
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual functional ruthenium arene complex induces differentiation and apoptosis of acute promyelocytic leukemia cells†
Hai
Huang‡
 a,
Kaiming
Cao‡
a,
Yaqiong
Kong
b,
Siming
Yuan
a,
Hongke
Liu
b,
Yucai
Wang
c and
Yangzhong
Liu
*a
a,
Kaiming
Cao‡
a,
Yaqiong
Kong
b,
Siming
Yuan
a,
Hongke
Liu
b,
Yucai
Wang
c and
Yangzhong
Liu
*a
aCAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: liuyz@ustc.edu.cn
bJiangsu Key Laboratory of Biofunctional Materials, College of Chemistry and Materials Science, Nanjing Normal University, Nanjing, Jiang Su 210046, China
cSchool of Life Sciences and Medical Center, University of Science and Technology of China, Hefei, Anhui 230027, China
First published on 28th August 2019
Abstract
Human acute promyelocytic leukemia (APL) is the most malignant form of acute leukemia. The fusion of PML and RARα genes is responsible for over 98% of cases of APL. In this work, we found that a Ru(II) arene complex, [(η6-p-bip)Ru(en)Cl][PF6] (Ru-1), can selectively react with PML, leading to zinc-release and protein unfolding. Consequently, the degradation of the fusion protein PML–RARα occurs, which causes the differentiation of APL cells. In addition, Ru-1 can also bind to DNA and trigger apoptosis of APL cells. Therefore, Ru-1 acts as a dual functional agent that inhibits the growth of APL cells and induces cell differentiation. In contrast, the other non-selective Ru(II) compound, though also highly reactive to PML, does not exhibit anti-APL activity. The selectivity of Ru-1 to PML suggests a new strategy for the development of anti-APL drugs using ruthenium agents.
Introduction
Human acute promyelocytic leukemia (APL), a distinctive subtype of acute myeloid leukemia (AML), is the most malignant form of acute leukemia.1 The most conspicuous genetic characteristic of APL is the translocation of chromosomes 15 and 17, by which the fusion of the promyelocytic leukemia (PML) gene and the retinoic acid receptor alpha (RARα) gene generates the fusion protein PML–RARα.2 The PML protein contains a zinc-bound ring-finger domain whose function is not fully understood; RARα is an important transcription factor that influences cell growth, differentiation, and death.3 The PML–RARα fusion protein affects retinoid signaling at multiple levels, which suppresses cell differentiation and senescence.4 Therefore, PML–RARα is crucial for the pathogenesis of APL, and is also an important drug target for the treatment of APL.5,6 All-trans retinoic acid (ATRA), a clinically used anti-APL drug, converts PML–RARα into an activator and restores differentiation of APL cells.7 Since PML contains a zinc-bound ring-domain, targeting the zinc-binding site of PML is an alternative strategy for the treatment of APL. Arsenic trioxide (ATO, As2O3, or NaAsO2 in solution), an effective anti-APL drug in clinic, cures APL by this approach. Arsenic can directly bind to the zinc-coordination site (cysteines) in the ring-finger domain of PML.8 The As(III) binding causes the unfolding and aggregation of PML protein, which induces the degradation of the PML–RARα fusion protein through SUMOylation.8Metal complexes remain an important resource for the generation of chemical diversity in the search for novel therapeutic agents, especially in anticancer drug development.9 Platinum complexes, such as cisplatin, have been widely used in clinics for the treatment of solid cancers. In addition, ruthenium complexes have shown promising antitumor activity and low side-effects.9–11 Two ruthenium complexes, NAMI-A and KP1019, have entered clinical trials. It is generally accepted that platinum drugs bind to DNA and the DNA damage triggers apoptosis of tumor cells. Although DNA is also proposed to be the target of ruthenium drugs, proteins are believed to be more pronounced in the reaction with ruthenium agents than in the case of platinum agents.12,13 A metallomics study suggests that ruthenium agents can bind to multiple proteins in cells.14
The successful application of arsenic trioxide in clinics inspired us to investigate more metallodrugs that could target the ring-finger domain of PML, as the protein is an important target for anti-APL drug development. Considering the coordination preference of metal agents, ruthenium complexes were chosen because of the low toxicity and potential medicinal application of these compounds. We hypothesized that the Ru(II) complexes can bind to the zinc-coordination site at the ring-finger domain of PML, in a way similar to arsenic trioxide. Ruthenium arene complexes (Fig. 1) were analyzed as these complexes exhibit promising anticancer activity in vitro and in vivo. In addition, the arene ring could modulate the ruthenium reactivity towards proteins.15 The effects of ruthenium complexes on growth inhibition, PML–RARα degradation and cell differentiation were verified.
