
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A Cu–Pd single-atom alloy catalyst for highly efficient NO reduction†
Feilong
Xing
 a,
Jaewan
Jeon
a,
Takashi
Toyao
ab,
Ken-ichi
Shimizu
ab and
Shinya
Furukawa
*ab
a,
Jaewan
Jeon
a,
Takashi
Toyao
ab,
Ken-ichi
Shimizu
ab and
Shinya
Furukawa
*ab
aInstitute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan. E-mail: furukawa@cat.hokudai.ac.jp; Fax: +81-11-706-9163
bElements Strategy Initiative for Catalysts and Batteries, Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 5th August 2019
Abstract
A series of Cu–Pd alloy nanoparticles supported on Al2O3 were prepared and tested as catalysts for deNOx reactions. XRD, HAADF-STEM, XAFS, and FT-IR analyses revealed that a single-atom alloy structure was formed when the Cu/Pd ratio was 5, where Pd atoms were well isolated by Cu atoms. Compared with Pd/Al2O3, Cu5Pd/Al2O3 exhibited outstanding catalytic activity and N2 selectivity in the reduction of NO by CO: for the first time, the complete conversion of NO to N2 was achieved even at 175 °C, with long-term stability for at least 30 h. High catalytic performance was also obtained in the presence of O2 and C3H6 (model exhaust gas), where a 90% decrease in Pd use was achieved with minimum evolution of N2O. Kinetic and DFT studies demonstrated that N–O bond breaking of the (NO)2 dimer was the rate-determining step and was kinetically promoted by the isolated Pd.
Introduction
The reactions of nitric oxide (NO) have garnered intense interest from researchers in the human health,1 and bioinorganic,2 industrial,3 and environmental chemistry fields.4 Specifically, NO removal has long been studied as an indispensable process for exhaust-gas purification.5 Platinum-group metals (PGMs) such as Pt, Pd, and Rh are known to be efficient catalysts for the reduction of NO using CO,6,7 H2,8 NH3,9 and hydrocarbons10 as reductants. The recent challenges in this field involve developing catalytic systems that function (1) at low temperatures under cold-start conditions,11 (2) with minimum use of PGMs,12,13 and (3) without emitting N2O,14–16 which is a potent greenhouse gas.17 These issues have been individually studied using different materials. The development of a single material that enables (1)–(3) is therefore highly desirable. To the best of our knowledge, no such material has been reported. In particular, achieving both (1) and (3) is difficult because N2O reduction to N2 on PGMs requires relatively high temperatures (>300 °C).18 Therefore, an appropriate catalyst design is needed to obtain not only outstanding catalytic activity toward NO reduction but also high selectivity to N2 with minimal incorporation of PGMs.A promising approach that overcomes these challenges is the single-atom alloying concept,19 which is relevant to single-atom chemistry.20,21 The dilution of PGM atoms with less active metal atoms not only substantially reduces the use of PGMs but also enables drastic modification of the electronic and geometric structures for enhanced catalysis.22 For example, the isolation of Pt or Pd with group 11 metals (Au, Ag, and Cu) enables molecular transformations that hardly proceed in the absence of single-atom alloying, such as selective hydrogenation,23–27 formic acid dehydrogenation,28 and hydrosilylation.29 In these systems, the group 11 metals act as inert elements but modify the electronic and geometric factors of the PGM and, thus, its catalytic properties. Conversely, for NO reduction, the group 11 elements are known to be capable of NO activation.30–32 Therefore, applying the single-atom alloying concept to NO reduction systems should provide an unprecedented synergistic effect for efficient NO conversion.
In this study, we focused on Cu as a main component because of its intrinsic activity toward NO reduction and its high earth abundance. We found that Cu–Pd/Al2O3 (Cu/Pd = 5) acts as a highly efficient catalyst for NO reduction at low temperatures (>150 °C), without generating N2O emissions. Herein, we report both an innovative catalytic system for efficient NO reduction and novel catalytic chemistry of single-atom alloys.
Experimental details
Catalyst preparation
Boehmite (γ-AlOOH) was supplied by SASOL Chemicals. γ-Al2O3 was prepared by the calcination of boehmite at 900 °C for 3 h. Pd/Al2O3 (Pd: 2 wt%) and Cu–Pd/Al2O3 (Cu: 6 wt%, Cu/Pd = 1) were prepared by a conventional impregnation method. The γ-Al2O3 support was added to a vigorously stirred aqueous solution containing Pd(NH3)2(NO2)2 (Kojima Chemicals, 4.765 wt% in HNO3) and/or Cu(NO3)2·3H2O (Sigma-Aldrich, 99%), followed by stirring for 3 h at room temperature (50 ml H2O per gram of Al2O3). The mixture was dried under a reduced pressure at 50 °C, followed by reduction under flowing H2 (30 ml min−1) at 400 °C (Pd) or 800 °C (CuPd) for 1 h. The Cu/Al2O3 (Cu: 6 wt%) and Cu–Pd/Al2O3 (Cu: 6 wt%, Cu/Pd = 3 and 5) catalysts were prepared by a deposition–precipitation method using urea. The γ-Al2O3 support was added to a vigorously stirred aqueous solution of Cu(NO3)2·3H2O (50 ml H2O per gram of Al2O3). Then, an aqueous solution of urea (Kanto, 99%) was added dropwise to the stirred mixture at room temperature (urea/Cu = 30). The mixture was sealed with a plastic film and kept with stirring at 70 °C for 10 h. After completing the precipitation of Cu(OH)2, the supernatant was decanted and the resulting crude product was washed with deionized water three times, followed by drying under a reduced pressure at 50 °C and calcination at 500 °C for 1 h. For Cu/Al2O3, the resulting CuO/Al2O3 was reduced under flowing H2 at 400 °C for 1 h. For Cu–Pd/Al2O3 (Cu/Pd = 3 and 5), the resulting CuO/Al2O3 was used for successive impregnation of Pd in a similar fashion to that mentioned above. The resulting Pd–CuO/Al2O3 was reduced under flowing H2 at 400 °C for 1 h.Reaction conditions
The catalyst (0.05 g) diluted with quartz sand (1.95 g, Miyazaki Chemical, 99.9%) was treated under flowing hydrogen (50 ml min−1) at 400 °C for 0.5 h prior to the catalytic reactions. NO reduction by CO was performed in a fixed-bed continuous flow system by feeding NO (5000 ppm), CO (5000 ppm), and He (balance) with a total flow rate of 96 ml min−1 (GHSV = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). The gas phase was analyzed using an online thermal conductivity detection gas chromatograph (Shimadzu GC-8A, column: SHINWA SHINCARBON ST) located downstream. A stability test was done using twice the amount of catalyst (0.10 g) under similar conditions (GHSV = 40000 h−1). After a time-on-stream of 24 h, the catalyst was regenerated by flowing hydrogen (50 ml min−1) at 400 °C for 0.5 h, followed by continuing the catalytic run. A kinetic study was performed by changing the concentration of NO and CO between 0.3 and 0.6% with that of the counterpart fixed at 0.5%. The reaction temperature was maintained at 150 °C so that NO conversion did not exceed 30%, and the reaction rate (mol s−1 molPd−1) was calculated on the basis of NO conversion. NO + CO + O2 and NO + CO + O2 + C3H6 reactions were performed under stoichiometric conditions as follows: NO (5000 ppm), CO (10000 ppm), O2 (2500 ppm), He (balance) with a total flow rate of 96 ml min−1 (GHSV = 80000 h−1), and NO (5000 ppm), CO (5000 ppm), O2 (5625 ppm), C3H6 (1250 ppm), and He (balance) with a total flow rate of 96 ml min−1 (GHSV = 80000 h−1), respectively.
