
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective access to either a doubly boron-doped tetrabenzopentacene or an oxadiborepin from the same precursor†
Julian
Radtke
a,
Kai
Schickedanz
a,
Marcel
Bamberg
a,
Luigi
Menduti
 a,
Dieter
Schollmeyer
b,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a and
Matthias
Wagner
*a
a,
Dieter
Schollmeyer
b,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a and
Matthias
Wagner
*a
aInstitut für Anorganische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Strasse 7, D-60438 Frankfurt (Main), Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bInstitute of Organic Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany
First published on 31st July 2019
Abstract
The well-known red emitter tetrabenzo[de,hi,op,st]pentacene (TBPA) has been transformed into a bright blue emitter (B2-TBPA; λem = 472 nm; c-hexane) via substitutional doping with two boron atoms. In contrast to the electron-rich TBPA, which forms endo-peroxides with O2 under daylight, the benchtop-stable B2-TBPA is a good electron acceptor and undergoes reversible reduction at a moderate half-wave potential of E1/2 = −1.73 V (vs. FcH/FcH+; THF). Although the size of B2-TBPA falls within the nanoscale, the helically twisted compound readily dissolves in c-hexane and does not require solubilizing substituents. The synthesis of B2-TBPA is based on the nickel-mediated Yamamoto-type dehalogenation of tetrabrominated 9,10-di(naphth-1-yl)-9,10-dihydro-9,10-diboraanthracene. This intramolecular C–C heterocoupling reaction shows a remarkable solvent dependence: B2-TBPA forms only in pyridine (79% yield), whereas an oxadiborepin is obtained from THF solutions (ODBE, 81%; the reaction mixture is quenched with air in both cases). Insight into the corresponding reaction mechanism was gained from the isolation of intermediates and an investigation of their chemical properties. ODBE is an interesting blue emitter in its own right. Furthermore, it can be ring-opened with excess BBr3 at the B–O–B moiety to afford a dimeric borabenzo[de]anthracene.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous building blocks for organic optoelectronic materials.1 Tuning of their frontier orbital energies, and, in turn, their light absorption/emission characteristics or redox properties, can be achieved by varying the degree of annulation, the edge structure, and the pattern of peripheral substituents.2 A fourth powerful tool – arguably the synthetically most challenging one – is substitutional doping, i.e., the replacement of selected C(sp2) atoms within the PAH scaffold by other p-block atoms.3 Among those, boron is a particularly useful electronically perturbative element, as it renders the resulting B-PAHs electron deficient and moderately Lewis acidic.4A fruitful strategy by which to obtain a deeper understanding of structure–property relationships in B-PAHs and to identify promising new lead structures is to compare PAHs of proven applicability directly with their B-doped counterparts. Given that a B atom possesses one valence electron less than a C atom, the transition from a stable closed-shell PAH to a closed-shell B-PAH requires the incorporation of an even number of B atoms. Examples include doubly B-doped acenes,5 pyrene,6 perylene,7–9 or bisanthene.10
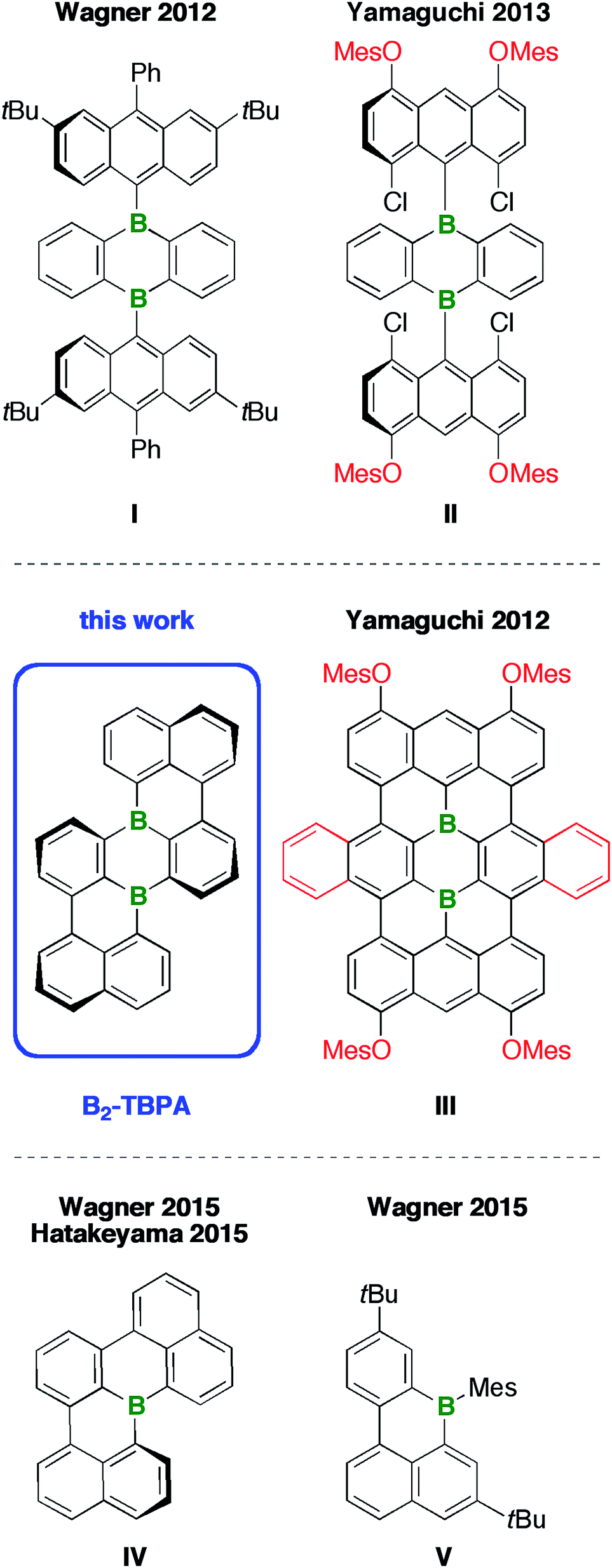
The bottom-up preparation of even larger structures, in the size regime of nanographene flakes, becomes increasingly difficult, mainly because of solubility issues and a lack of broadly applicable synthetic protocols. These problems can be illustrated by considering Yamaguchi's approach to the diborateranthene derivative III (Chart 1):11 Initial attempts to assemble the teranthene framework via oxidative cyclodehydrogenation of B-containing tri(9,10-anthrylene)s such as I (intramolecular Scholl reaction12) gave inseparable mixtures of unidentified products.13,14 The introduction of o,p-directing mesityloxy substituents15 at both 9-anthryl moieties resulted in an activation of four of the eight relevant C–H groups (exc FeCl3 in a CH3NO2/CH2Cl2 solvent mixture). However, chlorination rather than cyclization took place (cf.II).16 Only when four peripheral MesO groups and two extra annulated o-phenylene rings were simultaneously present at the anthryl fragments and the heterocyclic core, respectively, did the Scholl reaction proceed smoothly to afford the fully cyclized III. The purple-colored compound absorbs in the entire visible range of the electromagnetic spectrum and shows weak fluorescence across the visible/near-IR region.11 Remarkable turn-on fluorescence was observed upon exposure of III to various Lewis bases, among them NH3, indicating a potential use of this B-PAH as gas-sensor material.17 Moreover, prototype Li-ion batteries containing derivatives of III as electrodes have been fabricated. In this context, a conflict between the requirement for solubilizing side chains during the synthesis and purification of III and the obstructive impact of these substituents on the device performance emerged: large MesO groups led to a leaking of the electrode material into the battery electrolyte and decreased the specific capacity of the battery. Smaller n-BuO substituents resulted in lower yields of the B-PAH, because its poor solubility in common organic solvents made the purification procedure more laborious.17 Similar problems occur when organic light-emitting devices (OLEDs) or organic field-effect transistors (OFETs) are to be fabricated, because large, dangling substituents prevent tight packing of the materials in the solid state and thus reduce the extent of π–π interactions between the molecules.