 | ||
| Fig. 1 Molecular structures of ruthenium complexes. | ||
Results and discussion
Ruthenium complexes inhibit proliferation of APL cells
The proliferation of human APL NB4 cells was measured using the MTT assay. Four ruthenium(II) arene complexes (Fig. 1) were studied in terms of their inhibition of cell growth, and two clinical anti-APL drugs, ATRA and ATO, were used for comparison. ATRA and ATO can efficiently inhibit cell growth (IC50 11.9 μM and 1.2 μM, respectively, see Table 1). The two Ru-complexes, Ru-1 and Ru-2, demonstrated an anti-proliferation activity similar to that of ATRA (IC50 7.2 μM and 7.5 μM, respectively). The two di-nuclear complexes Ru-3 and Ru-4 showed very low activity towards NB4 cells (IC50 > 100 μM). Therefore, the two mono-nuclear complexes, Ru-1 and Ru-2, were chosen for further investigations.| Complex | IC50 (μM) |
|---|---|
| a Cells were treated with different drugs for 72 h before the MTT assay. | |
| ATRA | 11.9 ± 2.3 |
| As2O3 (ATO) | 1.2 ± 0.4 (in As) |
| [(η6-bip)Ru(en)Cl][PF6] (Ru-1) | 7.2 ± 4.0 |
| [(η6-p-cym)Ru(en)Cl][PF6] (Ru-2) | 7.5 ± 4.9 |
| [Ru2(η6-p-cym)2(p-bib)2Cl2]Cl2 (Ru-3) | >100 |
| [(η6-p-cym)Ru(en)Cl2]2 (Ru-4) | >100 |
Ruthenium complexes induce degradation of PML–RARα
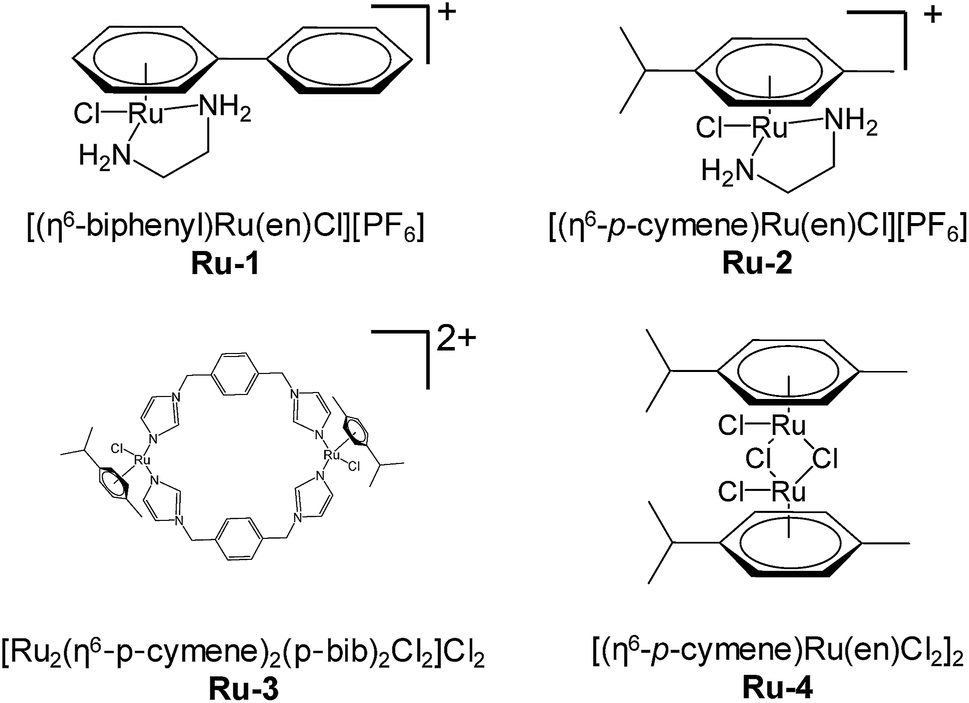
The degradation of PML–RARα in NB4 cells was assessed by western blotting with PML antibodies. The cells were treated with ATRA, ATO, Ru-1 or Ru-2. The result indicated that all these drugs can induce degradation of PML–RARα in a dose dependent manner, while ATO demonstrated the highest efficiency (Fig. 2A). 40 μM Ru-1 nearly completely cleared out PML–RARα, which is more effective than the treatment of ATRA. Ru-2 showed only moderate efficacy, which is similar to the effect of ATRA. This result revealed that the two ruthenium complexes can induce the degradation of PML–RARα in cells, and Ru-1 is much more potent than Ru-2. Therefore, Ru-1 was selected for further analysis.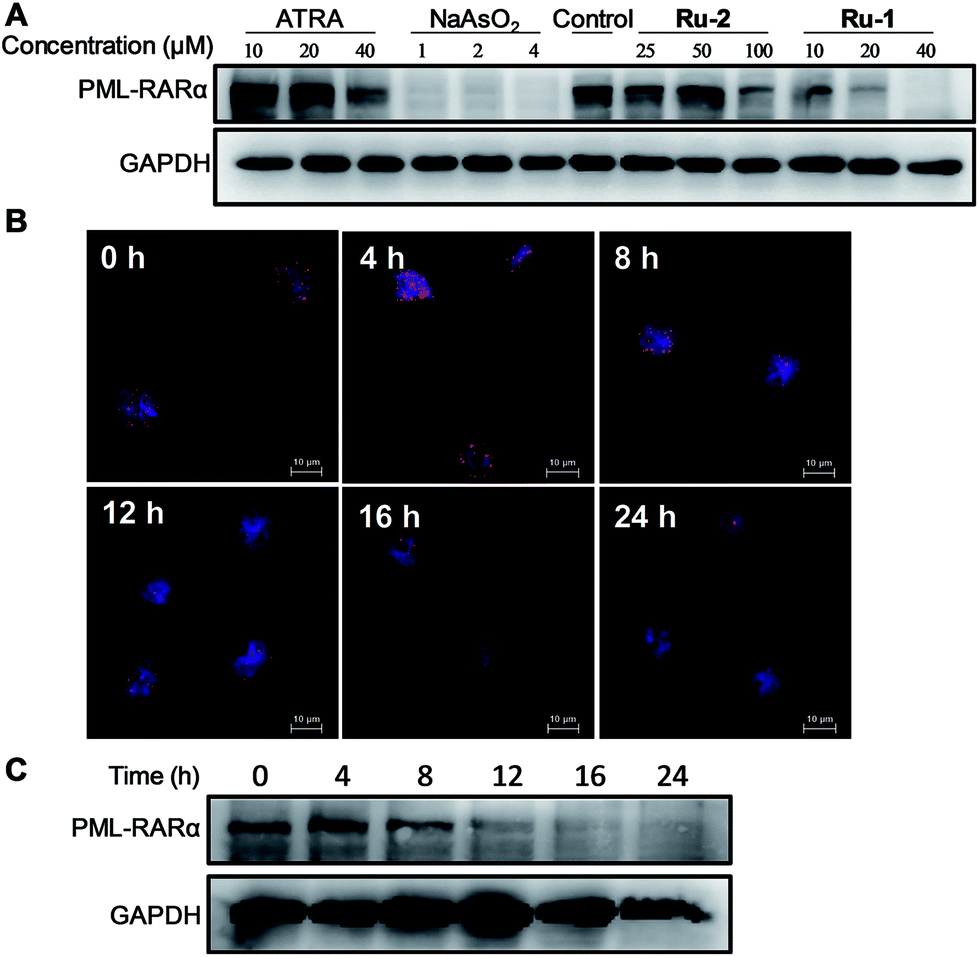 | ||
| Fig. 2 The degradation of PML–RARα in NB4 cells. (A) The PML–RARα level in NB4 cells upon treatment with different drugs for 24 h. The protein was assessed by western blotting with PML antibodies. (B) Time dependent degradation of PML–RARα in NB4 cells induced by 30 μM Ru-1. The PML–RARα protein in cells was measured using immunofluorescence staining with PML antibodies (red). The nuclei were stained with DAPI (blue). Scale bar, 10 μm. (C) Western blot measurement of the PML–RARα protein in NB4 cells after treatment with Ru-1 for different times. | ||
The degradation of PML–RARα in cells was also assessed using immunofluorescence staining with PML antibodies. Fluorescence measurement clearly showed the fusion protein PML–RARα in cells. The time dependent imaging showed that, after the treatment with 30 μM Ru-1, the amount of PML–RARα protein decreased gradually (Fig. 2B). In addition, the fusion protein in cells was also assessed by western blotting, confirming the gradual degradation of fusion protein (Fig. 2C). Both measurements showed that very little PML can be detected after 12 h of treatment, indicating that Ru-1 can efficiently induce the degradation of PML–RARα in NB4 cells.