000 h−1). The gas phase was analyzed using an online thermal conductivity detection gas chromatograph (Shimadzu GC-8A, column: SHINWA SHINCARBON ST) located downstream. A stability test was done using twice the amount of catalyst (0.10 g) under similar conditions (GHSV = 40000 h−1). After a time-on-stream of 24 h, the catalyst was regenerated by flowing hydrogen (50 ml min−1) at 400 °C for 0.5 h, followed by continuing the catalytic run. A kinetic study was performed by changing the concentration of NO and CO between 0.3 and 0.6% with that of the counterpart fixed at 0.5%. The reaction temperature was maintained at 150 °C so that NO conversion did not exceed 30%, and the reaction rate (mol s−1 molPd−1) was calculated on the basis of NO conversion. NO + CO + O2 and NO + CO + O2 + C3H6 reactions were performed under stoichiometric conditions as follows: NO (5000 ppm), CO (10000 ppm), O2 (2500 ppm), He (balance) with a total flow rate of 96 ml min−1 (GHSV = 80000 h−1), and NO (5000 ppm), CO (5000 ppm), O2 (5625 ppm), C3H6 (1250 ppm), and He (balance) with a total flow rate of 96 ml min−1 (GHSV = 80000 h−1), respectively.
Characterization
The crystal structure of the prepared catalyst was examined by powder X-ray diffraction (XRD) using a Rigaku MiniFlex II/AP diffractometer with Cu Kα radiation. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) was carried out using a JEOL JEM-ARM200 M microscope equipped with an energy dispersive X-ray (EDX) analyzer (EX24221M1G5T). STEM analysis was performed at an accelerating voltage of 200 kV. To prepare the TEM specimen, all samples were sonicated in ethanol and then dispersed on a Mo grid supported by an ultrathin carbon film.The Fourier-transformed infrared (FT-IR) spectra of adsorbed CO were obtained with a JASCO FTIR-4200 spectrometer equipped with an MCT detector in transmission mode (resolution 4 cm−1). The samples were prepared as self-supporting wafers (2.0 cm diameter, <0.5 mm thickness) and were placed inside an IR cell with CaF2 windows. A custom glass manifold was connected to the cell to control the gas for pretreatment and the amount of CO introduced. The cell was first purged with He, and the sample was reduced under flowing hydrogen (50 ml min−1) at 400 °C for 30 min. After reduction, the wafer was cooled to 40 °C under flowing He. The wafer was exposed to CO (0.5%) and He (balance) with a total flow rate of 50 ml min−1 for 20 min. After the CO exposure, He was flowed for 5 min to remove the gas phase and weakly adsorbed CO, followed by IR spectral measurements.
X-ray absorption fine structure (XAFS) spectra were recorded on the BL14B2 station at SPring-8 of the Japan Synchrotron Radiation Research Institute. A Si(311) double-crystal monochromator was used. Energy calibration was performed using Pd foil. The spectra were recorded at the edges of Pd K in a transmission mode at room temperature. The pelletized sample was pre-reduced with H2 at 400 °C for 0.5 h, and then sealed in a plastic pack under a N2 atmosphere together with an ISO A500-HS oxygen absorber (Fe powder). The obtained XAFS spectra were analyzed using Athena and Artemis software ver. 0.9.25 included in the Demeter package. The Fourier transformation of the k3-weighted EXAFS from k space to R space was performed over a k range of 3.0–15 Å−1. Some of the Fourier-transformed EXAFS spectra in the R range of 1.2–3.0 Å were inversely Fourier transformed, followed by an analysis using a usual curve fitting method in a k range of 3–15 Å−1. The back-scattering amplitude or phase shift parameters were simulated with FEFF 6L and used to perform the curve fitting procedure. For Pd–Cu scattering, intermetallic Cu3Pd with a Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure was considered for the FEFF simulation. The amplitude reduction factor (S02) of Pd was determined to be 0.775 by fitting the spectra of Pd black and then used for fitting of other EXAFS spectra.
m structure was considered for the FEFF simulation. The amplitude reduction factor (S02) of Pd was determined to be 0.775 by fitting the spectra of Pd black and then used for fitting of other EXAFS spectra.