 | ||
| Chart 1 Previously reported examples of B-doped PAHs (Mes = mesityl). | ||
Given this background, we decided to develop alternative routes to access soluble, benchtop-stable, nanoscale B-PAHs. The aim is to maintain the remarkable and useful properties of compounds such as III, but to have more possibilities for derivatization18 and avoid solubilizing or kinetically protecting substituents. In the new compounds, the two B atoms should still constitute integral parts of the PAH scaffold such that structural constraint19,20 protects them from adduct formation with O2/H2O and further shielding with bulky substituents is not required. A positive effect on the solubility of the B-PAH should be brought about by inducing a helical twist21 rather than by adding organyl substituents (cf. the hexane-soluble [4]helicene IV22vs. the planar B-PAH V;23,24Chart 1). As Scholl-type oxidative dehydrogenation reactions are usually associated with forcing conditions, milder and more selective reductive dehalogenation protocols, such as the Yamamoto reaction,25,26 will be applied (cf. our syntheses of IV22 and related twisted borepins18). As our specific target molecule, we selected compound B2-TBPA (Chart 1), a doubly B-doped congener of tetrabenzo[de,hi,op,st]pentacene (TBPA; cf.Chart 2). Pentacenes, in general, are among the most important compounds for organic electronics,27,28 and TBPA, in particular, has already been employed as a red emitter in OLEDs29,30 and also as an OFET material.31–33 Furthermore, TBPA has a low ionization potential34,35 and the two C atoms located at the cusps of both cove regions36 accumulate the largest HOMO electron density.37 B-Doping at these positions, as in the case of B2-TBPA, is therefore expected to have a profound influence on the optoelectronic properties of the molecule. Apart from being a pentacene derivative, B2-TBPA can be viewed as consisting of two diboraperylenes sharing the central B2C4 ring (Chart 2). B2-TBPA also contains two fragments resembling the [4]helicene IV, which has already proven its suitability as an emitter in a B-PAH-based OLED device.38,39 Last but not least, the B atoms formally divide the large π system of B2-TBPA into two smaller π systems of type V.
 | ||
| Chart 2 Substructures included in B2-TBPA. | ||
Herein, we report a high-yield synthesis of B2-TBPA, starting from readily available tetrahalogenated 9,10-di(naphth-1-yl)-9,10-dihydro-9,10-diboraanthracene precursors, by adapting the Yamamoto protocol. We also disclose the surprising result that a simple change of solvent from pyridine to THF changes the major product from B2-TBPA to an oxadiborepin ODBE (the reaction mixture is quenched with air in both cases). A mechanistic scenario rationalizing the underlying rearrangement reaction is proposed. Finally, we discuss key optoelectronic properties of B2-TBPA and ODBE in comparison to those of the reference systems TBPA, IV, and V.
Results and discussion
Syntheses
For the synthesis of the B-doped multiple helicene B2-TBPA, we started from the tetrahalogenated 9,10-dihydro-9,10-diboraanthracene (DBA) derivatives 1x, which are readily available from the corresponding known borinic acids and 6 equiv. of BBr3 (Scheme 1; cf. the ESI for full details).40 Treatment of 1x with 2 equiv. of (8-halogenonaphth-1-yl)lithium41,42 furnished syn/anti-isomer mixtures of 2X,Y in yields between 60% (2F,Br) and 89% (2Br,I). The compounds are benchtop-stable and can be purified by column chromatography; a separation of syn/anti-2X,Y, however, was not possible. | ||
| Scheme 1 (a) Synthesis of the 9,10-bis(8-halogenonaphth-1-yl)-DBAs 2X,Y. (b) Structure of syn-2Br,I in the solid state (C atoms: black, B atoms: green, Br atoms: brown, I atoms: purple; H atoms omitted for clarity).43 Reagents and conditions: (i) 2 equiv. of (8-halogenonaphth-1-yl)lithium, Et2O, 0 °C to rt. | ||
Our first attempt at forming the last two C–C bonds on the way from 2X,Y to B2-TBPA relied on an Li/I exchange at the naphthyl substituents of 2F,I and a subsequent intramolecular nucleophilic attack at the two C–F functionalities. Reactions of this type are well established for various fluoroarenes.44,45 In the particular case of 2F,I, the cyclization reaction should be facilitated by the fact that the B–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–F fragment is a Michael-type system.
C–F fragment is a Michael-type system.
However, 2F,I proved largely inert toward stoichiometric quantities of n-BuLi (2 equiv., Et2O, −78 °C to rt, 16 h; 83% of 2F,I re-isolated), but underwent complete degradation in the presence of excess lithiating reagent (4 equiv., Et2O, −78 °C to rt, 16 h; 74% of naphthalene isolated). A similar reaction outcome was observed for 2F,Br. Therefore, we modified the approach and treated 1F directly with 2 equiv. of 1,8-dilithionaphthalene. A complex reaction mixture was obtained from which the B-doped [4]helicene IV (Chart 1) was isolated in a yield of 11%. Even though the presence of C(Ph)–C(Naph) bonds in IV indicates that the desired nucleophilic substitution of F substituents is possible in principle, the lack of a selective conversion demanded a fundamentally different synthetic strategy for B2-TBPA.
We next focused on 2Br,Br/2Br,I and the Yamamoto-type C–C-coupling protocol that had successfully been applied to the targeted preparation of IV (1 equiv. of [Ni(COD)2]/bpy/COD per C–Br unit, rt, high dilution; COD = 1,5-cyclooctadiene, bpy = 2,2′-bipyridyl).22 The formation of paramagnetic Ni species in the course of the reaction precluded the NMR-spectroscopic monitoring of its progress. Thus, we routinely quenched the reaction mixtures after approximately 20 h by bubbling air through the vessel to precipitate all Ni-containing compounds (attempts at a workup under inert conditions were unsuccessful; cf. the ESI† for more details).
After filtration, the filtrates were free of paramagnetic impurities and suitable for NMR-spectroscopic investigations: 2Br,Br or 2Br,I were reproducibly converted to only one major product; however, the nature of this product depended heavily on the choice of solvent (Scheme 2). THF solutions, which are common media for Yamamoto reactions,46,47 contained ODBE instead of the multiple helicene B2-TBPA after quenching. The two C(Ph)–C(Naph) bonds in ODBE point toward successful debromination/C–C coupling, but the reaction also involved the formation of a third C–C bond and the cleavage of two B–C bonds. After replacement of THF with pyridine, an atypical solvent for Yamamoto-type transformations, B2-TBPA was isolated in high yields, whereas ODBE was not detected.
