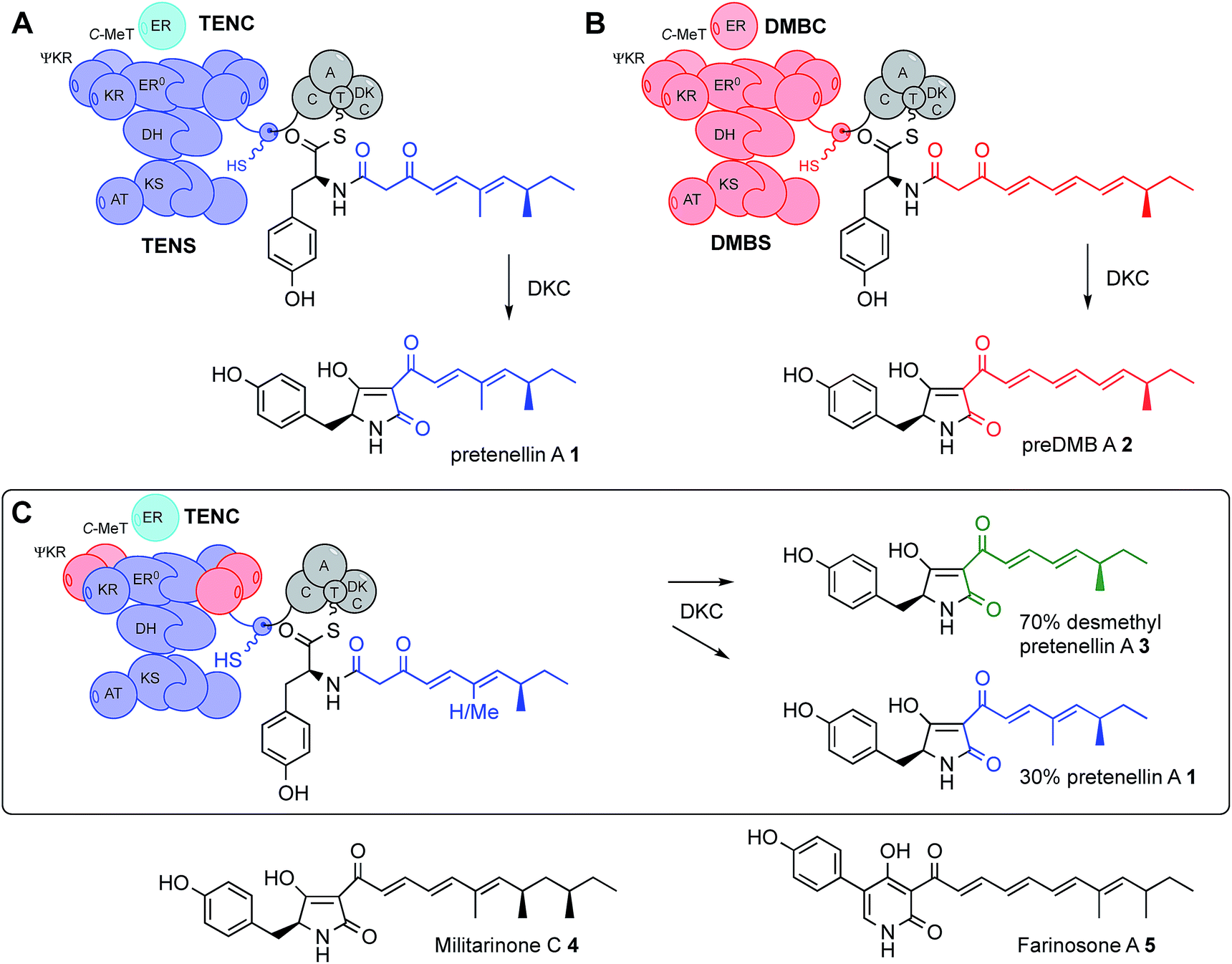 | ||
| Scheme 1 Domain structure and swaps of iterative polyketide synthases: cartoon views of (A), the tenellin PKS-NRPS (TENS) and (B), the desmethylbassianin PKS-NRPS (DMBS) and their key enzyme-bound intermediates and products; (C), cartoon view of a hybrid PKS-NRPS and its key intermediate and products. Abbreviations: as Fig. 1 and ER0, non-functional ER domain; C-MeT, C-methyltransferase; DKC, Dieckmann cyclase; C, condensation domain; A, adenylation domain and T, thiolation domain. | ||
Since hr-PKS are responsible for the biosynthesis of numerous biologically important compounds, e.g. the mycotoxic cytochalasans,7 the anticholesterol lovastatin8 and squalestatins9,10 and the antifungal strobilurins,11 discovering the mechanism of the programming is an important goal if the rational engineering of these systems is to be achieved. This goal is difficult to reach in the absence of structural data for complete fungal hr-PKS which have thus far resisted attempts to crystalise them – although structural data is available for a limited number of isolated or trans-acting domains.12,13 However, since fungal hr-PKS are closely related at the peptide sequence level to mammalian fatty acid synthases (mFAS, Fig. 1) for which complete structures are known,14 mFAS can be used as a useful guide.15
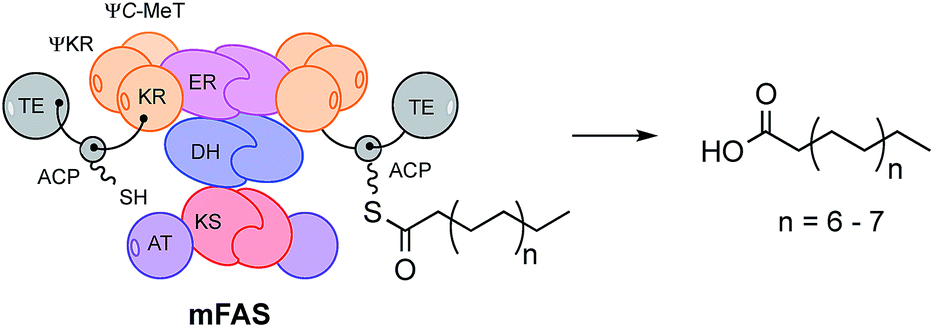 | ||
| Fig. 1 Cartoon model of the quaternary structure of mammalian fatty acid synthase (mFAS). Abbreviations: AT, acyltransferase; KS, ketosynthase; DH, dehydratase; ER, enoyl reductase; ΨC-MeT, non-functional C-methyltransferase; KR, ketoreductase; ΨKR, structural domain of KR; ACP, acyl carrier protein; TE, thiolesterase. | ||
In the first experiments to successfully probe the programming mechanism in hr-PKS, we showed that the exchange of entire functional domains between closely related (but differently programmed) hr-PKS could lead to the creation of new polyketides (Scheme 1).16 We used the pretenellin A 1 and predesmethylbassianin A (preDMB A, 2) synthetases (TENS and DMBS respectively). These PKS-NRPS proteins, each of approximately 600 kDa, consist of a typical fungal hr-PKS with an NRPS fused at the C-terminus. Pretenellin A 1 and predesmethylbassianin A 2 belong to a large family17 of fungal metabolites which also includes militarinone C 4 (produced by Paecilomyces militaris = Cordyceps militaris)18 which is a doubly-reduced trimethylated heptaketide and farinosone A 5 (Paecilomyces farinosus),19 a mono-reduced dimethylated heptaketide.
The tenellin and desmethylbassianin synthetases are 86% identical,16 but produce polyketides which differ in chain-length and methylation pattern: the dimethylated pentaketide in 1 and the monomethylated hexaketide in 2 respectively. By swapping individual and multi-domain fragments between TENS and DMBS, we determined that some catalytic domains such as β-ketoacyl synthase (KS), acyl transferase (AT) and acyl carrier protein (ACP) appear not to be involved in programming. In the cases of both TENS and DMBS, the PKS proteins are terminated with a non-ribosomal peptide synthetase (NRPS) module which catalyses the transfer of the polyketide via an amide linkage to tyrosine. A Dieckmann cyclisation domain (DKC) then effects chain-release by intramolecular cyclisation to form the tetramate moiety.20 Exchange of the TENS and DMBS NRPS domains also had no observed effect on programming (Scheme 1).16
TENS and DMBS both utilise trans-acting enoyl reductase proteins (ER, TenC and DmbC respectively) and these too can be exchanged without affecting programming. However, programming and fidelity changes are observed in these systems when the ketoreductase (KR) and C-MeT domains are swapped,13 and when the trans-acting ER is absent.8,21,22
In particular, it appears that a protein fragment of 503 residues encompassing the C-MeT and ΨKR domains (a structural component of the KR domain) affects both the frequency and fidelity of methylation. For example, creation of a chimeric PKS in which the DMBS C-MeT–ΨKR region is inserted into TENS gives a synthetase which produces 70% of the novel mono-methylated metabolite 3, along with 30% of the normal dimethylated TENS metabolite 1 (Scheme 1C). Swap of the entire KR domain results in a mixture of chain lengths, with hexaketides predominating. Similar reports of reprogramming in related PKS-NRPS systems have been more latterly reported by Schmidt and co-workers.23
Significantly no swap of a single domain gives more than 70% of monomethylated material, or hexaketides, but exchange of the entire C-MeT to KR region gives clean monomethylation and chain-length (hexaketide) selection suggesting that while the C-MeT and KR have some intrinsic selectivity (e.g. within their own active sites), extrinsic factors such as domain–domain interactions within the PKS may also be significant determinants of programming.