Computational details
Periodic DFT calculations were performed using the CASTEP code33 with Vanderbilt-type ultrasoft pseudopotentials34 and the Perdew–Burke–Ernzerhof exchange–correlation functional based on the generalized gradient approximation.35 The plane-wave basis set was truncated at a kinetic energy of 350 eV, and a Fermi smearing of 0.1 eV was utilized. Dispersion correlations were considered using the Tkatchenko–Scheffler method with a scaling coefficient of sR = 0.94 and a damping parameter of d = 20.36 The reciprocal space was sampled using a k-point mesh with a spacing of typically 0.04 Å−1, as generated by the Monkhorst–Pack scheme.37 Geometry optimization was performed on supercell structures using periodic boundary conditions. The surface was modeled based on Cu(211)-(2 × 3) (for NO and N2O related reactions), Cu(111)-(2 × 2) (for CO oxidation), and Cu(111)-(3 × 3) (for N2O decomposition) slabs that were six atomic layers thick with 13 Å of vacuum spacing. The convergence criteria for structural optimization and energy calculation were set to (a) an SCF tolerance of 1.0 × 10−6 eV per atom, (b) an energy tolerance of 1.0 × 10−5 eV per atom, (c) a maximum force tolerance of 0.05 eV Å−1, and (d) a maximum displacement tolerance of 1.0 × 10−3 Å. The transition state search was performed using the complete linear synchronous transit/quadratic synchronous transit (LST/QST) method.38,39 Linear synchronous transit maximization was performed, followed by energy minimization in the directions conjugate to the reaction pathway. The approximated TS was used to perform QST maximization with conjugate gradient minimization refinements. This cycle was repeated until a stationary point was found. Convergence criteria for the TS calculations were set to root-mean-square forces on an atom tolerance of 0.10 eV Å−1.Results and discussion
The monometallic Cu (6 wt%) and Pd (2 wt%) and the bimetallic Cu–Pd (Cu: 6 wt%, Cu/Pd = 1, 3, or 5; hereafter, CuxPd, x = 1, 3, or 5) catalysts were prepared using γ-Al2O3 as a support by deposition–precipitation and/or impregnation methods. X-ray diffraction (XRD) patterns of the prepared catalysts revealed that Cu–Pd solid-solution alloy phases with bimetallic compositions similar to the metal ratio in the feed were formed (Fig. S1† and Table 1).| a Estimated from Scherrer's equation using a Scherrer constant of 0.477 for the area-weighted mean diameter. b Area-weighted mean diameter obtained from TEM images. | |||||
|---|---|---|---|---|---|
| Pd | CuPd | Cu3Pd | Cu5Pd | Cu | |
| Pd loading (wt%) | 2.0 | 10.1 | 3.4 | 2.0 | 0 |
| Cu loading (wt%) | 0 | 6.0 | 6.0 | 6.0 | 6.0 |
| Cu fraction in the catalyst | 0 | 0.50 | 0.75 | 0.83 | 1.0 |
| Cu fraction in particles | 0 | 0.44 | 0.72 | 0.89 | 1.0 |
| Crystallite size/nma | 3.2 | 3.3 | 3.8 | 3.8 (4.2)b | 4.0 |
The crystallite sizes estimated using Scherrer's equation were 3–4 nm for all of the catalysts. Fig. 1a and b show a high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image of Cu5Pd/Al2O3 and the size distribution of the nanoparticles, respectively. A relatively narrow size distribution between 2 and 6 nm with an area-weighted mean diameter of 4.2 nm was obtained, consistent with the crystallite size estimated by XRD (Table 1).
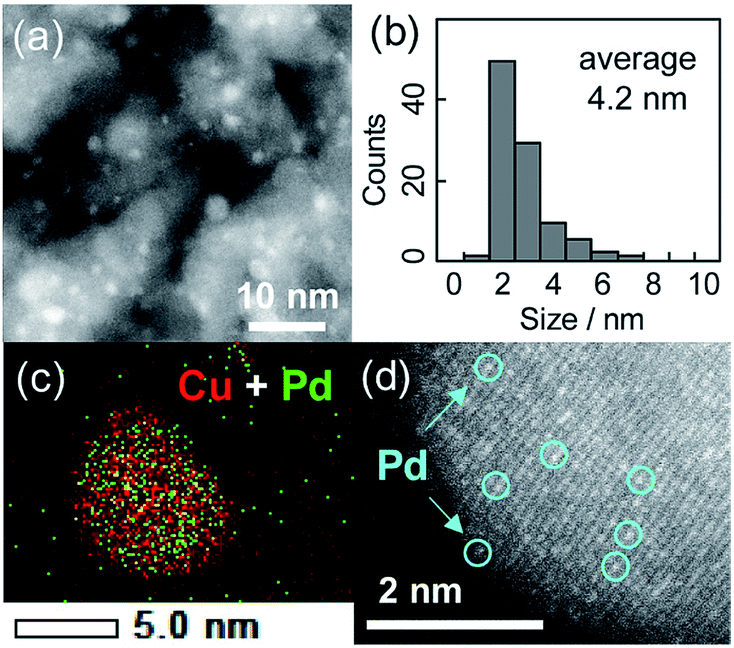 | ||
| Fig. 1 (a) HAADF-STEM image of Cu5Pd/Al2O3 and (b) size distribution of the nanoparticles. (c) Elemental map of the Pd + Cu overlayer, as acquired by EDS. (d) High-resolution image of a single nanoparticle. | ||
The energy-dispersive X-ray spectroscopy (EDS) analysis of a single nanoparticle revealed that the Cu and Pd atoms comprising the nanoparticle were homogeneously dispersed (Fig. 1c). The high-resolution HAADF-STEM image shows an fcc crystal structure viewed along the [100] direction, consistent with the formation of a solid-solution alloy (Fig. 1d). Moreover, the isolation of Pd atoms by Cu was indicated by the presence of atoms with distinct Z contrasts. Note that the corresponding HAADF-STEM image of Cu/Al2O3 showed a weak and uniform Z contrast compared with that of Cu5Pd/Al2O3 (Fig. S2†).
The degree of Pd isolation was further investigated by Fourier-transform infrared (FT-IR) and extended X-ray absorption fine structure (EXAFS) analyses (Fig. 2). As shown in Fig. 2a, the FT-IR spectra of CO adsorbed onto Pd/Al2O3 exhibited absorption peaks assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O adsorbed on top (2086 cm−1), bridge (1975 cm−1), and hollow sites (∼1880 cm−1).40 Similar absorption peaks were observed for CuPd/Al2O3, suggesting that the Pd–Pd ensembles largely remain even after 1:1 alloying. For Cu-rich samples, an absorption band assignable to CO adsorbed on metallic Cu was also observed at 2100 nm−1.41 The peak intensities for the bridge and hollow CO substantially decreased and disappeared in the spectra of Cu3Pd/Al2O3 and Cu5Pd/Al2O3, respectively, indicating that Pd atoms at the surface were isolated upon 5:1 alloying.