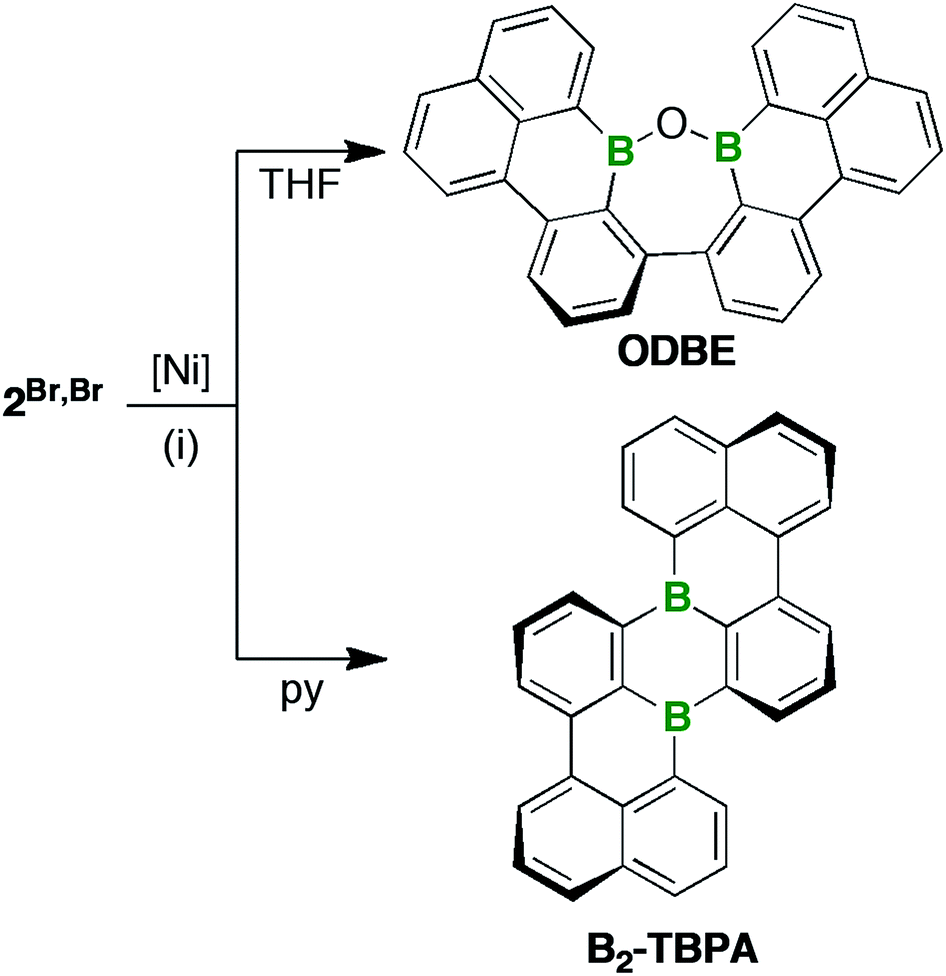 | ||
| Scheme 2 Synthesis of ODBE (in THF) and B2-TBPA (in pyridine) starting from 2Br,Br. Reagents and conditions: (i) 4 equiv. of [Ni(COD)2]/bpy/COD, rt, high dilution; COD = 1,5-cyclooctadiene, bpy = 2,2′-bipyridyl, py = pyridine. | ||
ODBE can be regarded as the intramolecular anhydride of two borinic acid moieties. Therefore, we explored the possibility of opening the ring through hydrolysis of the B–O–B bridge and, indeed, observed the emergence of an additional set of signals in the in situ1H-NMR spectrum (CDCl3 and two drops of added H2O; NMR tube, rt; cf. Fig. S3–46†). The number of these signals was the same as in the case of ODBE, but the chemical shift values differed slightly. After some time, the integral ratio of both sets of signals remained constant, which can be explained by assuming a dynamic equilibrium between ODBE and its open-chain hydrolysis product. In an attempt to cleave the B–O–B link quantitatively, we next treated ODBE with 5 equiv. of BBr3 (CDCl3; sealed NMR tube, 140 °C, 2 d). This time, the signals of ODBE completely vanished and new signals assignable to the ditopic bromoborane 3 appeared (Scheme 3; cf. the ESI† for the NMR data); the addition of H2O quantitatively reverted 3 to ODBE.
 | ||
| Scheme 3 Synthesis of 3 through treatment of ODBE with BBr3. Reagents and conditions: (i) 5 equiv. of BBr3, CDCl3, 140 °C, 2 d. | ||
NMR-spectroscopic and X-ray crystallographic characterization of 2X,Y, B2-TBPA, and ODBE
Each of the four derivatives 2X,Y gives rise to two similar sets of resonances in the 1H- as well as the 13C{1H}-NMR spectrum. All of these signals can be assigned to tetrahalogenated 9,10-bis(naphth-1-yl)-DBAs in which both phenylene halves and both naphthyl substituents are related by symmetry. Compounds 2X,Y thus exist as mixtures of syn- and anti-isomers, which do not interconvert due to restricted rotation about the exocyclic B–C bonds.14 One of these isomers (likely syn)43 dominates by a factor of 10 in the sterically most encumbered 2Br,I and by a factor of 2 in the second most congested derivative 2Br,Br. Samples of 2F,Br or 2F,I contain the syn- and anti-isomers in essentially equimolar distributions.The 11B{1H}-NMR spectra of 2X,Y are characterized by broad resonances with chemical shift values between 50 and 54 ppm. The 11B nuclei are therefore shielded by approximately 20 ppm compared to those of 9,10-bis(2-methylnaphth-1-yl)-DBA (71 ppm).14 This shielding effect likely results from (weak) intramolecular B⋯Y adduct bonds,16 which are also indicated by X-ray diffraction analysis of 2Br,I. Scheme 1b shows the solid-state structure of syn-2Br,I.43 Its B⋯I contacts of 2.78(1) and 2.82(1) Å are more than 1 Å shorter than the sum of the corresponding van der Waals radii (3.90 Å)48 and both boron centers are slightly pyramidalized (Σ(C–B–C) = 354.7 and 356.7°).
The successful dehalogenative cyclization to furnish B2-TBPA is evidenced by the following findings: (i) instead of the two signal sets characteristic of syn/anti-2Br,Br, only one set of resonances remains in the 1H- as well as 13C{1H}-NMR spectrum of B2-TBPA (this finding is also in line with the very low inversion barrier expected for the two planar-chiral [4]helicene fragments,49,50 which should otherwise give a pair of diastereomers); (ii) the numbers of 1H- and 13C-NMR signals are only compatible with a highly symmetric structure, thereby ruling out the possibility of just one C–C-coupling event; (iii) Br–C(ipso) resonances of syn/anti-2Br,Br at 135.2 and 126.8 ppm are absent in the 13C{1H}-NMR spectrum of B2-TBPA, whereas downfield-shifted signals at 140.5 and 132.7 ppm are present (assignable to the quaternary carbon atoms at the newly formed C–C bonds).