Here we have examined this phenomenon in more detail by making a large number of swaps of smaller domain fragments with the aim of separating intrinsic and extrinsic factors and in an attempt to obtain cleaner reprogramming effects. In total, the C-MeT, ΨKR and KR domains of TENS were divided into 14 sub-fragments and 28 different swaps achieved with the corresponding fragments of DMBS (Table 1). Additionally, three swaps of fragments from the KR domain of the heptaketide militarinone C 4 synthetase (MILS) were also achieved. Most of these experiments resulted in functional hybrid PKS systems and examination of the structures of the products leads to conclusions about the mechanism of programming within such PKS and the prospects for their rational engineering.
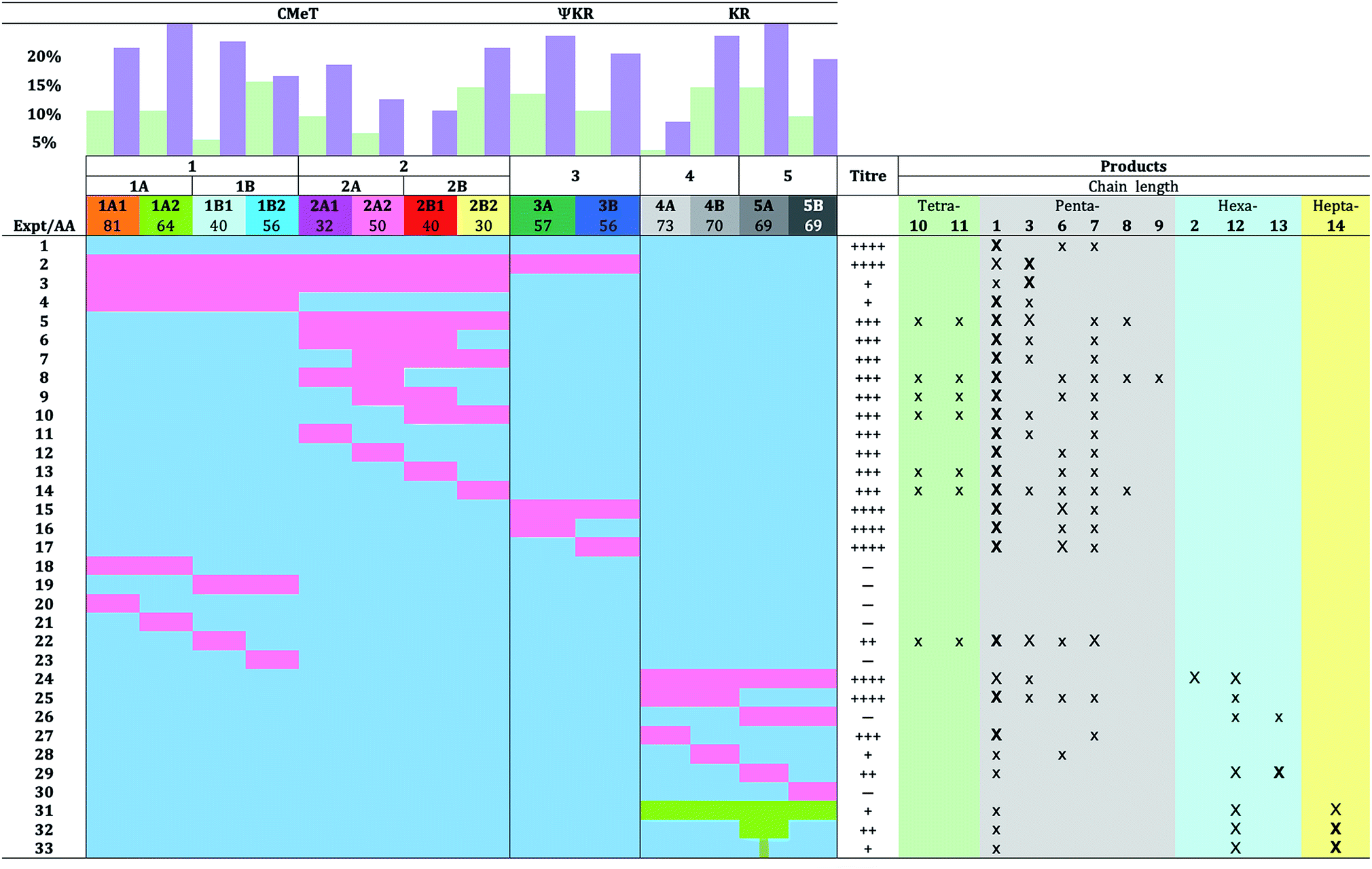
|
Results
TENS requires the presence of a trans-acting ER protein known as TenC.6 In previous work we had expressed tenS and tenC using two parallel expression systems in the fungal host Aspergillus oryzae.14 In order to accelerate the work, we cloned tenS and tenC into a single multi-gene fungal expression vector as described by Lazarus and coworkers.24 Transformation of this single vector into A. oryzae and induction of gene expression by fermentation in media containing maltose leads to the expected production of the dimethylated pentaketide pretenellin A 1 (Table 1, entry 1) as the major detected compound. The minor congeners prototenellin A 6 (in which reduction between C-11 and C-12 has not occurred and where methylation occurs at C-7 and C-11) and a methylation isomer of pretenellin A (most likely its 11,12-dihydro analogue 7 by MS and UV analysis) are each observed at less than 4% of the total.There is no existing structural information for a complete fungal hr-PKS. However sequence alignment (see ESI†) shows that mFAS consists of the same catalytic domains as hr-PKS arranged in the same order.14 Significant homologies within the predicted active sites and cofactor-binding regions, coupled with recent observation that stereochemical preferences for the catalysed reactions of mFAS and some hr-PKS are identical,25,26 suggest that mFAS is an appropriate model for fungal hr-PKS. While the mFAS structure gives good information regarding the likely quaternary arrangement and interactions of the hr-PKS catalytic domains (Fig. 3A), it was obtained at relatively low resolution (3.2 Å).
In particular the mFAS ΨC-MeT domain is represented poorly, and significant deletions and sequence variations from the PKS C-MeT domain are indicated by the sequence alignment (see ESI†). In order to obtain better quality models of the individual domains of mFAS, and the quaternary structure of TENS, we created a hybrid model. First a model of the TENS KR was developed using the X-ray structure of the amphotericin-B KR AmphB27 (to which TENS shows 27% homology) using SWISSMODEL28 which gave a satisfactory QMEAN score of −2.82.29–32 NADPH was built into the active site of the KR model using PyMOL33 and the model minimized in YASARA.34 Acetoacetyl pantetheine was docked into the active pocket using AutoDock VINA,35,36 again followed by YASARA minimisation. This revealed a substrate-binding helix which is not present in the mFAS KR structure (Fig. 5B and D).
The same procedure was used to generate a model of the TENS C-MeT using the CurJ C-MeT37 (to which it is 29% homologous) as the template. This also resulted in a satisfactory model structure with a QMEAN score of −3.16.38–40 The KR and C-MeT domain models were then used to replace the KR and ΨC-MeT domains of mFAS using PYMOL to generate an overall hybrid model of the quaternary structure. Such hybrid models have been previously used successfully, for example to generate a model of the quaternary structure of the CurJ PKS by Skiba and coworkers.38 The hybrid model indicates how the ΨKR forms a linking domain between the KR and C-MeT domains and it shows the possible quaternary structure of the β-processing region of TENS. This is illustrated for mFAS in Fig. 3A where the ΨC-MeT, ΨKR and KR are indicated in red, green and blue respectively. The hybrid model (Fig. 3B) shows that these domains probably take up a similar quaternary arrangement in TENS.