O adsorbed on top (2086 cm−1), bridge (1975 cm−1), and hollow sites (∼1880 cm−1).40 Similar absorption peaks were observed for CuPd/Al2O3, suggesting that the Pd–Pd ensembles largely remain even after 1:1 alloying. For Cu-rich samples, an absorption band assignable to CO adsorbed on metallic Cu was also observed at 2100 nm−1.41 The peak intensities for the bridge and hollow CO substantially decreased and disappeared in the spectra of Cu3Pd/Al2O3 and Cu5Pd/Al2O3, respectively, indicating that Pd atoms at the surface were isolated upon 5:1 alloying.
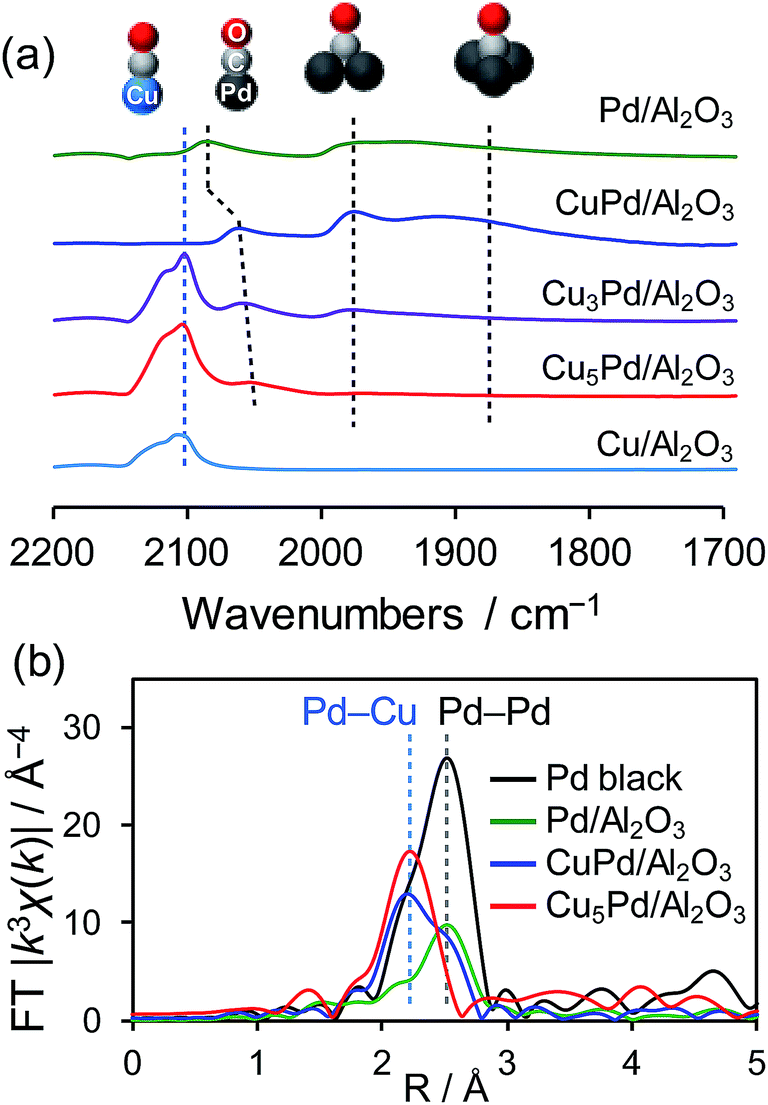 | ||
| Fig. 2 (a) FT-IR spectra of CO adsorbed on the prepared catalysts. (b) Fourier transforms of the Pd K-edge EXAFS spectra of the Pd-based catalysts. | ||
There remained a weak absorption band for linear CO on Pd in the spectrum for Cu5Pd/Al2O3, suggesting that the isolated Pd atoms are also present at the surface. Fig. 2b shows the Fourier transforms of the Pd K-edge EXAFS spectra of the Pd-based catalysts (the X-ray absorption near edge structure spectra, raw EXAFS oscillations, curve-fitting, and summary of EXAFS curve fitting are shown in Fig. S3–S5 and Table S1,† respectively). CuPd/Al2O3 showed both Pd–Pd and Pd–Cu bonds, while Cu5Pd/Al2O3 exclusively showed Pd–Cu bonds, suggesting that the Pd atoms in the bulk were also isolated by Cu upon 5:1 alloying. Thus, small Cu–Pd nanoparticles with a single-atom alloy structure were successfully formed on the Al2O3 support. Considering the limited sensitivity of EXAFS and FT-IR, we cannot completely exclude the presence of Pd–Pd interaction. However, only a small number of Pd–Pd sites, if any, seem not to contribute to the overall catalytic performance.
We next tested the catalytic activity of CuxPd/Al2O3 in NO reduction by CO (GHSV = 80000 h−1), as a model reaction for exhaust-gas purification. Fig. 3a shows the NO conversion to N2 (CN2) for the prepared catalysts as a function of reaction temperature. Here, CN2 was obtained by multiplying the NO conversion and the N2 selectivity (Fig. S6†). Pd/Al2O3 gave the lowest CN2, because of the poor N2 selectivity <40% (Fig. S6b†).