The molecular structure of B2-TBPA was confirmed by X-ray crystallography (Fig. 1b). B2-TBPA possesses C2 symmetry and two different kinds of axial chiral units: a cylindrical spiral substructure with its symmetry-related congener of the same helicity51 (cf. the blue screw axes) and one helical pentacene ribbon (cf. the red screw axis).52 Starting from the aromatic ring containing C(15), the five annulated rings contribute absolute values of 3.8(4), 12.4(3), 17.8(5), 12.4(3), and 3.8(4)° to the overall absolute twist of 50.2(5)°.53 The solid-state structures of B2-TBPA and TBPA are very much alike, apart from two significant differences: the end-to-end twist in the carbonaceous TBPA (40.9°; Fig. 1a)54 is less pronounced by almost 10°, which proves benzene rings to be more rigid than their B-doped counterparts. In a similar vein, the B–C bond lengths of B2-TBPA (1.540(6)–1.572(7) Å) are markedly longer than the corresponding C–C bonds of TBPA (1.404(5)–1.480(5) Å). The [4]helicene fragments of B2-TBPA and TBPA show essentially the same degree of distortion (skew angle between the C(1)–C(6) and C(11)–C(20) vectors = 38.0° and 39.4°, respectively). Monohelicene IV is remarkably less twisted (21.0°),22,38 which leads to the conclusion that the two helical units present in B2-TBPA (and TBPA) reinforce each other.
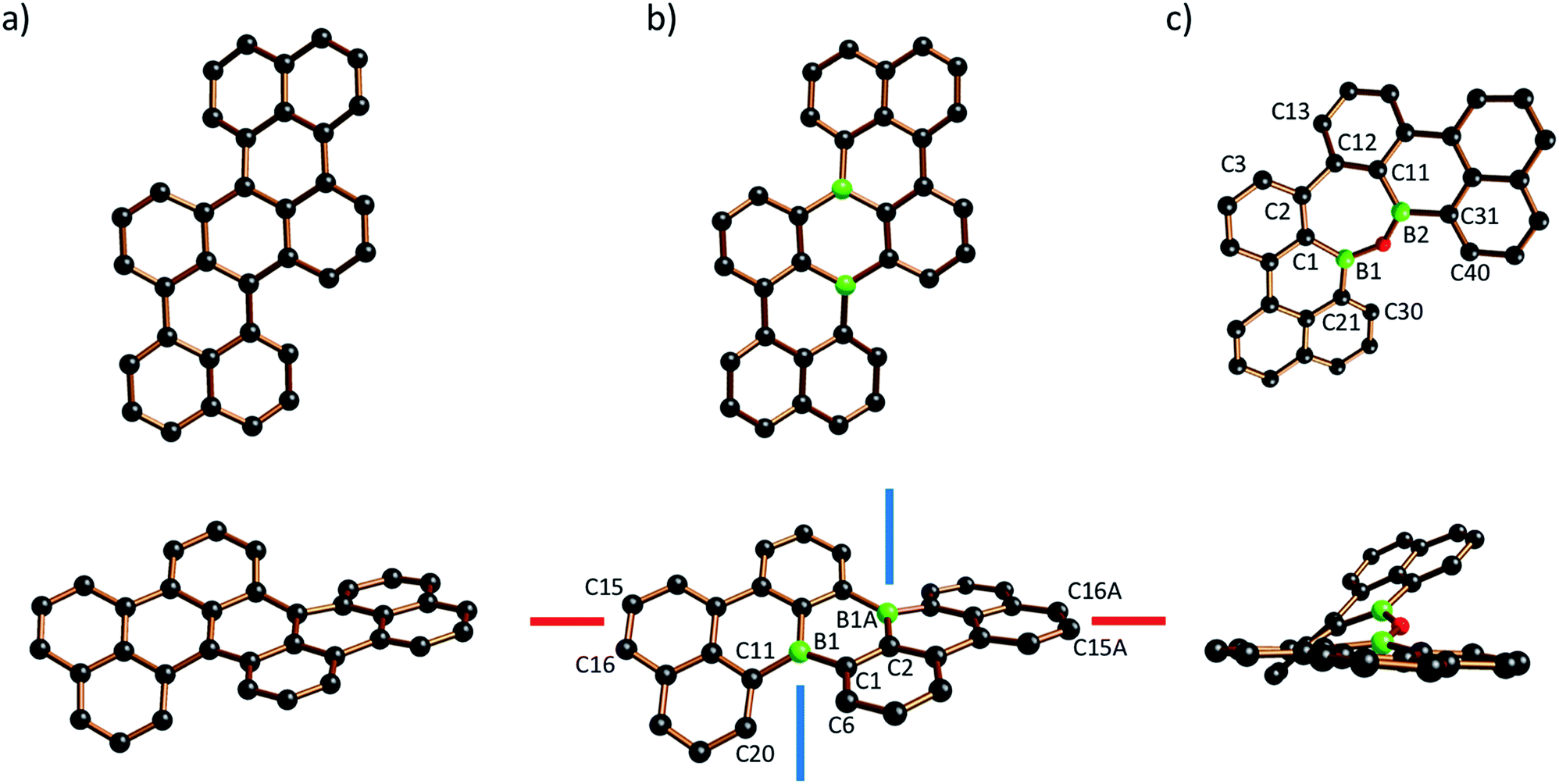 | ||
| Fig. 1 Two views of the crystallographically determined molecular structures of TBPA (a; CCDC 1216750, ref. 54), B2-TBPA (b), and ODBE (c) in the solid state (C atoms: black, B atoms: green, O atoms: red; H atoms omitted for clarity). Selected bond lengths (Å), bond angle (°), and torsion angles/skew angle between vectors (°): B2-TBPA: B(1)–C(1) = 1.572(7), B(1)–C(11) = 1.548(6), B(1)–C(2A) = 1.540(6); C(6)–C(1)–C(11)–C(20) = 38.0. ODBE: B(1)–C(1) = 1.567(13), B(1)–C(21) = 1.458(16), B(1)–O(1) = 1.418(13), B(2)–C(11) = 1.496(15), B(2)–C(31) = 1.564(14), B(2)–O(1) = 1.382(14); B(1)–O(1)–B(2) = 134.8(10); C(30)–C(21)–C(31)–C(40) = 33.3(10), C(1)–B(1)–B(2)–C(11) = 25.0(10), C(3)–C(2)–C(12)–C(13) = 37.4(16). | ||
The central benzene ring of TBPA is deformed to a boat conformation with an average flip angle of 14.9° between the bow/stern and the plane defined by the other four C atoms. It has been suggested that this deformation governs the reactivity of TBPA toward the electrophilic O2 molecule,54 which, under daylight, forms an endo-peroxide connecting the bow/stern C atoms.37 The B2C4 core of the boron isoster B2-TBPA also adopts a boat conformation, such that it should similarly be possible to bridge the B atoms at the bow/stern by a diatomic unit (flip angle = 17.1°). Given that B2-TBPA is isoelectronic with the TBPA2+ dication, suitable reaction partners would be ditopic Lewis bases, rather than electrophiles (cf. the triptycene-type adducts between DBA and 1,2-diacenes).55,56 Despite numerous efforts, we did not succeed in the crystallization of corresponding tricyclic scaffolds from B2-TBPA/phthalazine mixtures, even though titration experiments using DMAP showed B2-TBPA to possess considerable Lewis acidity (see below).