Design and implementation of fragment swaps
Using the hybrid model we divided the TENS C-MeT–ΨKR region (including proposed linker regions) into ten sub fragments of between 30 and 81 residues (Table 1, Fig. 3C). The KR of TENS was also divided into four sub-regions of approximately 70 amino acids each (Table 1, Fig. 3C). The fragment-exchange procedure consisted simply of combining overlapping DNA fragments constructed from tenS or dmbS templates with the linearised tenS cloning vector and transforming the mixture into yeast (Fig. 2A).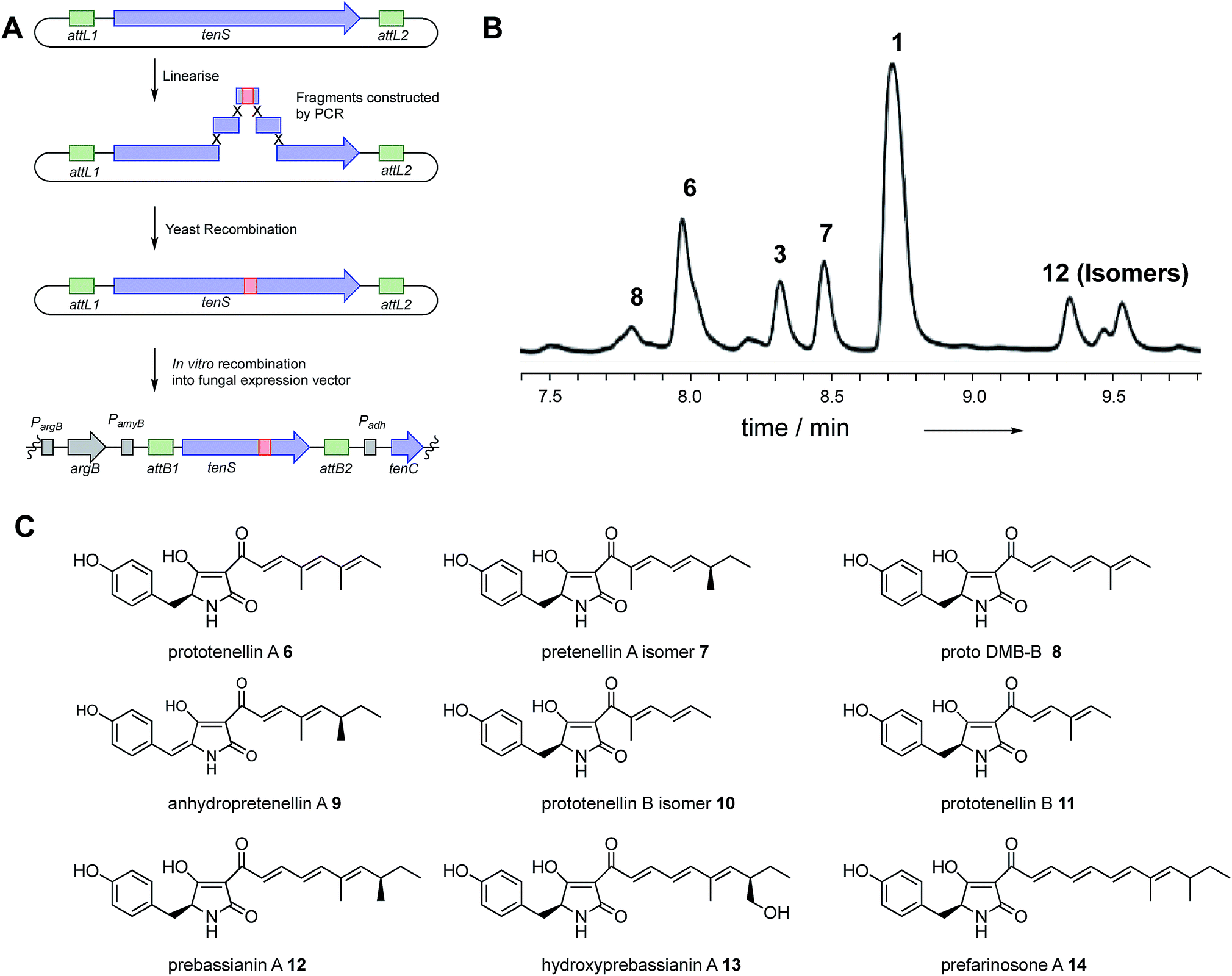 | ||
| Fig. 2 Cloning strategy for rapid fragment swaps of tenS and typical results: (A) cloning strategy; (B) typical HPLC trace (diode array detector) of pretenellin A 1 and analogs. See ESI† for full experimental data for each experiment; (C) structures of isolated compounds. | ||
For example, the exchange of the 32 AA fragment 2A1 (Table 1, exp. 11) into TENS was achieved using a 5′ PCR fragment obtained from tenS, a ca. 150 bp fragment spanning 2A1 from dmbS with ca. 30 bp tails matching the tenS sequence, and a 3′ fragment from tenS. The reconstructed plasmids were rescued in E. coli and then transferred by in vitro recombination (Gateway)41 methodology into the expression vector (which already contained tenC), and this was then transformed into A. oryzae.
Multiple A. oryzae transformants were selected and then grown in parallel on media containing maltose, which induces expression of the hybrid tenS gene, and organic extracts were prepared and examined using a standardised extraction and LCMS protocol (Fig. 2B). Sequencing of the A. oryzae host genomic DNA was performed in all cases to ensure the correct expected chimeric genes were integrated. All clones were obtained as expected.
In previous work we exchanged the entire 506-residue C-MeT–ΨKR fragment (Table 1, exp. 2), and this resulted in a switch to the biosynthesis of predominantly the pentaketide desmethylpretenellin A 3, although pretenellin A 1 itself was still produced (30%). In order to determine the role of the ΨKR in this effect we began here by swapping only the 393 residue C-MeT-domain itself (Table 1, exp. 3). This resulted in a significant reduction in titre, but the pentaketides desmethylpretenellin A 3 and pretenellin A 1 were synthesised as the observable products.
We next swapped the first 241 residues of the C-MeT (fragment 1, exp. 4) which led to a very low yield of predominantly 1, accompanied by some 3.
Closer investigation of fragment 1 using swaps of increasingly smaller sub-fragments was generally unproductive. Fragments 1A, 1B, 1A1, 1A2 and 1B2 (exp. 18 to 21, and 23, Table 1) gave no observable products. However, exchange of fragment 1B1 (exp. 22) consisting of 40 residues led to the production of at least six compounds in overall moderate titre. Pretenellin A 1 was the major component, with 3 and 7 also present as significant by-products with 6, and the tetraketides 10 and 11 as minor components.
Exchange of fragment 2, however, was much more productive. Exchange of the entire fragment 2 (exp. 5) resulted in good overall production of mainly demethylated 1 and some monomethylated 3, with 7 and 8, and again tetraketides 10 and 11 observed. Exchange of the first and second 3/4s of fragment 2 (exp. 6 and 7) gave a similar result: high titre of 1 and minor amounts of 3 and 7. Likewise, swap of fragment 2A (exp. 8) resulted in high titre of diverse compounds: again 1 was the dominant compound, but pentaketide 6–9 and tetraketide 10 and 11 methylation variants were observed as minor components.
Notably 3 was absent. The exchange of the central portion of fragment 2 (2A2 + 2B1, exp. 9) gave similar results, diverse compounds, but lacking 3. Exchange of fragment 2B (exp. 10) also gave high titres and all compounds – this time including a small amount of 3 – were observed.
Individual quarters of fragment 2 were then exchanged. Transfer of fragment 2A1 (exp. 11) resulted in a system almost identical to WT, but with an additional low content of 3, while swap 2A2 (exp. 12) gave a phenotype almost identical to WT. Swap of fragment 2B1 (exp. 13) gave a WT phenotype with additional tetraketide methylation isomers 10 and 11. Replacement of fragment 2B2 (exp. 14) resulted in high diversity with predominantly 1, with small amounts of pentaketides 3, 6, 7 and 8, and tetraketides 10 and 11 observed.
Next, the ΨKR and its sub-fragments were exchanged. Interchange of the entire 144 residue ΨKR itself (exp. 15) or the sub-fragments (exp. 16 and 17) resulted in no change in titre or production of new compounds, although the production of somewhat more triene 6 was observed compared to the WT phenotype in experiments 15 and 17. All products were pentaketides – no longer or shorter products were observed.
Ten swaps within the KR domain were also achieved. Previously we showed that swap of the entire TENS KR with its DMBS analog results in good titres of pentaketides 1 and 3 and the hexaketides 2 and 12, (exp. 24) with dimethylation (70% total product) and hexaketides (80%) predominating. Swaps of (and within) the first half of the KR were also productive (exp. 25, 27, 28), giving predominantly the pentaketides 1 and 6, but swap of the second half of the KR (exp. 26) gave low titres with few new compounds, while swap of the final quarter 5B resulted in an inactive synthase. Significantly, only swaps of regions containing fragment 5A (exp. 26 and 29) which contains the substrate-contacting helix, resulted in the production of hexaketides 12 and 13 (which is presumably created by adventitious oxygenation in A. oryzae).
Finally, (experiments 31–33) we swapped-in fragments from the militarinone PKS-NRPS (MILS) from C. militaris.42 Militarinone C 4 is heptaketide congener of pretenellin A 1 which has three methyl groups and is reduced twice by its trans-acting ER. Exchange of the entire KR domain resulted in small amounts of penta- 1, hexa- 12 and heptaketides 14 (Fig. 4), all of which are dimethylated and singly reduced, with a majority of the heptaketide. Compound 14 is presumably the precursor of farinosone A 5, and we therefore name it prefarinosone A.