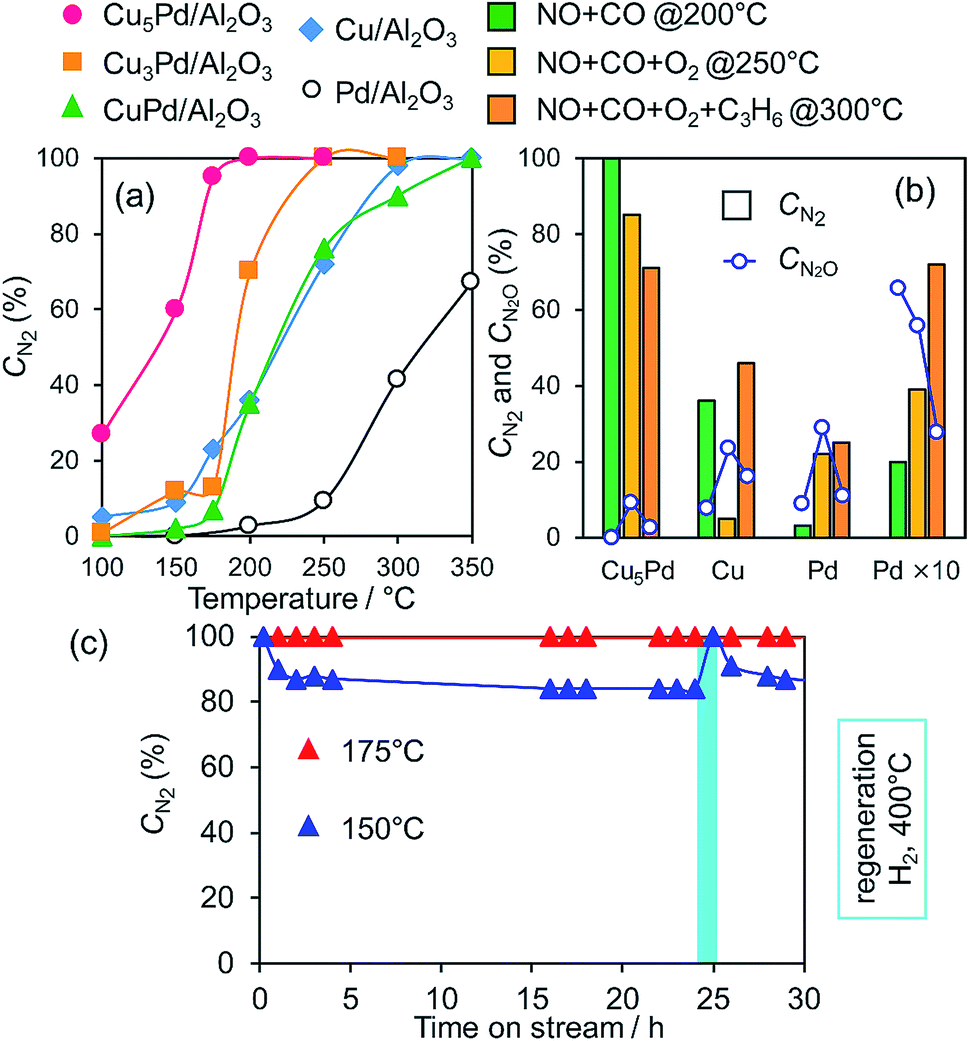 | ||
| Fig. 3 (a) NO conversion to N2 during the NO reduction by CO over Pd, Cu, and Cu–Pd catalysts as a function of reaction temperature (NO, CO: 0.5%, GHSV = 80000 h−1). (b) Comparison between CN2 and CN2O in NO reduction in the presence of O2 and C3H6. (c) Stability test for Cu5Pd/Al2O3 in the NO + CO reaction at low temperatures (NO, CO: 0.5%, GHSV = 40000 h−1). | ||
Cu/Al2O3 exhibited a higher CN2 than Pd/Al2O3 because of its much higher N2 selectivity (Fig. S6b†). CuPd/Al2O3 showed a CN2 trend similar to that of Cu/Al2O3 because of the consequence of increased NO conversion and decreased N2 selectivity (Fig. S6†). Thus, the 1:1 alloy of Cu and Pd gave an insufficient catalytic performance for selective NO reduction. Interestingly, however, both NO conversion and N2 selectivity increased when the alloying ratio was increased to 3:1 and 5:1 (Fig. S6†), which resulted in great enhancements in CN2 (Fig. 3a). NO was completely converted to N2 over Cu5Pd/Al2O3 without generating N2O emissions even at 200 °C, where Pd showed a CN2 of only 5%. Notably, on going from CuPd to Cu5Pd, the catalytic activity increased even though the Pd content was decreased to 1/5 (Table 1). Therefore, a specific synergistic effect between Cu and Pd likely contributed to the unique properties of the single-atom alloy catalyst. We emphasize that using an excess amount of Pd/Al2O3 (0.50 g) with 10 times the equimolar Pd included in Cu5Pd/Al2O3 (labeled as Pd × 10) still resulted in poor performance (Fig. 3b), highlighting the outstanding performance of the single-atom alloy catalyst. We also tested the long-term stability of Cu5Pd/Al2O3 in NO reduction by CO under standard conditions (GHSV = 40000 h−1), where 100% CN2 was maintained at 175 °C. Although a number of bimetallic catalysts for NO reduction have been reported thus far,12–16,42–48 to the best of our knowledge, the present work represents the first success in complete NOx removal at a temperature less than 200 °C. At 150 °C, although CN2 decreased slightly at the initial stage because of N2O formation, it recovered after a short H2 treatment. This result implies that the accumulation of oxygen species at the catalyst surface triggers the loss of N2 selectivity and that the catalytic performance could be recovered under rich conditions. We next examined the catalytic performance of Cu5Pd/Al2O in NO reduction in the presence of O2 and O2 + C3H6; these conditions more closely resemble those encountered in practical use. Cu/Al2O3 delivered poor performance under NO + CO + O2 conditions. By contrast, Cu5Pd/Al2O exhibited much higher performance than Cu/Al2O and Pd/Al2O. Notably, Cu5Pd/Al2O still exhibited a performance better than or comparable to “Pd × 10” even in the presence of O2 or O2 + C3H6, respectively (Fig. 3b and S7;† a comparison with temperature dependence and T50 is presented in Fig. S7†). Furthermore, N2O evolution was sufficiently suppressed over Cu5Pd, where the CN2O (NO conversion to N2O, Fig. 3b and S7†) was much lower than those for Pd. Thus, the single-atom alloy catalyst enabled not only a decrease in the noble metal use to 1/10 but also highly selective NOx removal. In the reactions conducted in the presence of O2 and O2 + C3H6, reaction temperatures greater than 200 °C were needed to achieve sufficient catalytic performance (Fig. 3 and S7†). A possible explanation is that the number of active sites for NO reduction decreased because of the involvement of other reactions such as CO and/or C3H6 oxidation.