The 1H- and 13C{1H}-NMR spectra of B2-TBPA and ODBE are deceptively similar. Yet, differences became apparent for the chemical shift values of the C–H vectors pointing into the cove regions of B2-TBPA and ending up in bay and gulf regions36 of ODBE. As a consequence of π-donation from the O bridge, the B nuclei of ODBE are more shielded by 13 ppm than those of B2-TBPA (δ(11B) = 43 vs. 56 ppm).
The solid-state structure of ODBE adopts C1 symmetry (Fig. 1c). The two B atoms are not directly attached to each other but linked through an O atom to afford a seven-membered oxadiborepin heterocycle. The B–O bond lengths amount to 1.382(14)/1.418(13) Å and the B–O–B bond angle is 134.8(10)°. ODBE differs significantly from planarity with absolute torsion angles of C(30)–C(21)–C(31)–C(40) = 33.3(10)° (gulf twist), C(1)–B(1)–B(2)–C(11) = 25.0(10)° (endocyclic twist), and C(3)–C(2)–C(12)–C(13) = 37.4(16)° (bay twist). A doubly benzannulated oxadiborepin model system, i.e., the intramolecular anhydride of 2,2′-bis(dihydroxyboryl)-1,1′-biphenyl, shows an even higher gulf (46.1(3)°) and endocyclic twist (39.6(2)°), whereas the bay twist remains largely the same (36.6(2)°).57,58 If the two annulated benzene rings of the model system are replaced with five-membered thiophene rings in a way that places the sulfur atoms at the opening of the bay region, the resulting oxadiborepin relaxes to a largely planar conformation.59
To assess the qualities of ODBE as a potentially ditopic Lewis acid, we grew crystals from an equimolar ODBE/phthalazine mixture. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct was obtained, in which, however, the phthalazine molecule acts only as monodentate N ligand (B(1)–N(1) = 1.686(14) Å, B(2)⋯N(2) = 2.945(15) Å; see the ESI for more details†).
:1 adduct was obtained, in which, however, the phthalazine molecule acts only as monodentate N ligand (B(1)–N(1) = 1.686(14) Å, B(2)⋯N(2) = 2.945(15) Å; see the ESI for more details†).
Optoelectronic properties of B2-TBPA and ODBE
The doubly B-doped tetrabenzo[de,hi,op,st]pentacene B2-TBPA and the oxadiborepin ODBE have been investigated by cyclic voltammetry (THF, [n-Bu4N][PF6], 0.1 M, 200 mV s−1, vs. FcH/FcH+). While the carbonaceous TBPA is electron-rich,34B2-TBPA undergoes a reversible one-electron reduction at a moderately cathodic half-wave potential of E1/2 = −1.73 V (Table 1). Electron injection into the reference compounds IV or V (Chart 1) requires more cathodic half-wave potentials of E1/2 = −1.87 (our measurement) and −2.11 V,23 respectively (Table 1). The differences between the monoboranes IV/V and B2-TBPA reflect the impact of the second electron-deficient B atom as is further corroborated by the increasingly cathodic E1/2(B2-TBPA) values in solvents of increasing Lewis basicities: −1.65 (CH2Cl2) < −1.73 (THF) < −2.16 V (pyridine). As outlined below, B2-TBPA tends to form monoadducts with pyridine donors, which basically eliminates the contribution of the tetracoordinate B atom to π-electron delocalization and renders the redox potential of B2-TBPA·pyridine similar to that of a borabenzo[de]anthracene moiety (Charts 1 and 2). Contrary to B2-TBPA, ODBE shows only one irreversible redox wave in its cyclic voltammogram, the cathodic peak potential of which is very similar to the half-wave potential of V (Epc = −2.19 V vs. E1/2 = −2.11 V).| λmax [nm] | λem [nm] | Φ PL [%] | Stokes shiftb [cm−1] | E 1/2 [V] | E G opt [eV] | E LUMO [eV] | |
|---|---|---|---|---|---|---|---|
| a Quantum yields were determined using a calibrated integrating sphere. b Stokes shifts represent the difference between each longest wavelength absorption maximum and the corresponding shortest wavelength emission maximum. c Optical band gap EGopt = 1240/λonset. λonset was determined by constructing a tangent on the point of inflection of the bathochromic slope of the most red-shifted absorption maximum. d E LUMO = −4.8 eV − E1/2Red1 (FcH/FcH+ = −4.8 eV vs. vacuum level). e c-Hexane. f C6H6. g CH2Cl2. h THF. i Pyridine. j Ref. 30. k Ref. 22. l Our measurements. m Ref. 23. n E pc value of an irreversible reduction event. | |||||||
| B2-TBPA | 456e | 472e | 69e | 719 | — | 2.63e | — |
| 462f | 484f | 65f | 984 | — | 2.58f | — | |
| 459g | 484g | 66g | 1125 | −1.65g | 2.60g | −3.15g | |
| 465h | 481h | 35h | 715 | −1.73h | 2.57h | −3.07h | |
| 354i | — | — | — | −2.16i | — | −2.64i | |
| TBPA | 628f | 690f | — | 1431 | — | — | — |
| IV | 462e | 485e | 81e | 1026 | −1.81g,l | 2.60e | −2.99g |
| −1.87h,l | −2.93h | ||||||
| V | 409e | 431e | 80e | 1248 | −2.11h | 2.88e | −2.69h |
| ODBE | 392e | 411e | 45e | 1179 | −2.19h,n | 2.55e | — |
TBPA is a blue compound, showing red to near-infrared photoluminescence (λmax = 628 nm, λem = 690 nm in C6H6; Table 1).30 Its analogue, B2-TBPA, in contrast, possesses a yellow color and emits in the blue region of the electromagnetic spectrum with an appreciable quantum efficiency of ΦPL = 69% (λmax = 456 nm, λem = 472 nm in c-hexane; Fig. 2a). In line with the rigid scaffold of B2-TBPA, its Stokes shift is small (719 cm−1) and the electronic spectra are vibrationally resolved. The absorption spectrum remains essentially the same upon going from c-hexane to C6H6 to CH2Cl2. This picture changes when donor solvents are used: in THF, the lower-energy band loses intensity with respect to the higher-energy band, and only the latter remains in pyridine (Fig. 2b). The solvatochromism of the fluorescence band in the solvent range from c-hexane to THF is also negligible (±10 nm), whereas pyridine quenches the emission. These observations agree with the results obtained from cyclic voltammetry, which indicated that THF is not completely innocent toward B2-TBPA and that pyridine can act as a ligand. To shed more light on the properties of B2-TBPA as a Lewis acid, we performed a titration with 4-dimethylaminopyridine (DMAP; Fig. 2c). During the addition of 0.2–1.0 equiv. of DMAP, the absorption at 462 nm gradually became smaller, but did not completely vanish. A full quenching was achieved after the addition of approximately 2 equiv. of DMAP; apart from that, no further qualitative changes occurred and two isosbestic points at 328 and 370 nm are present in the entire range between 0 and 2 equiv. of added DMAP. Thus, B2-TBPA still behaves as a Lewis acid, but only monoadducts B2-TBPA·DMAP seem to be formed, even with the strong donor DMAP. A second series of titrations, using [n-Bu4N]F as the source of Lewis-basic F− ions, gave analogous results (cf. the ESI†).
 | ||
| Fig. 2 (a) Normalized UV-vis absorption and emission spectra of B2-TBPA in c-hexane. (b) UV-vis absorption spectra of B2-TBPA in c-hexane, THF, and pyridine at the same concentration of 10−4 M. (c) UV-vis absorption spectra of B2-TBPA in the presence of various amounts of 4-dimethylaminopyridine (DMAP) in C6H6 at the same concentration of 10−4 M. | ||
A comparison of B2-TBPA with the reference systems IV and V reveals the [4]helicene to possess very similar absorption and emission characteristics, whereas both λmax and λem of the 7H-7-borabenzo[de]anthracene are hypsochromically shifted (Table 1). In particular, the differences between B2-TBPA and V demonstrate that the two halves of B2-TBPA are electronically coupled to an appreciable degree. In contrast, ODBE absorbs and emits further to the blue than B2-TBPA and even V. This hypsochromic shift is likely due to the electron-donating effect of the oxygen atom, which increases the HOMO–LUMO energy difference of ODBE with respect to B2-TBPA (cf. Table S6-5†). Furthermore, there is apparently little electronic communication across the B–O–B fragment or the twisted C–C bond of ODBE.