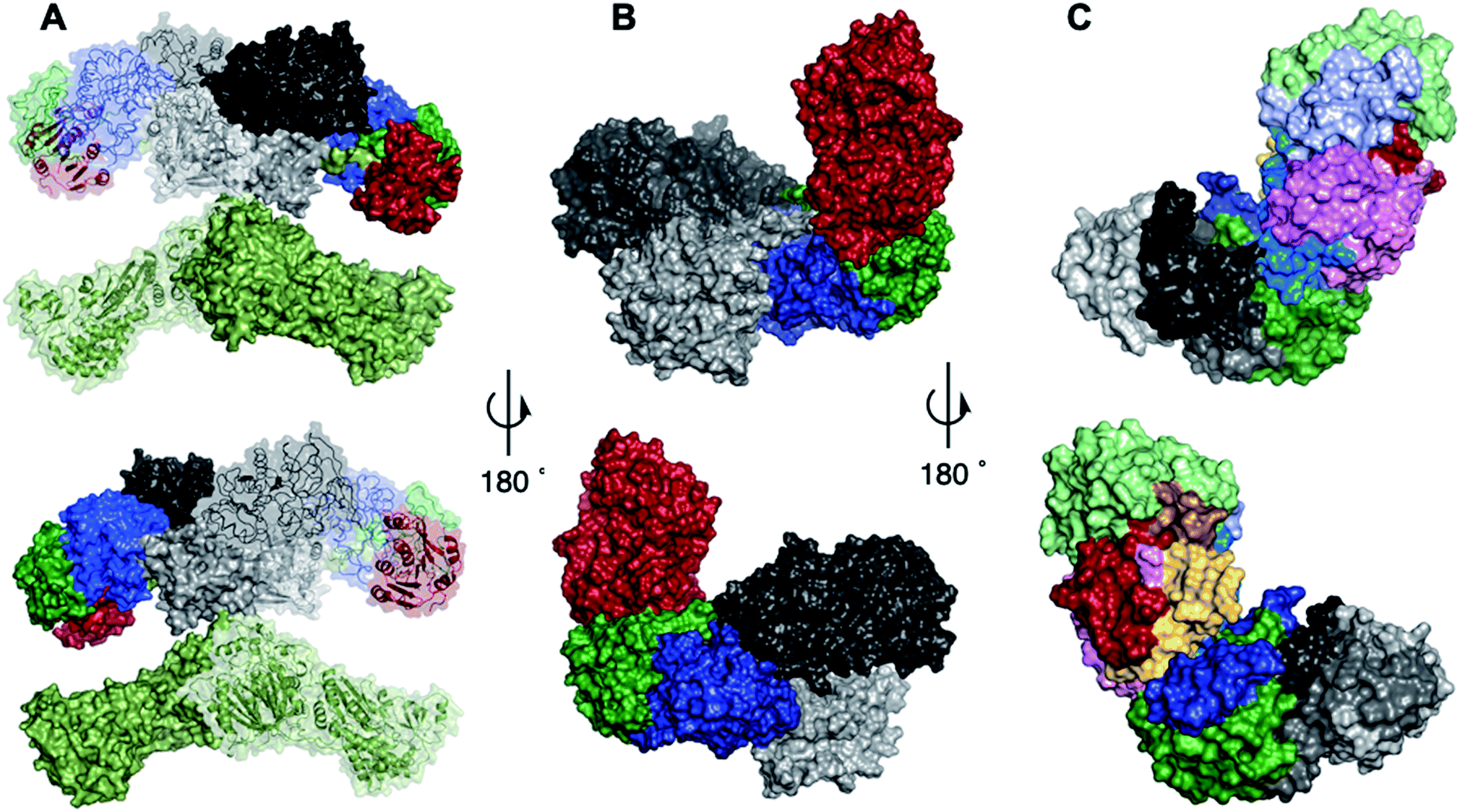 | ||
| Fig. 3 Structures and models: (A) overall domain architecture of mFAS. Colours: light green, KS, AT and linker regions; light grey, DH; red, ΨC-MeT; dark green, ΨKR; dark grey, ER; blue, KR. (B) Model quaternary structure of TENS β-processing domains. Colours: light grey, DH; dark grey, ER; red C-MeT; green, ΨKR; blue, KR. (C) Model of β-processing C-MeT, ΨKR and KR region of TENS showing the location of the swapped peptides. Colours: see Table 1. | ||
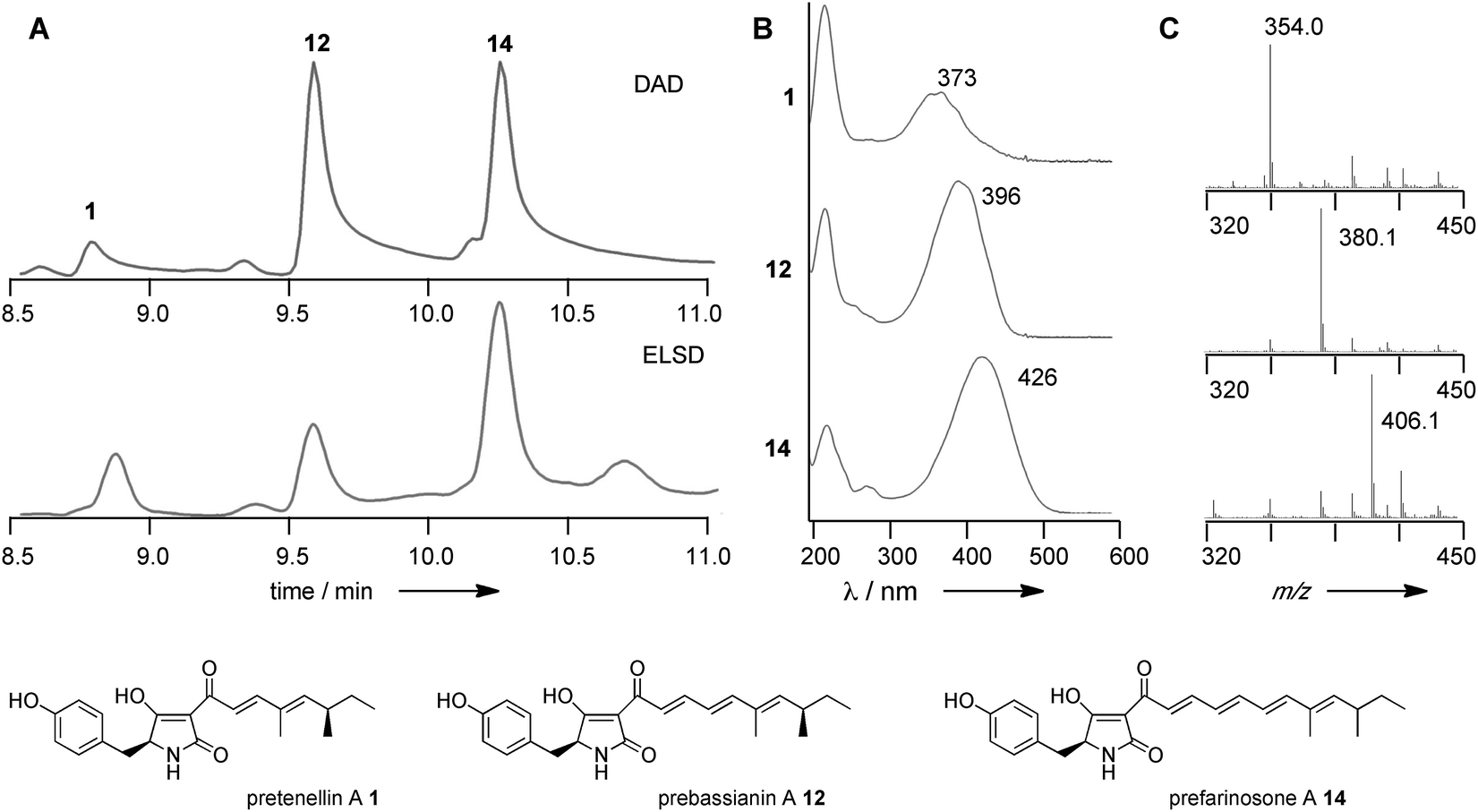 | ||
| Fig. 4 Results of MILS KR swap. (A) HPLC analysis of organic extracts resulting from experiment 31; (B) UV analysis of indicated compounds; (C) ESMS-analysis of indicated compounds. | ||
Swap of fragment 5A alone gave a similar distribution of products, but in around 5-fold higher titre, allowing isolation and full characterisation of the prefarinosone A 14 (ca. 1.0 mg L−1). As expected compounds 1, 12 and 14 show an increase in λmax from 373 nm for the dieneone 1 to 426 nm for the tetraeneone 14 (Fig. 4). In a final experiment a 36 bp sub-fragment of the KR domain corresponding to residues Q2398 to V2409 of TENS was exchanged. This produced the same distribution of 1, 12 and 14, but in overall low titre.