Ruthenium complexes trigger the differentiation of NB4 cells
The fusion of PML–RARα impairs neutrophilic differentiation and initiates development of APL;16 therefore, differentiation therapy is a key strategy for anti-leukemic chemotherapy, such as the function of ATRA. The degradation of PML–RARα by Ru-agents could also lead to differentiation of APL cells. To gain insight into the phenotypic changes of the NB4 cells induced by Ru-agents, the granulocytic differentiation marker CD11b was analyzed by flow cytometry. After the incubation of NB4 cells with various drugs, the cells were treated with FITC labelled CD11b antibody. The flow cytometry results showed that ATO significantly enhanced the CD11b level even at low concentration (5 μM) (Fig. S1†). ATRA, Ru-1 and Ru-2 demonstrated a concentration dependent enhancement of the CD11b level, while Ru-1 exhibited the highest effect among these three agents. The two di-nuclear ruthenium complexes Ru-3 and Ru-4 only marginally increased the CD11b level (Fig. 3A and S2†). This cell differentiation assay is consistent with the degradation of the fusion protein where Ru-1 demonstrated a strong effect against the leukemic factor of NB4 cells, and is more effective than ATRA. As the maturation of the granulocytic lineage is often associated with the generation of superoxides in cells,17 the reduction in NB4 cells was examined with the nitroblue tetrazolium (NBT) assay. The treatment with Ru-1 led to increased NBT reduction in NB4 cells (Fig. 3B), which further confirms the cell differentiation.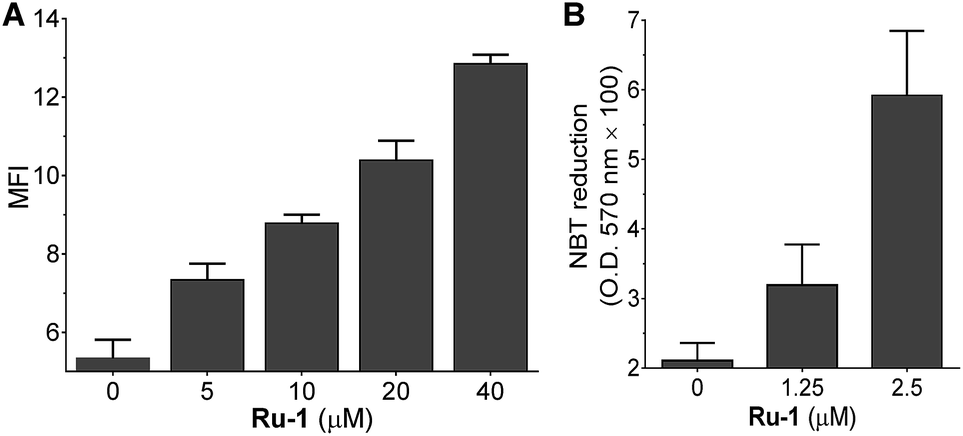 | ||
| Fig. 3 Ruthenium complexes induce the differentiation of NB4 cells. (A) The expression of the myeloid surface marker CD11b on NB4 cells after the treatment with Ru-1 for 3 days. The mean fluorescence intensity (MFI) was quantified by flow cytometry with CD11b-FITC antibody. (B) NBT reduction in NB4 cells after treatment with Ru-1 for 3 days. | ||
Ruthenium complex directly binds to PML and disrupts the protein folding
It has been reported that the anti-APL drug ATO induces the degradation of PML–RARα by directly binding to PML, which disrupts the protein folding and triggers SUMOylation. The ring-domain of PML is the arsenic binding site and the arsenic binding plays a central role in the PML SUMOylation.8 As Ru-1 efficiently induces the degradation of PML–RARα, we therefore verified whether the ruthenium complex can also bind to the ring-domain of PML. Fluorescence titration showed that Ru-1 quenched the fluorescence of PML (Zn-bound protein) in a concentration dependent manner (Fig. 4A). This observation is consistent with a recent report that Ru-binding leads to the fluorescence quenching of zinc-finger proteins.18 It has been reported that the binding of many metallodrugs, such as arsenic, platinum, and ruthenium agents, to zinc-finger proteins induces the release of zinc ions from the proteins.18–20 Here we observed that the reaction of Ru-1 also resulted in zinc release from PML (Fig. 4B). This result suggests that, consistent with the ATO reaction, Ru-1 also binds to the zinc-coordination site of PML. Electrospray ionization mass spectrometry (ESI-MS) analysis showed that the reaction of Ru-1 led to a mass increase of 183.2 Da in PML, providing direct evidence of binding of the (η6-bip)Ru(en) moiety to the protein with the release of zinc ions (Fig. 4C). As zinc coordination is crucial for the folding of PML, the structural perturbation of PML by Ru-1 binding was analyzed using circular dichroism (CD) spectra. The result showed that the Ru-1 reaction led to the decrease of the negative band at 228 nm, and the resulting spectra became more similar to those of apo-PML with an increased concentration of Ru-1 (Fig. 4D). In addition, 2D 1H–15N HSQC NMR spectra showed that a large amount of signals of PML disappeared after the Ru-1 reaction, while some new peaks appeared at around 8 ppm on the 1H dimension, confirming the protein unfolding upon the Ru-1 reaction (Fig. 4E). This result is similar to the arsenic binding induced unfolding of PML.8 | ||