Mechanistic considerations
As outlined above, the reaction between 1F and 2 equiv. of 1,8-dilithionaphthalene gave a complex product mixture, from which the B-doped [4]helicene IV was isolated in 11% yield. Three interconvertible modes of interaction, all of which are potentially relevant for the formation of IV, are conceivable between 1,8-dilithionaphthalene and the ditopic Lewis acid 1F (Scheme 4): the ligand can bind in a monodentate fashion such that one C–Li functionality remains uncoordinated (VIa), or in a chelating mode to create a four-membered BC3 ring (VIb), or in a B⋯B-bridging manner to furnish a tricyclic, triptycene-type system (VIc). Precedence for the proposed structural motifs of VIb and VIc exists in the form of the dimesityl-1,8-naphthalenediylborate anion60,61 and several diacene- or carboxylate-bridged DBAs55,56,62 that have been crystallographically characterized. The adduct VIa could undergo LiBr elimination and C–F substitution to afford the intermediate VII, which adds a second equivalent of 1,8-dilithionaphthalene to give the tricyclic compound VIII with two tetracoordinate B centers. It has recently been disclosed that certain DBA-based tetraarylborates are prone to reductive elimination with formation of biaryls and formal extrusion of diarylboryl anions (which subsequently stabilize themselves through intra- or intermolecular insertion reactions).63 A similar scenario involving the B atom ortho to the F substituent would establish the required C–C bond between the red and blue fragments. Subsequent cleavage of the only remaining exocyclic B–C bond (at the latest during chromatographic workup) opens a way to IV.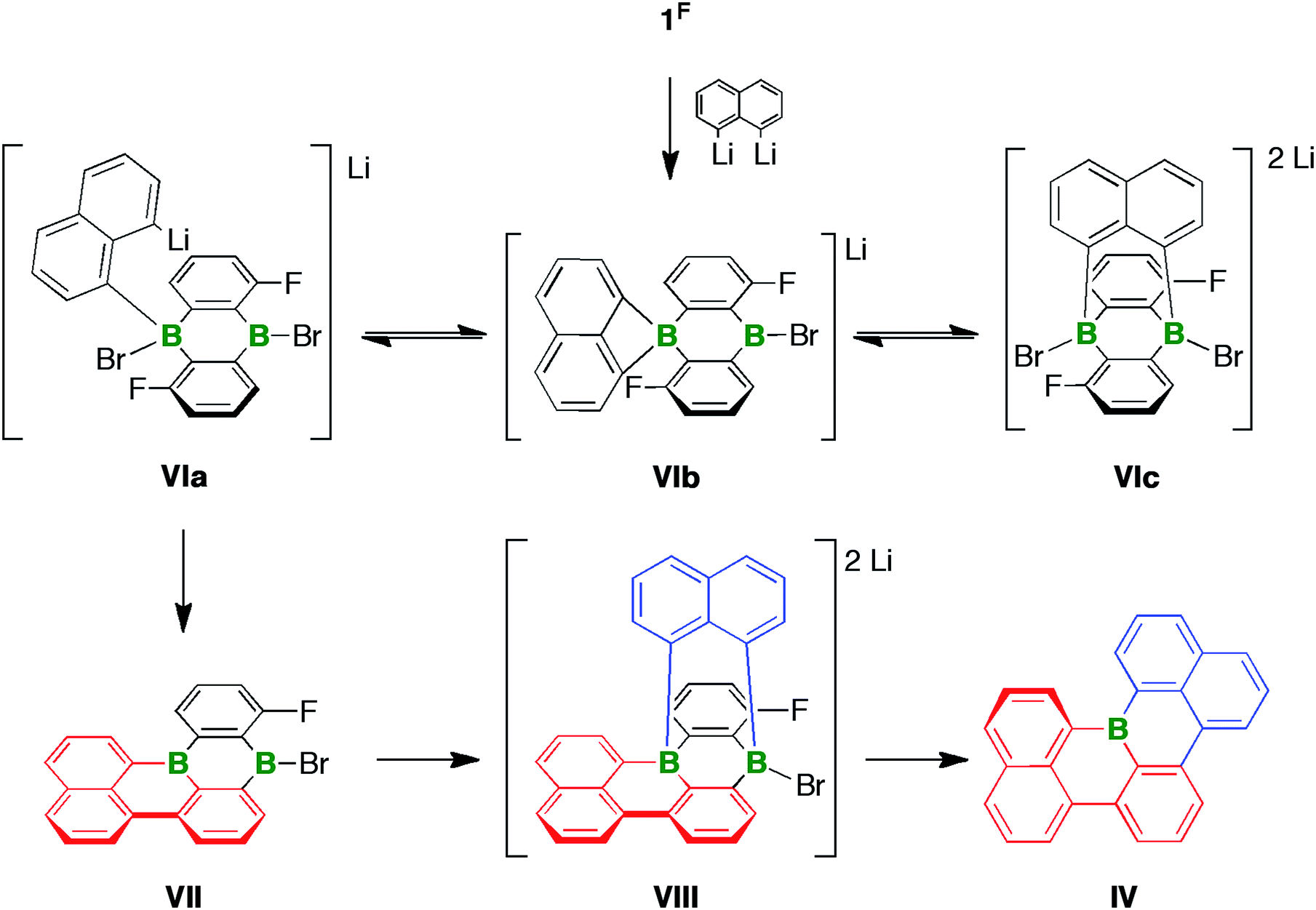 | ||
| Scheme 4 Three modes of interaction (VIa–VIc) between 1,8-dilithionaphthalene and the ditopic Lewis acid 1F. Formation of the [4]helicene IVvia the proposed intermediates VII and VIII. | ||
The Yamamoto-coupling reaction of 2Br,Br is remarkably solvent dependent, as it selectively leads to either B2-TBPA or ODBE when carried out in pyridine or THF, respectively. The mechanistic pathways underlying these transformations are difficult to elucidate, because even elementary steps of Ni-mediated Yamamoto-coupling reactions are still subject to debate and strongly influenced by the nature of the substrate and the choice of solvent.64 Originally, Semmelhack proposed two sequential oxidative addition steps, ultimately leading from one mononuclear [Ni0] complex and two aryl halides Ar–X to species of the form [Ar2NiIVX2]; subsequent reductive elimination gives the desired biaryl Ar–Ar and [NiIIX2] (in DMF).65 Tsou and Kochi suggested a [NiI]/[NiIII] radical-chain scenario for the coupling process (mainly in nonpolar solvents).66,67 Specifically for the cases of [(bpy)Ni(COD)]-mediated Ar–X coupling reactions, Yamamoto identified complexes [(bpy)NiII(X)Ar] as primary intermediates, which dismutate in the rate-determining step via dinuclear aggregates to give [(bpy)NiIIAr2] and [(bpy)NiIIX2]. Reductive elimination from [(bpy)NiIIAr2] then regenerates 1 equiv. of [(bpy)Ni0] and produces Ar–Ar (in DMF).25 None of these scenarios, however, accounts for empirical observations made by us and others,18,22,68–70 namely, that the addition of excess COD ligand and of 2 equiv. rather than 1 equiv. of [Ni(COD)2]/bpy per C–C bond to be formed results in improved yields of the Ar–Ar coupling products. We currently believe that one of the benefits of using more than the stoichiometrically required amount of [Ni(COD)2]/bpy traces back to the intramolecular nature of the cyclization reaction: according to Yamamoto's ligand metathesis mechanism, two Ni atoms have to insert at the same time into both C–Br bonds involved in dehalocoupling. An increase in the amount of available [Ni0] species will increase the chance that this prerequisite is fulfilled.