Structural analysis
Mapping of the residues which are different in the TENS and DMBS β-processing domains onto the model structure showed that the majority of these are located at solvent-exposed surfaces, and are thus unlikely to be involved in significant intrinsic effects (Fig. 5A). Changed residues which are not located at surfaces can be divided into two classes. First, those such as V1433I and V1437M in the C-MeT (1B1) and I2223L and V2336I in the KR (4AB) are in buried hydrophobic regions and unlikely to be in contact with active sites (Fig. 5B). The effect of changes such as S1350A (1A1) and A1504V (1B2) cannot be assessed as exchange of these fragments gives inactive synthases.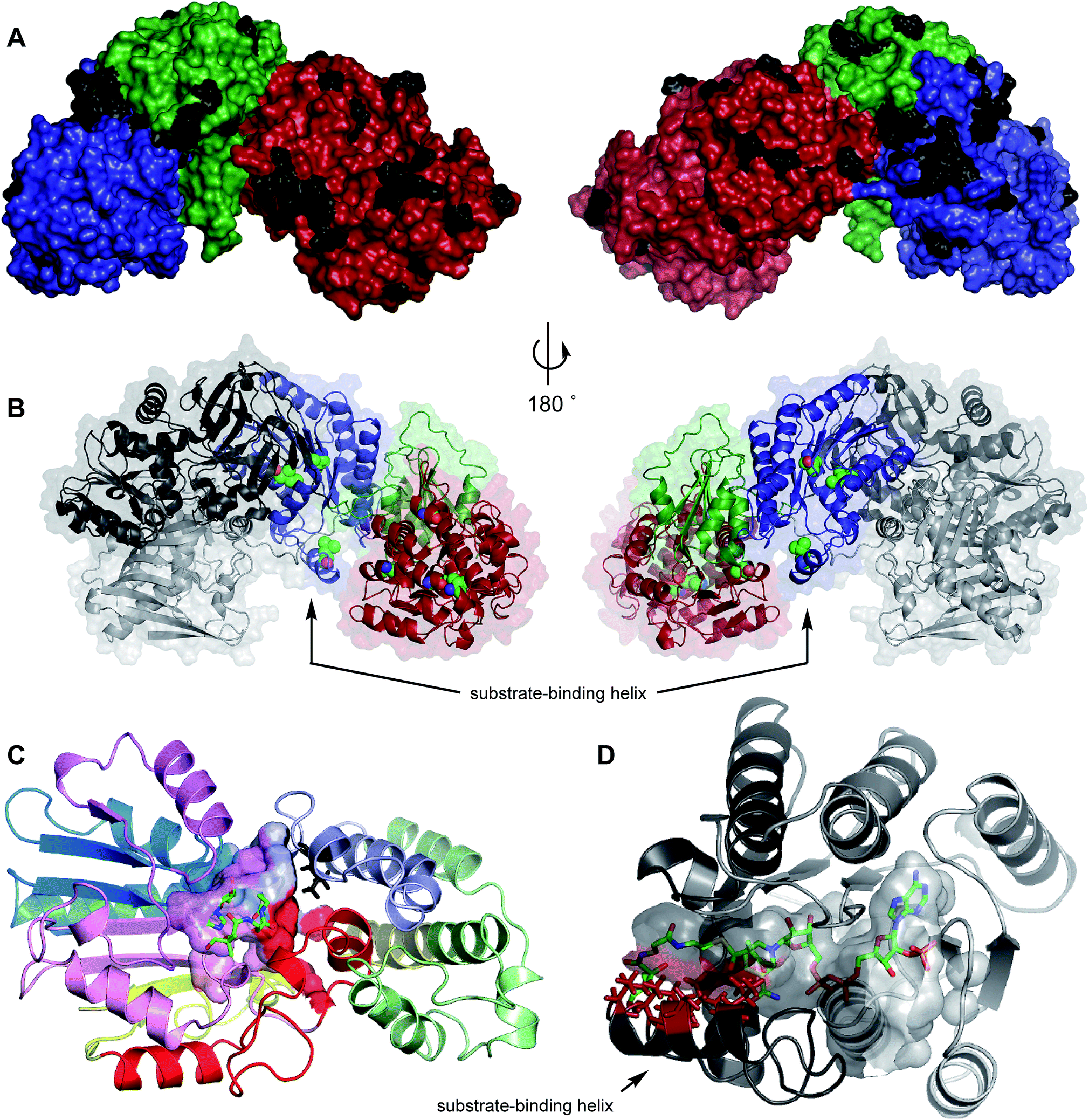 | ||
| Fig. 5 Model structures of: (A) TENS ΨKR, C-MeT and KR domains showing changed surface residues (black) in comparison to DMBS; (B) TENS β-processing domains showing non-surface mutations (spheres) in comparison to DMBS; (C) C-MeT domain of TENS with bound SAM and acetoacetylpantetheine showing likely active site and changed residue L1451 in black; (D) TENS KR domain with bound NADPH and 2′-methylacetoacetylpantetheine. Red residues indicate positions of V2397, S2400, L2401, T2404 and V2406. Cartoon colours (C and D): exchange peptide colours as Table 1. | ||
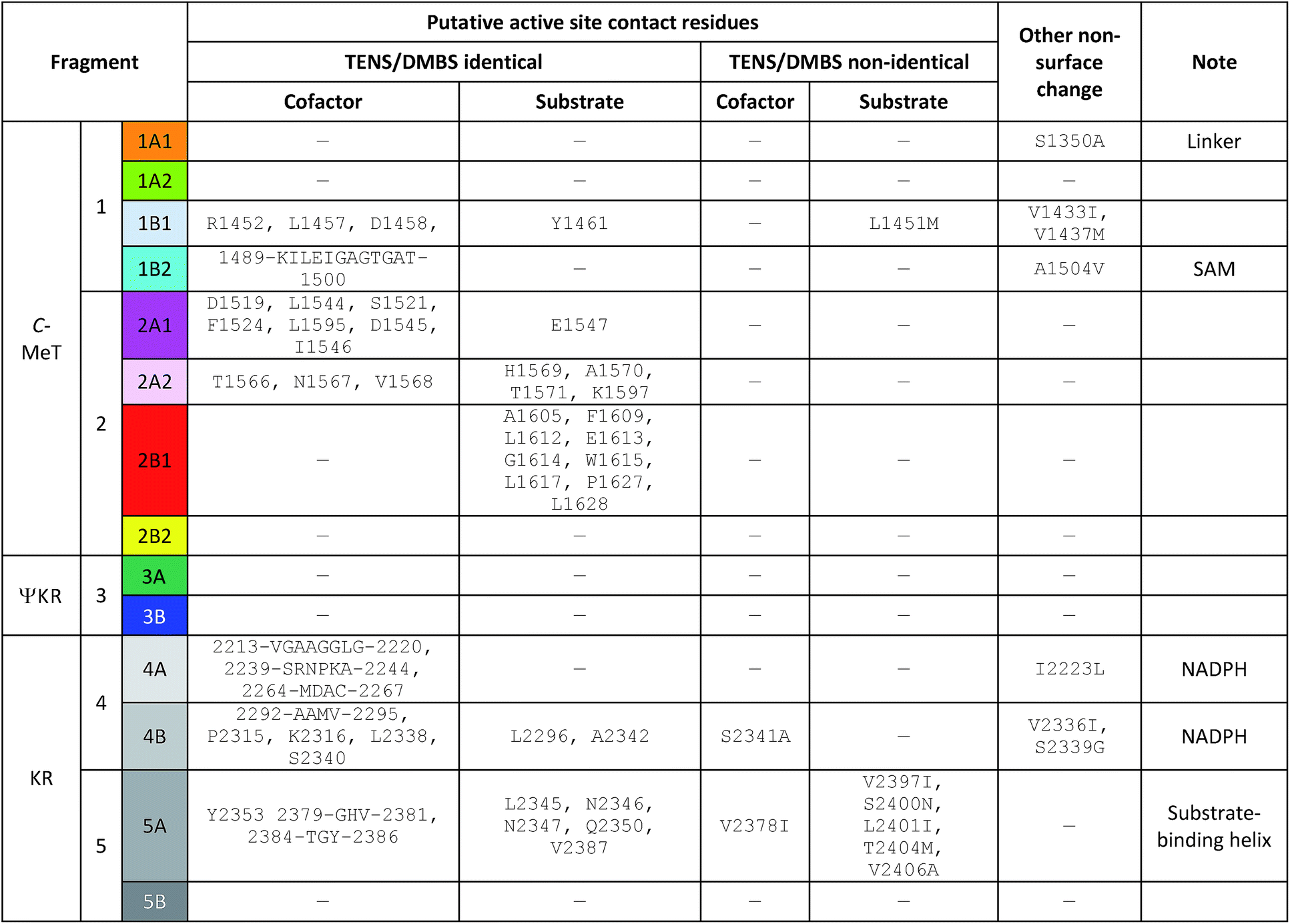
Secondly, non-surface residues likely to contact substrates and cofactors in the C-MeT and KR were identified (Table 2, Fig. 6). These are located on fragments 1B1, 1B2, 2A1, 2A2, 2B1, 4A, 4B and 5A (8 out of 14, Fig. 5C–E). However, of these, only fragments 1B1 (C-MeT), 4B and 5A (KR, Fig. 6) contain changes in these residues. All other fragments contain no changes in contact-residues and would not be expected to directly alter the active site upon exchange.
|
 | ||
| Fig. 6 Sequence alignment in the 4B–5A–5B region of the KR domain (grey bars). Yellow highlight = identity among PKS; green highlight = identity among mFAS; red text = identity among all; C = residues which contact cofactor; S = residues which contact substrate; B = other buried position with change. Box highlights substrate-binding helix most closely associated with chain-length selection. See ESI† for full alignment of all β-processing domains. | ||
In the case of fragment 1B1, the mutation is the highly conservative L1451M (Fig. 5C). While swap fragment 1B1 alone (experiment 22) leads to formation of some 7, which features a non-standard methylation pattern, this is no more significant than observations of 7 arising from swaps not involving fragment 1B1 (e.g. experiments 5–17, Table 1). For fragment 4B there is variation at position 2341 which contacts the cofactor only, and change of fragment 4B is not associated with a change in programming.