Fig. 4
Ru-1 binds to the ring-finger domain of PML and perturbs the protein folding. (A) Fluorescence spectra of PML with the titration of Ru-1. The measurement was conducted on 30 μM Zn-PML at 37 °C for 2 h in 20 mM phosphate buffer (pH = 7.0). The inset shows the decrease of fluorescence with the increase in the ratio of [Ru-1]/[Zn-PML]. (B) The zinc release from Zn-PML upon the reaction with Ru-1. After the reaction of Zn-PML (30 μM) with different ratios of Ru-1 at 37 °C for 1 h, 400 μM zinc dye (zincon) was added and the UV absorption at 620 nm was measured. (C) ESI-MS spectra of Zn-PML before and after the incubation with Ru-1 at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and 1:20 in 100 mM ammonium acetate buffer (pH = 7.0) at 37 °C for 2 h. (D) CD spectra of PML. The spectra were recorded on 30 μM Zn-PML before and after reaction at 37 °C for 2 h in 20 mM phosphate buffer (pH = 7.0). (E) 1H–15N HSQC NMR spectra of Zn-PML before (blue) and after (red) reaction with Ru-1. apo-PML was used for comparison. The reaction was performed using 0.2 mM 15N-labeled PML with 5 eq. of Ru-1 in 20 mM phosphate buffer (pH = 7.0) at 37 °C for 2 h. :5 and 1:20 in 100 mM ammonium acetate buffer (pH = 7.0) at 37 °C for 2 h. (D) CD spectra of PML. The spectra were recorded on 30 μM Zn-PML before and after reaction at 37 °C for 2 h in 20 mM phosphate buffer (pH = 7.0). (E) 1H–15N HSQC NMR spectra of Zn-PML before (blue) and after (red) reaction with Ru-1. apo-PML was used for comparison. The reaction was performed using 0.2 mM 15N-labeled PML with 5 eq. of Ru-1 in 20 mM phosphate buffer (pH = 7.0) at 37 °C for 2 h. | ||
Ruthenium complex binds to DNA and induces cell apoptosis
As DNA is the well-investigated target of metallodrugs (such as Pt- and Ru-drugs), the reaction of Ru-1 with DNA was also analyzed. The reaction was monitored by fluorescence using an EtBr probe, as the intercalation of EtBr generates fluorescence at 625 nm. The reaction of Ru-1 with DNA caused a gradual decrease of the fluorescence, showing the binding of Ru to DNA that expelled EtBr from DNA (Fig. 5A and B). This result confirmed that Ru-1 is capable of reacting with DNA. In addition, the binding of Ru-1 to DNA was also measured in cells. After the treatment of NB4 cells with Ru-1, the genomic DNA was extracted and the ruthenium in DNA was measured by ICP-MS (Fig. 5C). The result confirmed the binding of ruthenium to DNA, and more ruthenium was detected with a higher concentration of Ru-1 in the assay.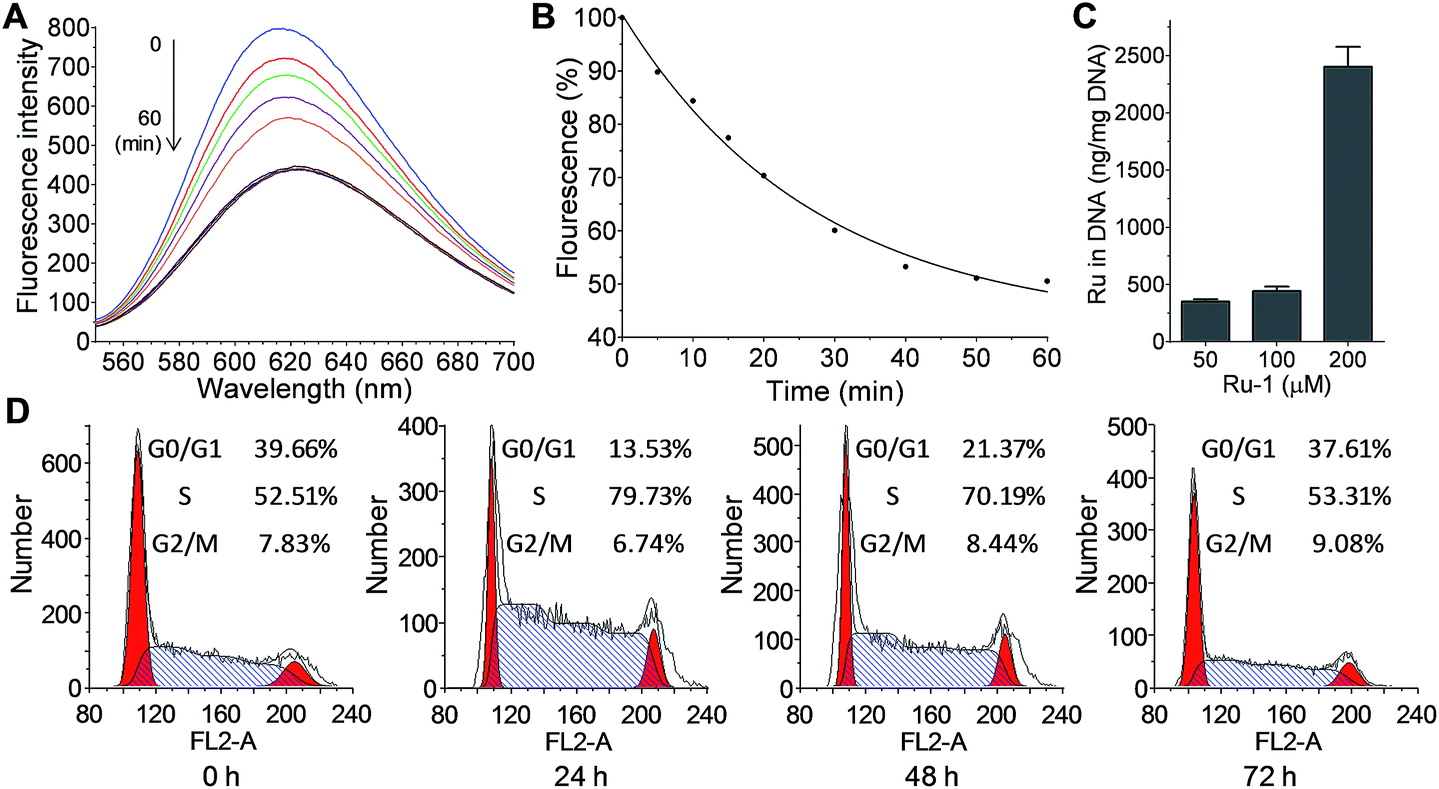 | ||
| Fig. 5
Ru-1 interacts with DNA and alters the cell cycles. Fluorescence measurement (A) and decrease (B) of the binding of Ru-1 when using an EtBr probe. Interactions were performed on extracted genomic DNA (20 ng μL−1) from NB4 cells with Ru-1 at a [Ru]/[nucleotide] ratio of 1:1 in 10 mM phosphate buffer (pH 7.4) containing 10 mM NaClO4 at 37 °C. EtBr (0.04 mg) was added before fluorescence measurements. (C) Ruthenium binding to DNA in NB4 cells. NB4 cells were treated with Ru-1 (50, 100 or 200 μM) for 8 h, then genomic DNA was extracted from the cells and the amount of ruthenium on DNA was determined using ICP-MS. (D) Flow cytometry analysis of the cell cycle profiles. NB4 cells were treated with 20 μM Ru-1 for different times, and the cells were stained with PI. | ||
The DNA damage caused by metallodrugs typically leads to cell cycle arrest; therefore, the alteration of the cell cycle by Ru-1 was measured (Fig. 5D). The results showed that the treatment of Ru-1 caused moderate cell cycle arrest at the S phase (∼50% increase) in 24 h. With longer treatment (48 and 72 h), the cell cycle progressed and the G0/G1 phase increased gradually while the S phase decreased. It implies that DNA repair could occur under the experimental conditions.