Next, to clarify the nature of the synergistic effect, we conducted a mechanistic study based on kinetic analysis and density functional theory (DFT) calculations. First, the apparent activation energy (EA) for NO reduction by CO was estimated for the representative catalysts. The corresponding Arrhenius-type plots and the resulting EA values are shown in Fig. 4 and Table 2, respectively. Cu5Pd gave an EA value lower than those of Pd and Cu, which is consistent with the observed catalytic activity. In addition, we estimated the reaction orders for NO and CO pressures (PNO and PCO, respectively) to consider the rate-determining step (RDS). Both Cu and Cu5Pd showed negative and positive orders for PNO and PCO, respectively (see Table 2 and Fig. S8† for details). Unlike the case for Pd-based catalysts,49 bond dissociation of N–O has been speculated to occur via (NO)2 dimer formation on Cu surfaces.50–52 Therefore, in the present study, an extended Langmuir–Hinshelwood model that includes (NO)2 dimer formation, the subsequent N–O scission (N2O formation), and N2O decomposition (N2 formation) was considered. We solved the rate equation of each step regarded as the RDS by using the overall site balance and equilibrium constants for the other steps (see the ESI,† kinetic analysis). In most cases, the reaction order for PNO is positive, which is inconsistent with the observed experimental results. Conversely, when the N–O scission of the (NO)2 is considered as the RDS, the orders for PNO and PCO range from −2 to 0 and from 0 to 2, respectively, in agreement with the experimental values. Thus, our kinetic study suggests that the N–O scission was the RDS in NO reduction by CO. Upon incorporation of Pd atoms into pure Cu, the order for PNO becomes less negative, while that for PCO decreases substantially. This result indicates that the strong adsorption of NO inhibits CO adsorption onto Cu, while the latter is enhanced in the presence of Pd. We performed DFT calculations for the relevant elemental steps on pure and Pd-doped Cu surfaces. On the basis of the literature,53 the step site of the (211) surface was considered the active site for N–O scission (Fig. S9†). The corresponding energy diagrams are shown in Fig. 5.
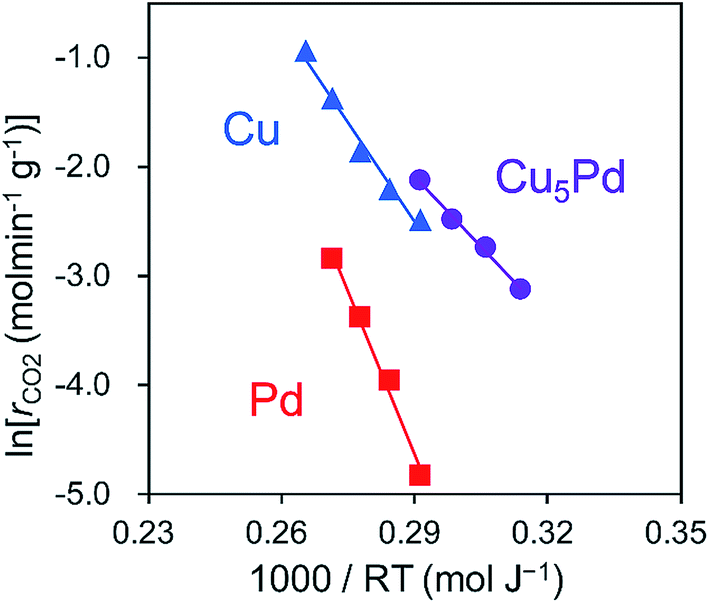 | ||
| Fig. 4 Arrhenius-type plots obtained in the NO + CO reaction over Cu5Pd/Al2O3, Cu/Al2O3, and Pd/Al2O3 catalysts. | ||
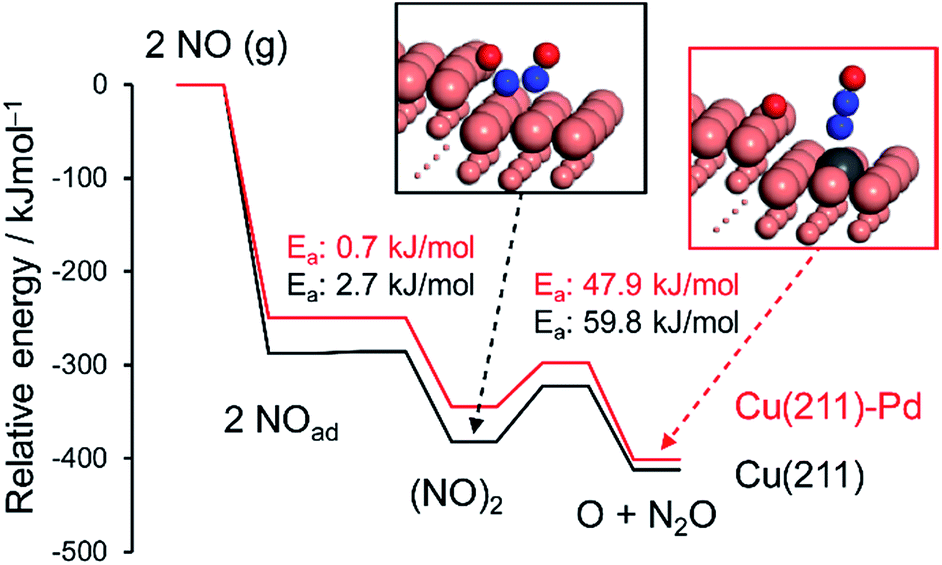 | ||
| Fig. 5 Energy diagrams of NO adsorption, dimerization, and the dimer's decomposition over pure and Pd-doped Cu(211) surfaces. The total energy of the slab and free NO was set to zero. | ||
NO adsorption was weakened by the addition of Pd, which is consistent with the change in the reaction orders (Table 2). Dimerization occurs at the terrace site adjacent to the step site, with a negligible energy barrier. The subsequent N–O scission is triggered by capture of an oxygen atom by the edge Cu atoms, resulting in the formation of an on-top N2O with EA values of 59.8 and 47.9 kJ mol−1 for pure and Pd-doped Cu, respectively. The calculated EA values agree with the experimental values (Table 2). The lower EA for the Pd-doped Cu appears to originate from the destabilized adsorption of the (NO)2 dimer by Pd.
We also considered the CO oxidation process (CO + O → CO2), which is necessary for the catalytic cycle (Fig. S10†). The CO + O reaction over pure and Pd-doped Cu(111) surfaces gave EA values of 60.9 kJ mol−1 and 34.1 kJ mol−1, respectively (Table 2 and Fig. S10†). These values are very similar to or lower than those for N–O scission, which is consistent with the RDS being the scission of N–O. We also simulated the N2O decomposition process (N2O → N2 + O) on Cu(211) and (111) surfaces to understand the intrinsic high N2 selectivity of Cu (Fig. 6 and S11; see S12† for the pictures of the optimized structures).