Irrespective of the general ambiguities regarding the mechanistic details of the Yamamoto reaction, we have succeeded in shedding light on some essential aspects of our transformations:
(i) B2-TBPA and ODBE result from rare Yamamoto-type heterocoupling reactions. Nevertheless, they proceed in high yields and we found no indications for competing intra- or intermolecular homocoupling.
(ii) B2-TBPA (in pyridine) or ODBE (in THF) are not only accessible from 2Br,Br and 4 equiv. of [Ni(COD)2]/bpy/COD, but also from the monocyclized derivative 4 (Scheme 5) and 2 equiv. of the Yamamoto reagent (under otherwise identical reaction conditions).
 | ||
| Scheme 5 (a) Synthesis of ODBE (in THF) and B2-TBPA (in pyridine) starting from 4. (b) Different behavior of B2-TBPA toward the Yamamoto reagent in THF vs. pyridine. (c) Transformation of B2-TBPA to ODBE with “spent Yamamoto reagent” and quenching with air. Reagents and conditions: (i) 2 equiv. of [Ni(COD)2]/bpy/COD. (ii) 4 equiv. of [Ni(COD)2]/bpy/COD per B2-TBPA. COD = 1,5-cyclooctadiene, bpy = 2,2′-bipyridyl, py = pyridine. | ||
(iii) In pyridine, [Ni(COD)2]/bpy/COD (4 equiv.) leaves B2-TBPA unaffected. From THF, a complex product mixture devoid ofODBE was obtained after the usual workup process.
(iv) A “spent Yamamoto reagent”, prepared via homocoupling of 2 equiv. of Ph–Br with 2 equiv. of [Ni(COD)2]/bpy/COD in THF, transformed 0.5 equiv. of added B2-TBPA to ODBE in 50% yield (after quenching; yield of re-isolated B2-TBPA: 16%, yield of biphenyl: 85%).
The results (ii)–(iv) do not exclude that the DBA rearrangement occurring in THF can already be initiated at the stages of 2Br,Br or 4. Yet, as unequivocally demonstrated by experiment (iv), we can most easily explain all observations by assuming that B2-TBPA is formed as the primary product in both solvents, but only in THF is it further converted to ODBE. This conversion is promoted by the “spent Yamamoto reagent” and uses the O2/H2O from the quenching step as oxygen sources (recall that solutions of B2-TBPA alone are benchtop-stable).71
Taking all available information together, we arrive at the following working hypothesis regarding the B2-TBPA → ODBE rearrangement: the overall process, which requires the cleavage of two juxtaposed B–C bonds and connects the resulting borabenzo[de]anthracene halves via both previously B-bonded C atoms, starts with the coordination of OH− to one B atom (Scheme 6; cf. the F− titration of B2-TBPA). The corresponding adduct IX is an active transmetalating reagent and shifts the phenyl ligand onto the [(bpy)NiBr2] present in the “spent Yamamoto reagent”. The repetition of this process at the second B atom ultimately leads to the diaryl nickel species X (Scheme 6). A subsequent reductive elimination from X establishes the new C–C bond. Intramolecular condensation of the ditopic borinic acid according to Scheme 3 ultimately furnishes the oxadiborepin ODBE.
 | ||
| Scheme 6 Proposed mechanism for the Ni-mediated B2-TBPA → ODBE rearrangement. [Ni] = [(bpy)NiII]. | ||
Why is this reaction sequence suppressed in pyridine? In view of the DMAP-titration results, it is conceivable that pyridine coordination to boron prevents (or at least delays) the attack of OH− ions. In case that single-electron transfer processes contribute to the rearrangement reaction, it is also important to recall that B2-TBPA is harder to reduce by 430 mV in pyridine vs. THF solutions. As a third option, reactive Ni species may be tamed if pyridine is present as a strong ligand.
Conclusions
We have shown that a mild intramolecular C–C-heterocoupling reaction of the Yamamoto type is an ideal tool for the synthesis of nanoscale doubly B-doped polycyclic aromatic hydrocarbons, such as the tetrabenzopentacene derivative B2-TBPA. However, special care must be taken to choose the proper solvent: the otherwise identical synthesis protocol produces high yields of B2-TBPA when the reaction is carried out in pyridine, whereas in THF solution, the oxadiborepin ODBE is formed almost quantitatively. Mechanistic investigations revealed an oxidative rearrangement B2-TBPA → ODBE, taking place in THF during the O2/H2O quench, as the most straightforward explanation. As THF solutions of B2-TBPA are inert toward air and moisture, the rearrangement reaction is obviously promoted by nickel species present in the “spent Yamamoto reagent”. We assume that pyridine suppresses the formation of ODBE because of its ligand properties, which annihilate the residual Lewis acidity of B2-TBPA and also tame the nickel complexes.With regard to the materials properties of carbonaceous TBPAvs.B2-TBPA, the most notable effects of boron doping are a shift in the emission wavelengths from red to blue and a change in the redox behavior from electron-donating to electron-accepting. Similar to B2-TBPA, ODBE is a benchtop-stable blue fluorophore, but possesses a lower electron affinity, due to its π-donating oxygen atom.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by the Bundesministerium für Wirtschaft und Energie through the WIPANO grant number 03THW10F04. Donations of lithium organyls by Albemarle Lithium GmbH are gratefully acknowledged.Notes and references
-
(a) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed
; (b) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS
-
Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jaekle, John Wiley & Sons, Ltd., Chichester, 1st edn., 2018 Search PubMed
-
(a) S. K. Mellerup and S. Wang, Trends in Chemistry, 2019, 1, 77–89 CrossRef
-
(a) A. John, S. Kirschner, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2019, 48, 1871–1877 RSC
- D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969–7977 RSC
- J. M. Farrell, D. Schmidt, V. Grande and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 11846–11850 CrossRef CAS PubMed
- J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed
- T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2019, 58, 11379–11384 CrossRef CAS PubMed
- V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 CrossRef CAS PubMed
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed
- P. Kovacic and M. B. Jones, Chem. Rev., 1987, 87, 357–379 CrossRef CAS
- C. Hoffend, F. Schödel, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2012, 18, 15394–15405 CrossRef CAS PubMed
- C. Hoffend, M. Diefenbach, E. Januszewski, M. Bolte, H.-W. W. Lerner, M. C. Holthausen and M. Wagner, Dalton Trans., 2013, 42, 13826–13837 RSC
- B. T. King, J. Kroulík, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, J. Org. Chem., 2007, 72, 2279–2288 CrossRef CAS PubMed
- C. Dou, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 9346–9349 CrossRef CAS PubMed
- S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Chem. Sci., 2016, 7, 219–227 RSC
- K. Schickedanz, J. Radtke, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2017, 139, 2842–2851 CrossRef CAS PubMed
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed
- K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808–15810 RSC
- V. M. Hertz, H.-W. Lerner and M. Wagner, Org. Lett., 2015, 17, 5240–5243 CrossRef CAS PubMed
- V. M. Hertz, J. G. Massoth, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 13181–13188 CrossRef CAS PubMed
- T. Yamamoto, S. Wakabayashi and K. Osakada, J. Organomet. Chem., 1992, 428, 223–237 CrossRef CAS
- T. Yamamoto, Bull. Chem. Soc. Jpn., 2010, 83, 431–455 CrossRef CAS
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed
- B. X. Mi, Z. Q. Gao, M. W. Liu, K. Y. Chan, H. L. Kwong, N. B. Wong, C. S. Lee, L. S. Hung and S. T. Lee, J. Mater. Chem., 2002, 12, 1307–1310 RSC
- M. M. Rauhut, B. G. Roberts, D. R. Maulding, W. Bergmark and R. Coleman, J. Org. Chem., 1975, 40, 330–335 CrossRef CAS
- T. Asahi, M. Yudasaka, K. Nakanishi, S. Iwashima, J. Aoki, N. Sato and H. Inokuchi, Phys. Status Solidi, 1990, 122, K97–K100 CrossRef
-
S. Toguchi, A. Oda, H. Ishikawa, US 6747287 B1, 2004
-
T. Fujiyama, Y. Toya, M. Nakatsuka, JP 2008098360, 2008
- N. Sato, H. Inokuchi, K. Seki, J. Aoki and S. Iwashima, J. Chem. Soc., Faraday Trans. 2, 1982, 78, 1929–1936 RSC
- R. S. Becker and W. E. Wentworth, J. Am. Chem. Soc., 1963, 85, 2210–2214 CrossRef
- C. L. Hilton, J. M. Crowfoot, P. Rempala and B. T. King, J. Am. Chem. Soc., 2008, 130, 13392–13399 CrossRef CAS PubMed
- K. Sakai, S. Ohshima, A. Uchida, I. Oonishi, S. Fujisawa and U. Nagashima, J. Phys. Chem., 1995, 99, 5909–5913 CrossRef CAS
- F. Miyamoto, S. Nakatsuka, K. Yamada, K. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS PubMed
- A. John, M. Bolte, H.-W. Lerner, G. Meng, S. Wang, T. Peng and M. Wagner, J. Mater. Chem. C, 2018, 6, 10881–10887 RSC
- S. Brend’amour, J. Gilmer, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2018, 24, 16910–16918 CrossRef PubMed
- H. E. Katz, Organometallics, 1986, 5, 2308–2311 CrossRef CAS
- R. Tsuji, K. Komatsu, Y. Inoue and K. Takeuchi, J. Org. Chem., 1992, 57, 636–641 CrossRef CAS
-
2Br,I crystallizes with two symmetry-independent molecules in the asymmetric unit. One of them (shown in Scheme 1 and discussed in the main text) adopts a syn-configuration, the other one is disordered with a syn:anti ratio of approximately 9:1. Given this background and the results of 1H-NMR spectroscopy, it is reasonable to assume that the syn-conformation is preferred over the anti-conformation in 2Br,I.
- J. M. Birchall and R. N. Haszeldine, J. Chem. Soc., 1961, 3719–3727 RSC
- M. Schulte and F. P. Gabbaï, J. Organomet. Chem., 2002, 643–644, 164–167 CrossRef CAS
- E. C. Rüdiger, M. Porz, M. Schaffroth, F. Rominger and U. H. F. Bunz, Chem.–Eur. J., 2014, 20, 12725–12728 CrossRef PubMed
- T. Yamamoto, Prog. Polym. Sci., 1992, 17, 1153–1205 CrossRef CAS
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed
- S. Grimme and S. D. Peyerimhoff, Chem. Phys., 1996, 204, 411–417 CrossRef CAS
- K. Kato, Y. Segawa and K. Itami, Synlett, 2019, 30, 370–377 CrossRef CAS
- The absolute configuration (i.e., M,M or P,P) could not be determined due to the absence of anomalous scatterers.
- The so far most highly twisted PAH is a hexacene with an end-to-end twist of 184°: R. G. Clevenger, B. Kumar, E. M. Menuey and K. V. Kilway, Chem.–Eur. J., 2018, 24, 3113–3116 CrossRef CAS PubMed
- R. A. Pascal, Chem. Rev., 2006, 106, 4809–4819 CrossRef CAS PubMed
- A. Izuoka, K. Wakui, T. Fukuda, N. Sato and T. Sugawara, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 900–902 CrossRef
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC
- Ö. Seven, S. Popp, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2014, 43, 8241–8253 RSC
- A. Das, A. Hübner, M. Weber, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2011, 47, 11339–11341 RSC
- In this particular case, the degree of distortion of the oxadiborepin ring is likely also influenced by extensive intermolecular O–H⋯O hydrogen bonding.
- Q. Yan, M. Yin, C. Chen and Y. Zhang, J. Org. Chem., 2018, 83, 9096–9102 CrossRef CAS PubMed
- J. D. Hoefelmeyer and F. P. Gabbaï, Organometallics, 2002, 21, 982–985 CrossRef CAS
- J. D. Hoefelmeyer, S. Solé and F. P. Gabbaï, Dalton Trans., 2004, 4, 1254–1258 RSC
- E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2018, 57, 16491–16495 CrossRef CAS PubMed
- S. Kirschner, S.-S. Bao, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Org. Biomol. Chem., 2019, 17, 5060–5065 RSC
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed
- M. F. Semmelhack, P. M. Helquist and L. D. Jones, J. Am. Chem. Soc., 1971, 93, 5908–5910 CrossRef CAS
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 7547–7560 CrossRef CAS
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS
- E. C. Rüdiger, S. Koser, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chem.–Eur. J., 2018, 24, 9919–9927 CrossRef PubMed
- A. F. Alahmadi, R. A. Lalancette and F. Jäkle, Macromol. Rapid Commun., 2018, 39, 1800456 CrossRef PubMed
- Y. Fu, K. Zhang, E. Dmitrieva, F. Liu, J. Ma, J. J. Weigand, A. A. Popov, R. Berger, W. Pisula, J. Liu and X. Feng, Org. Lett., 2019, 21, 1354–1358 CrossRef CAS PubMed
- Even though unlikely, we cannot exclude THF as source of the O atom incorporated into ODBE.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1922188–1922192. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03115d |
| This journal is © The Royal Society of Chemistry 2019 |