DNA damage is a well-known mechanism of cisplatin; the failure of DNA repair triggers cell apoptosis. To verify the cell apoptosis resulting from the DNA binding of Ru-1, the expression of apoptotic pathway proteins, including caspase-3, cleaved caspase-3, PARP-1 and cleaved PARP-1, was measured (Fig. 6A). Western blot results indicate that, after the treatment of NB4 cells with 40 μM Ru-1, the caspase-3 level decreased significantly, while its cleavage product level increased. The cleavage of caspase-3 indicates the activation of enzymes in apoptotic cells. The alteration of the levels of these proteins occurred in a time dependent manner, and significant changes occurred after the drug treatment for 4–8 h. In the meantime, the cleavage of the DNA repair protein PARP-1, which is the substrate of caspase-3, was observed. To further analyze the cell apoptosis induced by Ru-1, the cells were stained with PI and Annexin V (Fig. 6B and S4†). The results clearly show that Ru-1 induced apoptosis of NB4 cells. The ratio of the apoptotic cells increased with time, and higher concentration of Ru-1 caused more apoptosis. This result clearly confirmed that the treatment with Ru-1 induced DNA damage causing the apoptosis of NB4 cells.
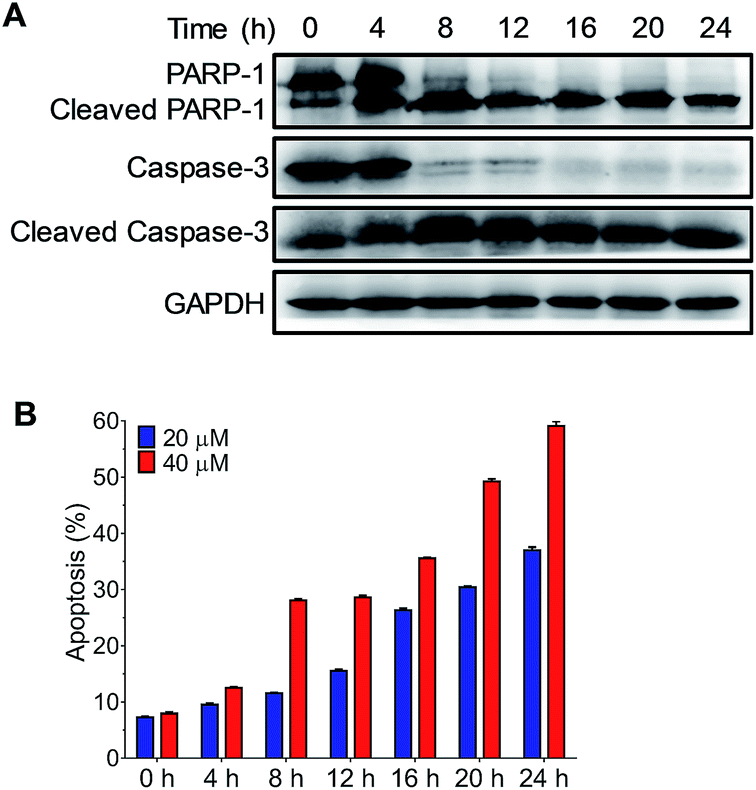 | ||
| Fig. 6 Ru-1 induced apoptosis of NB4 cells. (A) The time dependent expressions of caspase-3, cleaved caspase-3, PARP-1 and cleaved PARP-1 in NB4 cells. The proteins were detected by western blotting after the treatment with 40 μM Ru-1. (B) The percentage of apoptotic cells after the treatment with Ru-1 for different times obtained by flow cytometry with Annexin V-FITC/PI dual-staining. | ||
The fusion protein PML–RARα is responsible for the occurrence of over 98% of cases of APL.2In vitro and in vivo studies have shown that PML–RARα is a crucial target for the treatment of APL.6 Interestingly, the two effective anti-APL drugs used in clinics, ATRA and arsenic trioxide, target the two components of the fusion protein, RARα and PML, respectively. Arsenic trioxide can directly bind to the ring-domain of PML at the zinc-binding site, as As(III) possesses high coordination preference to thiol groups. The coordination of zinc with the ring-domain of PML is vital for maintaining PML–RARα stability in APL cells.21 In this regard, the ring-domain of PML could also be the target of other metallodrugs that favor cysteine residues if these ruthenium compounds can bind selectively to PML in APL cells. On the other hand, the development of new metallodrugs might circumvent the side-effects of arsenic trioxide.
Here in this work, we have examined four ruthenium arene complexes as they have the potential to bind to zinc-finger proteins and result in zinc ejection. The results indicate that the complex [(η6-bip)Ru(en)Cl][PF6] (Ru-1) can efficiently bind to PML, degrade PML–RARα and induce the differentiation of APL cells. The other ruthenium complex [(η6-p-cym)Ru(en)Cl][PF6] (Ru-2) demonstrates lower reactivity to PML. Consistent with the different chemical reactivity to PML, Ru-1 shows much higher efficiency in the degradation of the fusion protein and induces cell differentiation. This result suggests the positive correlation of PML reaction and cell differentiation. Nevertheless, the two dinuclear Ru-agents, Ru-3 and Ru-4, which are highly reactive to PML in solution (Fig. S2†), showed negligible activity in the cell-based assays. This divergence could be due to either the different cellular uptake or the nonspecific reactions of the two dinuclear Ru-agents in cells. To verify this hypothesis, the cellular uptake of these Ru-agents was measured by ICP-MS. The result indicated that Ru-1 is internalized more efficiently than Ru-2, but less than Ru-3 (Fig. S4†). This result suggests that efficient drug uptake is necessary, but it is not the only factor determining the drug efficacy.