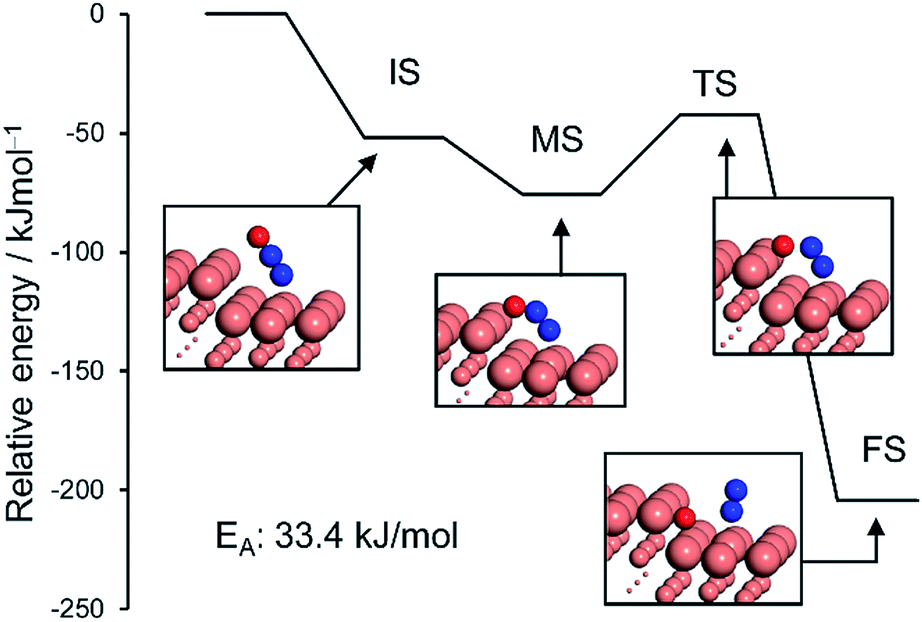 | ||
| Fig. 6 Energy diagrams of N2O bending (initial (IS) to intermediate (MS) states) and its subsequent decomposition to N2 and O (MS to final state (FS)) over the Cu(211) surface. The total energy of slab and free N2O was set to zero. | ||
The monodentate linear N2O was bent to form a bidentate N2O at the edge site of the Cu(211) plane without an energy barrier. The bidentate N2O was subsequently decomposed into N2 and O with a low EA of 33.4 kJ mol−1, indicating that the N2O once formed could be smoothly decomposed into N2 to afford high N2 selectivity. Although the Cu(111) surface was also active for N2O decomposition in a similar fashion, the energy barrier was higher than that of Cu(211) (51.6 kJ mol−1, Fig. S11†). Because large Cu–Cu ensembles are present on the surface of the Cu and Cu-rich catalysts (Cu5Pd and Cu3Pd), N2O decomposition could also be enhanced on these catalysts. However, for CuPd, this effect is limited because of the dilution of Cu–Cu ensembles and the increase of Pd–Pd ensembles. Thus, our calculation rationalized the substantial enhancement in catalytic activity on the basis of the formation of the Cu–Pd single-atom alloy and the origin of the excellent selectivity for N2 formation. The elucidated mechanism differs completely from those proposed for other bimetallic alloy systems. For example, for the Pt–Co system, Sato et al. reported that alloying with Co makes Pt electron-rich, which enhances back donation to adsorbed NO, inducing bond breaking.13 Therefore, Co likely acts as a promoter for Pt. By contrast, in our system, the isolated Pd acts as an efficient promotor for Cu.
Conclusion
We prepared a series of Cu–Pd/Al2O3 catalysts for selective NO reduction at low temperatures. Alloying of Pd with a large amount of Cu (Cu/Pd = 5) isolates Pd and drastically improves both the catalytic activity and N2 selectivity, affording outstanding catalytic performance. In the NO reduction by CO, NO is completely converted to N2 even at 175 °C, with long-term stability for at least 30 h. The high catalytic performance is also achieved in the presence of O2 and C3H6, where the amount of Pd needed for a comparable performance can be reduced to 1/10, with minimum evolution of N2O. For Cu/Al2O3 and Cu5Pd/Al2O3, the N–O bond scission of the (NO)2 dimer is the RDS in NO reduction by CO. This step is kinetically facilitated by the isolated Pd atoms. N2O decomposition to N2 smoothly proceeds on the Cu surface, which contributes to the excellent N2 selectivity observed for Cu and Cu-rich catalysts. The key to this efficient catalysis is the sufficient isolation of Pd atoms by Cu, highlighting the importance of catalyst design based on single-atom alloy structures. The insights gained in this study provide not only a highly efficient deNOx system with substantially reduced noble-metal content, but also open a new path for the chemistry of single-atom alloys.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant Numbers 17H01341 and 17H04965) and by MEXT within the projects “Integrated Research Consortium on Chemical Sciences (IRCCS)” and “Elements Strategy Initiative to Form Core Research Center”, as well as by the JST CREST project JPMJCR17J3. We deeply appreciate Dr H. Asakura of Kyoto University for help with XAFS measurement. We thank the technical staff of the Research Institute for Electronic Science, Hokkaido University for help with HAADF-STEM observation. Computation time was provided by the supercomputer systems at the Institute for Chemical Research, Kyoto University. X-ray absorption measurements were carried out at the BL-14B2 beamline of SPring-8 at the Japan Synchrotron Radiation Research Institute (JASRI; 2018A1757).References
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- B. C. Berks, S. J. Ferguson, J. W. B. Moir and D. J. Richardson, Biochim. Biophys. Acta, Bioenerg., 1995, 1232, 97–173 CrossRef.
- F. Rezaei, A. A. Rownaghi, S. Monjezi, R. P. Lively and C. W. Jones, Energy Fuels, 2015, 29, 5467–5486 CrossRef CAS.
- C. Monn, Atmos. Environ., 2001, 35, 1–32 CrossRef CAS.
- J. H. Wang, H. Chen, Z. C. Hu, M. F. Yao and Y. D. Li, Catal. Rev., 2015, 57, 79–144 CrossRef CAS.
- J. H. Holles, R. J. Davis, T. M. Murray and J. M. Howe, J. Catal., 2000, 195, 193–206 CrossRef CAS.
- A. Srinivasan and C. Depcik, Catal. Rev., 2010, 52, 462–493 CrossRef CAS.
- J. Shibata, M. Hashimoto, K. Shimizu, H. Yoshida, T. Hattori and A. Satsuma, J. Phys. Chem. B, 2004, 108, 18327–18335 CrossRef CAS.
- W. Z. An, Q. L. Zhang, K. T. Chuang and A. R. Sanger, Ind. Eng. Chem. Res., 2002, 41, 27–31 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283–303 CrossRef CAS.
- V. Tomasic, Catal. Today, 2007, 119, 106–113 CrossRef CAS.
- S. Hosokawa, K. Matsuki, K. Tamaru, Y. Oshino, H. Aritani, H. Asakura, K. Teramura and T. Tanaka, Mol. Catal., 2017, 442, 74–82 CrossRef CAS.
- K. Sato, A. Ito, H. Tomonaga, H. Kanematsu, Y. Wada, H. Asakura, S. Hosokawa, T. Tanaka, T. Toriyama and T. Yamamoto, ChemPlusChem, 2019 Search PubMed.
- T. Tanabe, T. Imai, T. Tokunaga, S. Arai, Y. Yamamoto, S. Ueda, G. V. Ramesh, S. Nagao, H. Hirata, S. Matsumoto, T. Fujita and H. Abe, Chem. Sci., 2017, 8, 3374–3378 RSC.
- T. Imai, S. Ueda, S. Nagao, H. Hirata, K. R. Deepthi and H. Abe, RSC Adv., 2017, 7, 9628–9631 RSC.
- J. Jeon, K. Kon, T. Toyao, K. Shimizu and S. Furukawa, Chem. Sci., 2019, 10, 4148–4162 RSC.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- P. Granger, C. Dujardin, J. F. Paul and G. Leclercq, J. Mol. Catal. A: Chem., 2005, 228, 241–253 CrossRef CAS.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65 CrossRef CAS.
- M. J. Hülsey, J. Zhang and N. Yan, Adv. Mater., 2018, 30, 1802304 CrossRef PubMed.
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbruster, A. Knop-Gericke and R. Schlogl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed.
- P. Aich, H. J. Wei, B. Basan, A. J. Kropf, N. M. Schweitzer, C. L. Marshall, J. T. Miller and R. Meyer, J. Phys. Chem. C, 2015, 119, 18140–18148 CrossRef CAS.
- G. X. Pei, X. Y. Liu, A. Q. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. L. Pan, X. F. Yang, X. D. Wang, Z. J. Tai, K. Wilson and T. Zhang, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS.
- X. X. Cao, A. Mirjalili, J. Wheeler, W. Xie and B. W. L. Jang, Front. Chem. Sci. Eng., 2015, 9, 442–449 CrossRef CAS.
- F. R. Lucci, J. L. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2015, 6, 8550 CrossRef PubMed.
- J. L. Liu, J. J. Shan, F. R. Lucci, S. F. Cao, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Catal. Sci. Technol., 2017, 7, 4276–4284 RSC.
- M. D. Marcinkowski, J. L. Liu, C. J. Murphy, M. L. Liriano, N. A. Wasio, F. R. Lucci, M. Flytzani-Stephanopoulos and E. C. H. Sykes, ACS Catal., 2017, 7, 413–420 CrossRef CAS.
- H. Miura, K. Endo, R. Ogawa and T. Shishido, ACS Catal., 2017, 7, 1543–1553 CrossRef CAS.
- P. Bera, K. C. Patil and M. S. Hegde, Phys. Chem. Chem. Phys., 2000, 2, 3715–3719 RSC.
- P. Miguel, P. Granger, N. Jagtap, S. Umbarkar, M. Dongare and C. Dujardin, J. Mol. Catal. A: Chem., 2010, 322, 90–97 CrossRef.
- H. Iwamoto, S. Kameoka, Y. Xu, C. Nishimura and A. P. Tsai, J. Phys. Chem. Solids, 2019, 125, 64–73 CrossRef CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, Comput. Mater. Sci., 2003, 28, 250–258 CrossRef CAS.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- W. K. Kuhn, J. Szanyi and D. W. Goodman, Surf. Sci., 1992, 274, L611–L618 CrossRef CAS.
- O. Dulaurent, X. Courtois, V. Perrichon and D. Bianchi, J. Phys. Chem. B, 2000, 104, 6001–6011 CrossRef CAS.
- S. Zhou, B. Varughese, B. Eichhorn, G. Jackson and K. McIlwrath, Angew. Chem., Int. Ed., 2005, 44, 4539–4543 CrossRef CAS PubMed.
- M. Fernández-Garcia, A. Martinez-Arias, C. Belver, J. Anderson, J. Conesa and J. Soria, J. Catal., 2000, 190, 387–395 CrossRef.
- F. Gao, Y. Wang and D. W. Goodman, J. Catal., 2009, 268, 115–121 CrossRef CAS.
- T. Hirano, Y. Ozawa, T. Sekido, T. Ogino, T. Miyao and S. Naito, Catal. Commun., 2007, 8, 1249–1254 CrossRef CAS.
- A. Hungrıa, A. Iglesias-Juez, A. Martınez-Arias, M. Fernández-Garcıa, J. Anderson, J. Conesa and J. Soria, J. Catal., 2002, 206, 281–294 CrossRef.
- T. Komatsu, H. Kobayashi, K. Kusada, Y. Kubota, M. Takata, T. Yamamoto, S. Matsumura, K. Sato, K. Nagaoka and H. Kitagawa, Chem.–Eur. J., 2017, 23, 57–60 CrossRef CAS.
- A. Hungría, M. Fernández-García, J. Anderson and A. Martínez-Arias, J. Catal., 2005, 235, 262–271 CrossRef.
- D. R. Rainer, S. M. Vesecky, M. Koranne, W. S. Oh and D. W. Goodman, J. Catal., 1997, 167, 234–241 CrossRef CAS.
- W. Brown, R. Sharma, D. King and S. Haq, J. Phys. Chem., 1996, 100, 12559–12568 CrossRef.
- A. Bogicevic and K. Hass, Surf. Sci., 2002, 506, L237–L242 CrossRef CAS.
- N. Takagi, K. Ishimura, H. Miura, T. Shishido, R. Fukuda, M. Ehara and S. Sakaki, ACS Omega, 2019, 4, 2596–2609 CrossRef CAS.
- R. Burch, S. Daniells and P. Hu, J. Chem. Phys., 2004, 121, 2737–2745 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of characterization, kinetic analysis, and DFT calculations. See DOI: 10.1039/c9sc03172c |
| This journal is © The Royal Society of Chemistry 2019 |