In the case of fragment 5A there are five changes (Table 2, Fig. 6) in residues which appear to contact the substrate in the structural model (Fig. 5D) when compared to DMBS. These are clustered on the substrate-contacting-helix which appears to be absent from mFAS. Some of these residues (e.g. V2406A) are not conservative and could be expected to expand the volume of the active site. Significantly, swap of fragment 5A alone (experiment 29) leads to production of hexaketides and in particular 12/13 in which a hexaketide with double methylation is formed as the major product. This observation is emphasised for the MILS 5A swap (experiments 31 + 32) where these residues are also changed and a heptaketide becomes the major product. Swap of as few as 12 residues from MILS reproduces this effect, albeit in lower titre (exp. 33).
Discussion
Overall observations
The programming of the TENS, DMBS and MILS PKS systems appears to be controlled by four catalytic domains: KS, C-MeT, KR and the trans-acting ER (Summarised in Scheme 2). Other domains appear to have their selectivity controlled chemically: that is, they act on substrates which are chemically competent, so that for example, the DH domain always acts on ACP-bound β-alcohols and the AT always supplies extender units. When no other domain can act, the NRPS C-domain can remove ACP-bound β-ketothiolesters and release substrates from the synthetase via the DKC to form the observed tetramic acids.This is supported by the limited number of reported in vitro investigations of hr-PKS catalytic domains. We recently showed that the functional ER domain from squalestatin tetraketide synthase has low selectivity and is able to effectively reduce a wide range of enoyl-pantetheines, even including unnatural isomers and stereoisomers.26 Meanwhile, Vederas and coworkers have shown that the C-MeT domain from the lovastatin nonaketide synthase does display tight selectivity for its substrate, while the KR in the same system is less selective.43
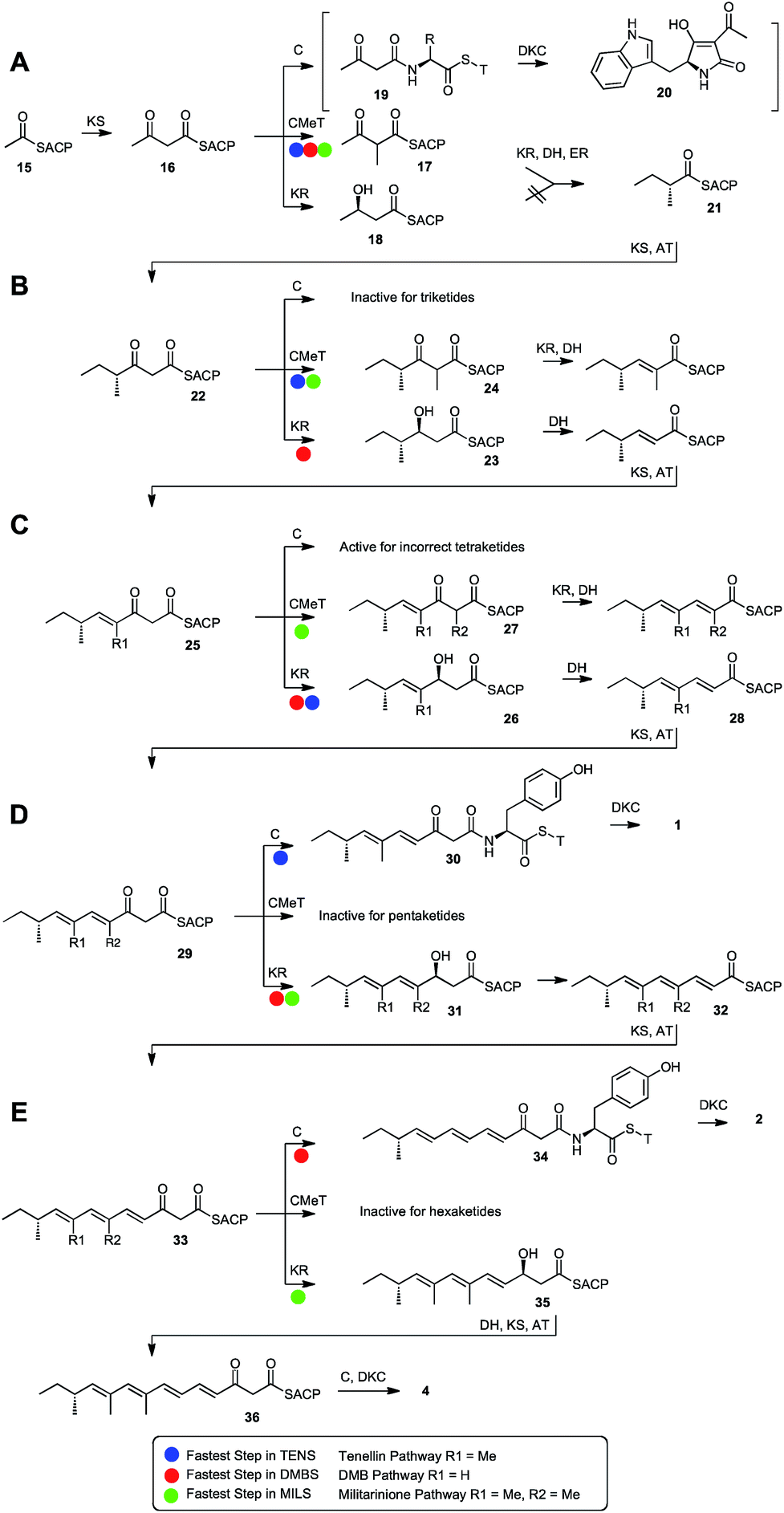 | ||
| Scheme 2 Course of polyketide biosynthesis catalysed by hybrid PKS-NRPS: (A), first condensation and subsequent selectivity; (B) second condensation and subsequent selectivity; (C) third condensation and subsequent selectivity; (D) fourth condensation and subsequent selectivity; (E) fifth condensation and subsequent selectivity. T = NRPS thiolation domain. | ||
These observations are consistent with a programming mechanism which arises by competition between the selective domains for the ACP-bound substrate. For example, after the first chain extension of acetyl ACP 15 to acetoacetyl ACP 16, four domains (KS, KR, C-MeT and the NRPS C domain) are catalytically competent to react next. In the cases of TENS, DMBS and MILS the C-MeT must be significantly faster than KR to give 17, but this is still a valid substrate for the KR which reduces it (more slowly) to the β-alcohol, which is dehydrated and enoyl-reduced to give 21. A similar mechanism has very recently been proposed in nr-PKS systems where C-MeT must compete with KS for β-keto–ACP species.44 It appears that the NRPS C-domain is selective for β-ketothiolesters so the only option left for diketide 21 is chain-extension by KS to provide the triketide 22. The C-domain must be slower than both C-MeT and KR in competition for diketides in these cases, as diketide shunts are never observed. However in some systems, such as cyclopiazonic acid PKS–NRPS45 the C-catalysed offloading of a diketide via19 to 20 is the fastest reaction.
The C-MeT domains of TENS and MILS must be faster than their KR domains after the second round of extension to give 24 (Scheme 2B), but for DMBS the KR is faster, giving 23. This is consistent with the C-MeT domain swap (experiments 2 + 3) in which the TENS C-MeT provides mostly monomethylated products.
The TENS and DMBS trans-acting ERs cannot reduce triketides, and thus the unsaturated chain is extended again by KS/AT to provide tetraketide 25. The TENS and DMBS C-MeT domains cannot methylate tetraketides (although presumably the MILS C-MeT can, Scheme 2C), so here KR is again fast and DH must eliminate H2O to form 28 which is again extended by KS/AT to form the pentaketide 29. At this stage (Scheme 2D) the TENS KR cannot reduce the β-carbonyl and the default pathway is offloading by the NRPS C domain to give pentaketide 30 and subsequently 1. However the MILS and DMBS KR domains are able to reduce pentaketides quicker than off-loading, to give 31, which is then dehydrated to 32.