It has been reported that ATO preferentially binds to PML over other zinc-finger proteins, such as ERα and NCp7,20 and this selectivity is crucial for the anti-APL activity of ATO since there are many zinc-finger proteins in cells. Therefore, we further analyzed the selectivity of Ru-complexes to PML over other zinc-finger proteins. Fluorescence titration showed that Ru-1 had higher binding affinity to PML than to NCp7 and ERα, while Ru-3 was highly reactive to both PML and zinc-finger proteins (Fig. S5†). The zinc-release assay showed that Ru-1 more efficiently caused zinc-release from PML than Ru-2 and Ru-3, whereas Ru-3 more efficiently induced zinc-release from NCp7 than Ru-1 and Ru-2 (Fig. S6†). These results indicate that these two types of anti-APL agents, ATO and Ru-1, possess similar selectivity to the PML protein and their reactions cause the unfolding of PML. Therefore, both ATO and Ru-1 demonstrate similar biological functions by inducing the degradation of the PML–RARα fusion protein in APL cells. However, the reactivity of these two agents to PML is not exactly the same. It has been reported that ATO binds to zinc-finger protein NCp7 under weak acidic conditions or in the presence of other coordination active agents.22 Here we found that acidic conditions are also required for the reaction of ATO with PML (Fig. S5C†). Nevertheless, Ru-1 can bind to PML under neutral conditions without other assistants. Therefore, ATO and Ru-1 possess similar selectivity and binding manner to PML, but their reactivities are not identical. Although Ru-3 is also reactive to PML, it is more reactive to zinc-finger proteins. This reactivity could lead to the non-specific reactions of Ru-3 in cells. These results reveal that the selectivity to PML is important for the development of new anti-APL drugs. On the other hand, the different selectivity of ruthenium complexes indicates that the ligands and coordination manner significantly affect the reactivity and selectivity of Ru-agents to proteins, which consequently determines their cellular response and drug efficacy. These different activities provide the opportunity to design more selective ruthenium agents towards specific target proteins.
The binding of Ru-1 to PML in other leukemia cells, including HL-60 and MOLM-13 cells, was also investigated. The western blot and immunofluorescence imaging measurements showed that Ru-1 can efficiently induce degradation of PML in HL-60 and MOLM-13 cells in a dose dependent manner (Fig. S9†), confirming the binding of Ru-1 to the PML protein in these cells as well. However, the differentiation assay showed that Ru-1 could not alter the level of CD11b, the myeloid differentiation marker (Fig. S10†). These results indicate that Ru-1 cannot induce the differentiation of HL-60 and MOLM-13 cells, although it can efficiently bind to PML in these cells. It is worth noting that the fusion of PML–RARα occurs only in NB4 cells, so NB4 cells are the genuine promyelocytic leukemia cell line,23 while HL-60 represents a discrete stage of differentiation between the late myeloblast and the promyelocyte, and MOLM-13 is an AML cell line.24 This cellular assay confirmed that the reaction of Ru-1 with PML observed in vitro can occur in different types of cells; Ru-1 is able to bind to the fusion protein PML–RARα in NB4 cells, and to the PML protein in HL-60 and MOLM-13 cells as well. Nevertheless, only NB4 carries the t(15;17) fusion gene for the expression of the PML–RARα fusion protein, which blocks promyelocyte differentiation and leads to the proliferation of leukemia blasts.25 The PML protein is not involved in the pathogenesis of the leukemia associated with HL-60 and MOLM-13 cells. Therefore, the binding of Ru-1 to the PML–RARα in NB4 cells leads to the differentiation of NB4 cells, but not HL-60 and MOLM-1. These results highlight the significance of selective targeting in drug design.
Ruthenium agents have shown promising activity in the treatment of solid cancers.26 Ru(II)-arene complexes are unique types of complexes that effectively inhibit the growth of a number of tumor cells.27 It is generally believed that, similar to cisplatin, these Ru-agents also bind to DNA and induce the apoptosis of tumor cells.28 This DNA binding activity is indeed observed for Ru-1 and Ru-2 in the cell proliferation assay in this work, which is consistent with the literature report.29 The binding of Ru-1 to DNA activates the apoptotic pathway, subsequently triggering the apoptosis of NB4 cells. On the other hand, Ru-1 has been found to be also highly reactive to the protein PML, the specific drug target of leukemia cells. These results indicate that this ruthenium complex is a dual-targeting agent that is active towards both the PML protein and DNA in APL. Consequently, it induces the differentiation and apoptosis of APL cells.30 Dual-functional treatment is of great interest in the design of metallodrugs. In order to achieve dual-functional effects, an additional functional group is typically tethered in the metal complexes.31 In this work, the demonstrated Ru-1 complex is a dual-functional agent without further ligand modifications. Specific targeting of proteins is a proposed strategy to design Ru(II)-based drugs.32 The findings of this work revealed a new function of existing ruthenium complexes, that is, in addition to the widely-studied apoptotic activity on solid tumors, they can also be used for the treatment of APL by selectively targeting PML.
Conclusions
In summary, four ruthenium arene complexes have been investigated against the growth of acute promyelocytic leukemia (APL) cells. One of the complexes, [(η6-p-bip)Ru(en)Cl][PF6] (Ru-1), can effectively degrade the oncogenic fusion protein PML–RARα in APL cells, leading to cell differentiation. The mechanism study showed that Ru-1 can directly bind to the ring-domain of PML and cause zinc-ejection from the protein. The zinc-release leads to structural perturbation of the protein, which can further induce the PML–RARα degradation and cell differentiation. This effect is similar to the function of arsenic trioxide, the anti-APL drug used in clinics. On the other hand, Ru-1 can also bind to genomic DNA in the APL cells. The DNA damage activates the apoptotic pathway and triggers apoptosis of APL cells. Therefore, Ru-1 is a dual functional agent that can inhibit the growth of APL cells by targeting DNA, and induce cell differentiation by targeting the oncoprotein PML–RARα. As PML–RARα is crucial for the pathogenesis of APL, the selectivity of Ru-1 to PML suggests a new strategy for the development of anti-APL drugs. Another Ru-agent, [Ru2(η6-p-cym)2(p-bib)2Cl2]Cl2 (Ru-3), is highly reactive to both PML and zinc-finger proteins, and therefore does not exhibit anti-APL activity due to non-selectivity. The result identified a new therapeutic function of ruthenium complexes, and the work paves the way for designing novel anti-APL drugs.Experimental
Materials
NaAsO2 was purchased from Sigma-Aldrich (St. Louis, MO). Ruthenium agents were synthesized according to a previous report.33 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4-(2-pyridylazo)resorcinol (PAR) and 2-carboxy-2′-hydroxy-5′-sulfoformazylbenzene sodium salt (Zincon) were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). Nitroblue tetrazolium chloride (NBT) and phorbol 12-myristate 13-acetate (PMA) were purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China). RPMI Medium 1640, trypsin–EDTA and fetal bovine serum (FBS) were obtained from Biological Industries (Kibbutz Beit-Haemek, Israel). Anti-PML and anti-CD11b(FITC) antibodies were obtained from Abcam (Cambridge, US). Anti-GAPDH, anti-PARP-1 and anti-caspase-3 antibodies were purchased from Proteintech (Wuhan, China). The apoptosis detection kit and DNA Content Quantitation Assay (Cell Cycle) were purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China). Ultra-purified water was prepared using a Milli-Q Synthesis System (Millipore, Bedford, MA). All other solvents and reagents were used as received.Cell culture
The human acute promyelocytic leukemia NB4 cells were maintained in RPMI 1640 medium containing 10% FBS in a humidified atmosphere containing 5% CO2 at 37 °C.Cell cytotoxicity assays
NB4 cells were seeded in 96-well plates at 4000 cells per well in 50 μL of RPMI 1640 medium containing 10% FBS. And 50 μL of drug media solutions at the desired concentrations were distributed in each well. The cells were further incubated for 72 h, and then 10 μL of 5 mg mL−1 MTT solution was added. The cells were incubated for another 4 h to allow viable cells to reduce the yellow tetrazolium salt (MTT) into dark blue formazan crystals. Finally, 100 μL of lysis buffer was added to the wells and the cells were incubated for another 4 h at 37 °C. The absorbance was measured at 490 nm using a Bio-Rad 680 microplate reader. The IC50 values were calculated using GraphPad Prism software (version 6.01) based on data from three parallel experiments.Cell differentiation assays
NB4 cells cultured in 12-well plates at a density of 200 × 104 cells per well were treated with different drugs at the corresponding concentration for 3 days. After then, the cells were harvested and immunofluorescence stained with FITC-labeled CD11b antibody. CD11b expression was determined using a BD FACS Calibur Flow Cytometer. The data were analyzed using FlowJo software. Differentiation of NB4 cells was also assessed through the ability of the cells to produce superoxides. This was measured by the degree of reduction of nitroblue-tetrazolium (NBT, Solarbio) achieved by the cells over a 30 minute period at 37 °C in the presence of phorbol myristate acetate (PMA, Solarbio). NBT reduction was detected via absorbance at 570 nm using a visible spectrophotometer.Cell cycle analysis
NB4 cells were seeded in 12-well plates at a density of 100 × 104 cells per well in 2 mL of RPMI 1640 medium containing 20 μM Ru-1 and incubated in a humidified 5% CO2 atmosphere for 24, 48 or 72 hours. After then, the cells were harvested for DNA content analysis using the cell cycle detection kit according to the manufacturer's instructions. Cell cycle distributions and DNA contents were determined using a BD FACS Calibur Flow Cytometer. The results were analyzed using ModFit LT 5.0 software. Percentages of cells in G0/G1, S and G2/M phases were recorded.Apoptosis analysis
NB4 cells cultured in 12-well plates at a density of 100 × 104 cells per well were treated with 20 or 40 μM Ru-1 for 4, 8, 12, 16, 20 or 24 hours. Apoptotic cells were detected by flow cytometry after staining with Annexin V and propidium iodide (PI) using the Annexin V-FITC apoptosis detection kit and determined using a BD FACS Calibur Flow Cytometer. The data were analyzed using FlowJo software.Western blot
NB4 cells cultured in 12-well plates at a density of 200 × 104 cells per well were treated with different drugs at the corresponding concentration for 4, 8, 12, 16 or 24 hours. After then, the cells were harvested and lysed in RIPA lysis buffer. Cell lysates with equal amounts of protein were resolved on 10% or 12% SDS-PAGE and transferred to a polyvinylidene fluoride membrane, then detected with various antibodies. The proteins were visualized with a chemiluminescence system (Thermo Fisher).Immunofluorescence
NB4 cells cultured in 24-well plates at a density of 100 × 104 cells per well were treated with 30 μM Ru-1 for 4, 8, 12, 16, 20 or 24 hours. After then, the cells were harvested and pelleted onto slides. The slides were quickly air-dried and the cells were immobilized using 4% paraformaldehyde (PFA). After permeabilization and blocking treatment, immunofluorescence staining of the PML was performed using PML antibodies and AF594 labeled secondary antibodies. All slides were examined under a LSM 710 CLSM (Carl Zeiss, Jena, Germany).The interaction between Ru-1 and DNA
Interactions were performed with extracted genomic DNA (20 ng μL−1) from NB4 cells, and at a Ru-1/nucleotide ratio of 1:1 in 10 mM phosphate buffer (pH = 7.4) containing 10 mM NaClO4 at 37 °C. EtBr (0.04 mg) was added before fluorescence measurements.