Triene 32 is extended again by KS/AT to give hexaketide 33. Again, this is consistent with the DMBS KR swap (experiment 24) which can convert TENS to a hexaketide synthase. This swap gives a mixture of pentaketide and hexaketide products showing that the donated KR presumably competes poorly with the off-loading C-domain. However this is still faster than the TENS KR which cannot reduce pentaketides. The DMBS KR and C-MeT domains cannot react with 33 so the default pathway is again C-catalysed offloading to give hexaketide 34 and then 2 (Scheme 2E).
Finally, if the KR can reduce hexaketides to give 35 (Scheme 2E), after dehydration the KS/AT will extend the chain again to 36, but the resulting heptaketide can only be off-loaded to produce 4. This is consistent with the MILS KR swap (experiments 31–33) which are the only swaps to produce heptaketide products from the TENS chassis. Significantly no tetraketides were produced in these experiments, and pentaketide product was a minority. This also suggests that the MILS KR usually always outcompetes off-loading for shorter chains.
These results also illustrate that the TENS KS/AT and DH domains appear to have little selectivity (forming all chains up to heptaketides) and the NRPS offloading module most likely acts on β-ketothiolesters waiting on the ACP. This would suggest that off-loading is the slowest step of the entire process, becoming the default when no other domain can intercept the ACP-bound substrate, and is unselective with respect to chain-length and methylation pattern. This is in contrast to kinetic studies of Type II iterative PKS in which off-loading is not rate-limiting.46
Mechanisms of selectivity and programming
Thus a mechanism by which competition for substrates by a limited number of catalytic domains can explain the observed programming outcomes. The results also show that programming in these iterative PKS arises due to a combination of intrinsic and extrinsic factors rather than simply as a result of direct substrate-selectivity of each catalytic domain.The clearest evidence for intrinsic selectivity of a catalytic domain for its substrate comes from the KR domain. Here, our modelled structure indicates that five key residues on a substrate-binding helix appear to form part of the substrate binding pocket, and the difference of these residues between TENS, DMBS and MILS appears to account for the observed change in KR substrate selectivity and hence chain-length control. Here swap of fragment 5A for the corresponding fragment from DMBS gave hexaketides, while the same fragment from MILS specifies chain-lengths up to heptaketide. This also corresponds with additional variations in the MILS KR active site (e.g. L2345A). This result is reinforced by the observation that exchange of a 12-residue fragment corresponding to Q2398 to V2409 of the TENS KR (Fig. 6) reprogrammes the system to produce hexa and heptaketides. In-turn, this swap in particular validates the modelling of the TENS PKS which predicts that these residues are most likely in contact with the substrate in the KR active site (Fig. 5D). This sequence is absent or of very low homology in the FAS and AmphB KR sequences which correlates with the absence of programming in these systems – in both cases the mFAS and AmphB KR domains need only display an unselective ability to reduce the β-ketothiolesters supplied to them.
This clear evidence for intrinsic selectivity in the KR domain, however, is lacking for the C-MeT domain where there appears to be no significant changes in the active site itself. Evidence for the operation of extrinsic programming effects in the TENS PKS is widespread.
First, programming changes are usually not limited to changing a single structural factor of the product. For example, swap of C-MeT fragment 2A (experiment 8) leads to the production of tetra- and pentaketides with a range of methylation patterns (6, 7, 8, 9, 10 and 11) indicative of changes in both C-MeT and KR selectivity (i.e. their relative rates). Even within the KR domain, very precise exchanges, such as those enacted in experiment 33 in which only 12 residues were changed, lead to mixtures and lowered titres.
Second, unreduced compounds 6, 8, 10 and 11 must arise through inactivity of the trans-acting ER TenC which we did not change in these experiments. This suggests that TenC does not interact with a ‘free’ substrate-bound ACP, but with a substrate-bound ACP intimately associated with the rest of the PKS. Swaps which result in failure of TenC to reduce correctly are associated with the C-MeT and ΨKR domains, although not with the KR, suggesting that the trans-acting ER associates with C-MeT and ΨKR.
Third, all active swaps still produce the native product 1, either as the major product or as part of a mixture, again indicating that there is no simple control of overall selectivity. Finally, swaps within non-catalytic domains such as the ΨKR (experiments 15–17) also lead to changes in methylation pattern (e.g. compound 7), although not to changes in chain-length.
All these factors suggest that extrinsic protein–protein interactions between the catalytic domains can affect the rates at which they compete for ACP-bound intermediates. This is supported by our previous results16 in which the cleanest change in selectivity was observed when the complete C-MeT–ΨKR–ER0–KR fragment was exchanged, consistent with the limitation of introduction of deleterious extrinsic protein–protein interactions.
The importance of extrinsic selectivity effects is highlighted by observation of the results from swaps of the fragments with the lowest number of changes. For example, fragment 2B1 at the C-terminus of C-MeT is 100% similar and 90% identical (e.g. 4 conservative changes in 40 AA) while fragment 4A is 92% identical and 99% similar over 73 residues at the N-terminus of the KR. Swap of fragment 4A (experiment 27) makes almost no change to the programme; but swap of 2B1 (experiment 13) produces tetraketides, compounds with changes to the methylation programme and unreduced shunts.
Close examination of the modelled structures of the TENS β-processing domains shows that almost all mutations are located at surface-exposed positions (Fig. 5A). Some of these may account for the observed reduction in TenC ER activity by impeding correct docking of this trans-acting component. These variations may also contribute to altered domain–domain interactions and hence changes in the relative rates of C-MeT and KR domains. We also looked for the occurrence of non-surface variations of residues away from the active sites (Fig. 5B). Six residues fall into this class. Of these, two pairs (V1433I, V1437M and I2223L, V2336I) appear to be in contact in hydrophobic core regions of the C-MeT and KR respectively, and two others do not seem to be located at domain interfaces.
Conclusion
Our results support a programming mechanism for hr-PKS in which individual catalytic domains such as the C-MeT and KR clearly have a component of intrinsic selectivity for their substrates. But, this selectivity is not the only feature determining overall programming when competition for different catalytic domains is possible. This is the case for the β-keto ACP thiolester intermediates formed by the KS which can be substrates for four different catalytic domains (KR, C-MeT, C and KS) in hybrid PKS-NRPS systems and it must be the ratios of the intrinsic and extrinsic selectivities which determine the final outcome. Thus the overall programme appears to be based on a complex balance of relative selectivities displayed by several catalytic domains. These selectivities must change as the chain grows and such effects can clearly lead to complex outcomes – for example the skipped methylation patterns observed in prototenellin A 6, fusarin C 3747 and the ACE1 shunt compound 3848 presumably means that for some intermediates the C-MeT can again out-compete the KR (Fig. 7) in these systems.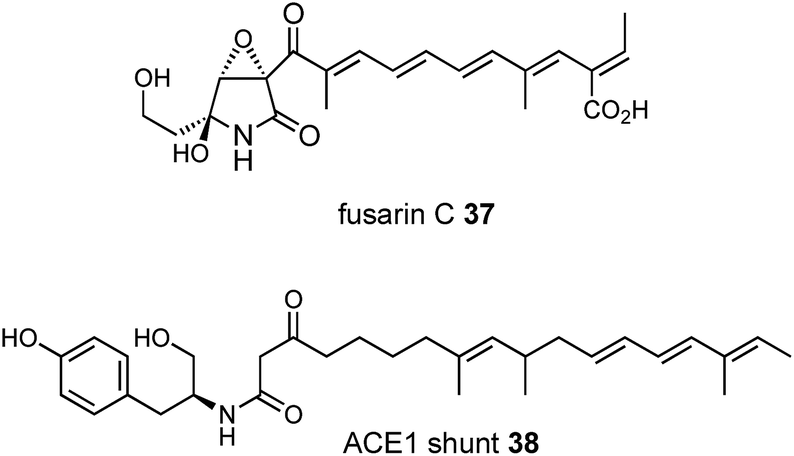 | ||
| Fig. 7 Fungal polyketides produced by hybrid PKS-NRPS enzymes with skipped methylation patterns. | ||
These conclusions also give hope that it may be possible to predict the product of hr-PKS if the relative selectivities of competing domains can be determined. Studies to determine the selectivities of DH and ER domains are already underway in our laboratories.25,26
Our results also suggest possible mechanisms by which programming selectivity in fungal hr-PKS could evolve. For example, fragment 2B1 of DMBS differs from fragment 2B1 of TENS by only 3 conservative changes (i.e. it is 100% similar and 92% identical). It contains 9 residues which likely make up the substrate binding site, although none of these is mutated in the DMBS sequence, but its exchange into TENS results in a significant erosion of fidelity such that five different compounds are produced (exp. 13). Thus a small number of amino acid changes in the synthase can lead to the production of several new compounds. We have previously observed how other physical changes such as the levels of gene expression and protein production can similarly affect product diversity in the tenellin and related synthases.20,49
Similar observations of erosion of fidelity during biosynthesis have been recently reported by Cane, Christianson and coworkers in the arena of terpene cyclases.50 The creation of a range of secondary metabolites of varied structure could give an organism the possibility to rapidly respond to changing environmental conditions. Production of a small library of related products could more rapidly access compounds with evolutionary utility than single step changes through a series of compounds. Once a useful new compound is produced the system could then more slowly optimise the library by restoring fidelity to the newly selected secondary metabolite by mutations of more distant residues. Our observations of this effect in iterative highly reducing fungal polyketide synthases suggests it may be a phenomenon which occurs more generally.
Thus the results of this study strongly suggest that domain swaps of individual domains, swaps of sub-domains, or even possibly mutations of single amino acid residues, are unlikely to lead to the predictable and clean production of desired compounds without considering cognate mutations in competing domains to alter their selectivities. In the short and medium terms, and in the absence of high quality crystallographic data, it is likely to be more productive to exchange multidomain fragments with the aim of preserving their delicate balances of intrinsic and extrinsic selectivities, or make precise and very limited changes to active sites with the hope of changing substrate selectivity without influencing protein–protein interactions between domains. Such experiments, and associated site-directed mutations are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
RJC, X-LY, KW and OP thank the University of Hannover for funding. LCMS and NMR instrumentation were funded by the University of Hannover and DFG (INST 187/621, INST 187/686-1, CO 1328/3-1). SY thanks the Chinese Scholarship Council for funding (CSC 2016 06210136). Dr Colin Lazarus, University of Bristol, is thanked for the gift of C. militaris genomic DNA.Notes and references
- (a) R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC; (b) Y. H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS PubMed.
- Y. Ma, L. H. Smith, R. J. Cox, P. Beltran-Alvarez, C. J. Arthur and T. J. Simpson, ChemBioChem, 2006, 7, 1951–1958 CrossRef CAS PubMed.
- D. A. Herbst, C. R. Huitt-Roehl, R. P. Jakob, J. M. Kravetz, P. A. Storm, J. R. Alley, C. A. Townsend and T. Maier, Nat. Chem. Biol., 2018, 14, 474–479 CrossRef CAS PubMed.
- (a) A. L. Vagstad, A. G. Newman, P. A. Storm, K. Belecki, J. M. Crawford and C. A. Townsend, Angew. Chem., Int. Ed., 2013, 52, 1718–1721 CrossRef CAS PubMed; (b) J. M. Crawford, P. M. Thomas, J. R. Scheerer, A. L. Vagstad, N. L. Kelleher and C. A. Townsend, Science, 2008, 320, 243–246 CrossRef CAS PubMed.
- T. P. Nicholson, C. Winfield, J. Westcott, J. Crosby, T. J. Simpson and R. J. Cox, Chem. Commun., 2003, 686–687 RSC.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS PubMed.
- E. J. Skellam, Nat. Prod. Rep., 2017, 34, 1252–1263 RSC.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- K. E. Lebe and R. J. Cox, Chem. Sci., 2019, 10, 1227–1231 RSC.
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L. C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef PubMed.
- L. Wang, M. Yuan and J. Zheng, Synth. Syst. Biotechnol., 2019, 4, 10–15 CrossRef PubMed.
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W.-H. Li, J. C. Vederas, Y. Tang and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef CAS PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef CAS PubMed.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef CAS PubMed.
- H. Jessen and K. Gademann, Nat. Prod. Rep., 2010, 27, 1168–1185 RSC.
- K. Schmidt, U. Riese, Z. Li and M. Hamburger, J. Nat. Prod., 2003, 66, 378–383 CrossRef CAS PubMed.
- Y. Cheng, B. Schneider, U. Riese, B. Schubert, Z. Li and M. Hamburger, J. Nat. Prod., 2004, 67, 1854–1858 CrossRef CAS PubMed.
- Z. Wasil, K. Pahirulzaman, C. P. Butts, T. J. Simpson, C. M. Lazarus and R. J. Cox, Chem. Sci., 2013, 4, 3845–3856 RSC.
- M. Heneghan, A. A. Yakasai, K. Williams, K. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC.
- S. M. Ma, J. W. H. Li, J. W. Choi, H. Zhou, M. K. K. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS PubMed.
- T. Kakule, Z. Lin and E. Schmidt, J. Am. Chem. Soc., 2014, 136, 17882–17890 CrossRef CAS PubMed.
- K. Pahirulzaman, K. Williams and C. M. Lazarus, Methods Enzymol., 2012, 517, 241–260 CAS.
- E. Liddle, A. Scott, L. C. Han, D. Ivison, T. J. Simpson, C. L. Willis and R. J. Cox, Chem. Commun., 2017, 53, 1727–1730 RSC.
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2016, 8, 1116–1126 RSC.
- J. Zheng, C. A. Taylor, S. K. Piasecki and A. T. Keatinge-Clay, Structure, 2010, 18, 913–922 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46(W1), W296–W303 CrossRef CAS PubMed.
- S. Bienert, A. Waterhouse, T. A. P. de Beer, G. Tauriello, G. Studer, L. Bordoli and T. Schwede, Nucleic Acids Res., 2017, 45(D1), D313–D319 CrossRef CAS PubMed.
- N. Guex, M. C. Peitsch and T. Schwede, Electrophoresis, 2009, 30(Suppl 1), S162–S173 CrossRef PubMed.
- P. Benkert, M. Biasini and T. Schwede, Bioinformatics, 2011, 27(3), 343 CrossRef CAS PubMed.
- M. Bertoni, F. Kiefer, M. Biasini, L. Bordoli and T. Schwede, Sci. Rep., 2017, 7(1), 10480 CrossRef PubMed.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Proteins: Struct., Funct., Bioinf., 2009, 77(Suppl 9), 114–122 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11(5), 905–919 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455–461 CAS.
- D. Seeliger and B. L. de Groot, J. Comput.-Aided Mol. Des., 2010, 24(5), 417–422 CrossRef CAS PubMed.
- W. D. Fiers, G. J. Dodge, D. H. Sherman, J. L. Smith and C. C. Aldrich, J. Am. Chem. Soc., 2016, 138, 16024–16036 CrossRef CAS PubMed.
- M. A. Skiba, A. P. Sikkema, W. D. Fiers, W. H. Gerwick, D. H. Sherman, C. C. Aldrich and J. L. Smith, ACS Chem. Biol., 2016, 11, 3319–3327 CrossRef CAS PubMed.
- D. L. Akey, J. R. Razelun, J. Tehranisa, D. H. Sherman, W. H. Gerwick and J. L. Smith, Structure, 2010, 18, 94–105 CrossRef CAS PubMed.
- A. E.-L. Busche, D. Gottstein, C. Hein, N. Ripin, I. Pader, P. Tufar, E. B. Eisman, L. Gu, C. T. Walsh, D. H. Sherman, F. Löhr, P. Güntert and V. Dötsch, ACS Chem. Biol., 2012, 7, 378–386 CrossRef CAS PubMed.
- H. Shafran, I. Miyara, R. Eshed, D. Prusky and A. Sherman, Fungal Genet. Biol., 2008, 45, 1147–1154 CrossRef CAS PubMed.
- M. D. Koziol, Ph.D. thesis, Investigating Programming and Production of Fungal Polyketide Synthases, University of Bristol, 2014 Search PubMed.
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef CAS PubMed.
- P. A. Storm, P. Pal, C. R. Huitt-Roehl and C. A. Townsend, ACS Chem. Biol., 2018, 13, 3043–3048 CrossRef CAS PubMed.
- Y. Seshime, P. R. Juvvadi, M. Tokuoka, Y. Koyama, K. Kitamoto, Y. Ebizuka and I. Fujii, Bioorg. Med. Chem. Lett., 2009, 19, 3288–3292 CrossRef CAS PubMed.
- P. Beltran-Alvarez, R. J. Cox, J. Crosby and T. J. Simpson, Biochemistry, 2007, 46, 14672–14681 CrossRef CAS PubMed.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS PubMed.
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. M. Khalid, J. Collemare, E. J. Skellam, D. Tharreau, M. H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC.
- A. A. Yakasai, J. Davison, Z. Wasil, L. M. Halo, C. P. Butts, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 10990–10998 CrossRef CAS PubMed.
- (a) M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Biochemistry, 2002, 41, 1732–1741 CrossRef CAS PubMed; (b) R. Li, W. Chou, J. Himmelberger, K. Litwin, G. Harris, D. E. Cane and D. W. Christianson, Biochemistry, 2014, 53, 1155–1168 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Including all experimental, bioinformatic and characterisation details. See DOI: 10.1039/c9sc03173a |
| ‡ Present address: School of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, P. R. China. |
| This journal is © The Royal Society of Chemistry 2019 |