Genomic DNA ruthenation in NB4 cells
NB4 cells cultured in 6-well plates at a density of 1000 × 104 cells per well were treated with 50, 100 or 200 μM Ru-1 for 8 hours. After then, the cells were harvested and washed with PBS for 3 times. The genomic DNA was extracted from the harvested cells with the MiniBEST Universal Genomic DNA Extraction Kit (Takara). The extracted DNA was digested with nitric acid for ICP-MS measurement. The DNA concentrations were measured via an ultraviolet spectrophotometer before digestion.Preparation of recombinant PML
The PML ring-finger domain (aa 49–104) was expressed in pET28a with a SUMO tag in E. coli BL21(DE3). The protein was purified using Ni-NTA affinity chromatography, and the SUMO-tag was removed using small ubiquitin-like modifier protease. PML was further purified through gel filtration chromatography on a Superdex 75 PG column (GE Healthcare) with phosphate buffer at pH 7.0. The protein concentration was determined through UV absorption.Circular dichroism (CD)
CD measurements were performed with a Jasco J-810 CD spectrometer from 190 to 260 nm, and flashed with high purity nitrogen. All spectra were recorded with a scan speed of 100 nm min−1 and 1 nm data pitch. CD spectra of 30 μM Zn-PML-R were measured with the titration of Ru-1. The fluorescence titrations were conducted at 37 °C for 2 hours in 20 mM phosphate buffer (pH = 7.0).Electrospray ionization mass spectrometry
The mass spectrometry experiments were conducted on an Exactive Plus (Thermo Fisher Scientific, CA, USA) mass spectrometer. Zn-PML was incubated with Ru-1 in 100 mM ammonium acetate buffer (pH = 7.0) at 37 °C for 2 hours. The positive ion mode was used in the ESI-MS experiments. Data were processed using Xcalibur software (version 2.0, Thermo Finnigan).NMR spectroscopy
1H–15N heteronuclear single quantum coherence (HSQC) spectra were collected on a Bruker 600 MHz NMR spectrometer at 25 °C. NMR samples were prepared using 0.2 mM 15N-labeled Zn-PML interacted with 5 eq. of Ru-1 in 20 mM phosphate buffer containing 100 mM NaCl and 10% D2O (v/v). The data were processed and analyzed using TopSpin software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0505400), the National Natural Science Foundation of China (21877103, 21573213) and the Collaborative Innovation Center of Suzhou Nano Science and Technology. A portion of this work was performed at the Steady High Magnetic Field Facilities, High Magnetic Field Laboratory, CAS.Notes and references
- C. C. Coombs, M. Tavakkoli and M. S. Tallman, Blood Cancer J., 2015, 5, e304 CrossRef CAS.
- A. Kakizuka, W. H. Miller, K. Umesono, R. P. Warrell, S. R. Frankel, V. V. V. S. Murty, E. Dmitrovsky and R. M. Evans, Cell, 1991, 66, 663–674 CrossRef CAS.
- A. di Masi, L. Leboffe, E. De Marinis, F. Pagano, L. Cicconi, C. Rochette-Egly, F. Lo-Coco, P. Ascenzi and C. Nervi, Mol. Aspects Med., 2015, 41, 1–115 CrossRef CAS.
- S. Saeed, C. Logie, H. G. Stunnenberg and J. H. A. Martens, Br. J. Cancer, 2011, 104, 554–558 CrossRef CAS.
- Z. Wang and Z. Chen, Blood, 2008, 111, 2505–2515 CrossRef CAS.
- H. de The, P. P. Pandolfi and Z. Chen, Cancer Cell, 2017, 32, 552–560 CrossRef CAS.
- H. de The and Z. Chen, Nat. Rev. Cancer, 2010, 10, 775–783 CrossRef CAS.
- X. Zhang, X. Yan, Z. R. Zhou, F. Yang, Z. Wu, H. Sun, W. Liang, A. X. Song, V. Lallemand-Breitenbach, M. Jeanne, Q. Y. Zhang, H. Yang, Q. Huang, G. Zhou, J. Tong, Y. Zhang, J. Wu, H. Hu, H. de The, S. Chen and Z. Chen, Science, 2010, 328, 240–243 CrossRef CAS.
- M. Frezza, S. Hindo, D. Chen, A. Davenport, S. Schmitt, D. Tomco and Q. P. Dou, Curr. Pharm. Des., 2010, 16, 1813–1825 CrossRef CAS.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. Nazarov, C. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. Dyson and C. Davey, Nat. Commun., 2014, 5, 3462 CrossRef.
- A. R. Timerbaev, TrAC, Trends Anal. Chem., 2016, 80, 547–554 CrossRef CAS.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 38, 4764–4776 RSC.
- A. A. Lane and T. J. Ley, Cell, 2003, 115, 305–318 CrossRef CAS.
- B. Zakhireh and R. K. Root, Blood, 1979, 54, 429–439 CAS.
- Y. Sheng, K. Cao, J. Li, Z. Hou, S. Yuan, G. Huang, H. Liu and Y. Liu, Chem.–Eur. J., 2018, 24, 19146–19151 CrossRef CAS.
- S. Yuan, X. Ding, Y. Cui, K. Wei, Y. Zheng and Y. Liu, Eur. J. Inorg. Chem., 2017, 12, 1778–1784 CrossRef.
- K. Cao, Y. Sheng, S. Zheng, S. Yuan, G. Huang and Y. Liu, Metallomics, 2018, 10, 1564–1569 RSC.
- B. Zhu, J. Wang, J. Zhou, F. Zhou, W. Cheng, Y. Liu, J. Wang, X. Chen, D. Chen, L. Luo and Z. Hua, J. Inorg. Biochem., 2017, 175, 92–100 CrossRef CAS.
- L. Zhao, Z. Wang, Z. Xi, D. Xu, S. Chen and Y. Liu, Inorg. Chem., 2014, 53, 3054–3061 CrossRef CAS.
- H. Drexler, H. Quentmeier, R. MacLeod, C. Uphoff and Z. Hu, Leuk. Res., 1995, 19, 681–691 CrossRef CAS.
- Y. Matsuo, R. MacLeod, C. Uphoff, H. Drexler, C. Nishizaki, Y. Katayama, G. Kimura, N. Fujii, E. Omoto, M. Harada and K. Orita, Leukemia, 1997, 11, 1469–1477 CrossRef CAS.
- R. Carmody and K. Keeshan, Cancer Cell, 2017, 31, 612–613 CrossRef CAS.
- E. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS.
- G. Suss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- V. Brabec and O. Novakova, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS.
- R. Morris, R. Aird, P. Murdoch, H. Chen, J. Cummings, N. Hughes, S. Parsons, A. Parkin, G. Boyd, D. Jodrell and P. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS.
- M. Ganeshpandian, R. Loganathan, E. Suresh, A. Riyasdeen, M. Akbarsha and M. Palaniandavar, Dalton Trans., 2014, 43, 1203–1219 RSC.
- R. Kenny and C. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS.
- S. Thota, D. Rodrigues, D. Crans and E. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS.
- R. Morris, R. Aird, P. del Socorro Murdoch, H. Chen, J. Cummings, N. Hughes, S. Parsons, A. Parkin, G. Boyd and D. Jodrell, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CD11b expression in cells, fluorescence titration, apoptosis and cellular accumulation of Ru(II). See DOI: 10.1039/c9sc03110c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |