
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light harvesting pyrene-based MOFs as efficient ROS generators†
Yingmu
Zhang
a,
Jiandong
Pang
a,
Jialuo
Li
 a,
Xinyu
Yang
a,
Mingbao
Feng
c,
Peiyu
Cai
a and
Hong-Cai
Zhou
*ab
a,
Xinyu
Yang
a,
Mingbao
Feng
c,
Peiyu
Cai
a and
Hong-Cai
Zhou
*ab
aDepartment of Chemistry, Texas A&M University, College Station, Texas 77842-3012, USA. E-mail: zhou@chem.tamu.edu
bDepartment of Materials Science and Engineering, Texas A&M University, College Station, Texas 77842, United State
cDepartment of Environmental and Occupational Health, School of Public Health, Texas A&M University, College Station, TX 77843, United State
First published on 31st July 2019
Abstract
The utilization of reactive oxygen species (ROS) in organic transformations is of great interest due to their superior oxidative abilities under mild conditions. Recently, metal–organic frameworks (MOFs) have been developed as photosensitizers to transfer molecular oxygen to ROS for photochemical synthesis. However, visible-light responsive MOFs for oxygen activation remains scarce. Now we design and synthesize two porous MOFs, namely, PCN-822(M) (M = Zr, Hf), which are constructed by a 4,5,9,10-(K-region) substituted pyrene-based ligand, 4,4′,4′′,4′′′-((2,7-di-tert-butylpyrene-4,5,9,10-tetrayl)tetrakis(ethyne-2,1-diyl))-tetrabenzoate (BPETB4−). With the extended π-conjugated pyrene moieties isolated on the struts, the derived MOFs are highly responsive to visible light, possessing a broad-band adsorption from 225–650 nm. As a result, the MOFs can be applied as efficient ROS generators under visible-light irradiation, and the hafnium-based MOF, PCN-822(Hf), can promote the oxidation of amines to imines by activating molecular oxygen via synergistic photo-induced energy and charge transfer.
Introduction
Reactive oxygen species (ROS) are highly active molecules that are commonly formed by normal metabolism of oxygen in aerobic life.1,2 With their superior oxidative abilities, ROS present great potential in various applications, such as industrial wastewater treatment, photochemical synthesis and photodynamic therapy (PDT).3–6 So far, efforts have been devoted to explore artificial chemical systems to produce ROS under environmentally benign conditions. The biomimetic process usually requires a photosensitizer (PS), which can harvest solar energy and then generates ROS via energy transfer and/or electron transfer.7 In particular, organic dyes and transition metal complexes have been extensively studied as photosensitizers to create ROS for organic transformations.8–12 However, most photosensitizers suffer from aggregation-cause quenching (ACQ) at high concentrations, which undoubtedly hampers their performance in solar-energy conversion.13Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), are an emerging class of crystalline porous materials that are assembled with inorganic secondary building units (SBUs) and organic linkers via coordination bonds.14,15 Owing to their intrinsic porosity and infinite tunability, MOFs have shown a vast and ever-expanding array of applications, including gas storage, separation, heterogeneous catalysis, chemical sensing, drug delivery, and proton conduction.15–25 Recently, MOFs have been exploited as a unique platform to integrate photosensitizers for light harvesting and photocatalysis.26–31 For light-driven ROS generation, many MOF-based systems employ predesigned organic and metal–organic chromophores as linkers to impart MOFs with photoactivities.32–34 The confinement and isolation effect of the frameworks prevent the undesired self-aggregation of the photosensitizers, reducing the non-productive decay process of the excited states. Meanwhile, the porous feature of the MOFs facilitates the diffusion of ROS, enhancing their further interaction with incoming reactants.35–37
Among various photochromic compounds, pyrene and its derivatives with the four-ring-fused planar electron-enriched skeleton are well-known for their unique optical and photoelectronic properties.38 They have been demonstrated to possess a low energy triplet state as well as significant O2-induced enhanced intersystem crossing (EISC), which are favourable to generate ROS, especially singlet oxygen (1O2), through energy transfer.39–41 Several pyrene-based MOFs have been investigated for 1O2 generation.37,42–44 However, as the absorption bands of the reported materials mainly reside in the UV region, they exhibit inferior performance upon visible-light irradiation. Therefore, the development of a MOF-based system that can take advantage of the merits of pyrene to produce 1O2, while utilizing visible light, is desirable.
Bearing these considerations in mind, we report two visible-light responsive MOFs, PCN-822(Zr/Hf), consist of a judiciously designed pyrene-based ligand, 4,4′,4′′,4′′′-((2,7-di-tert-butylpyrene-4,5,9,10-tetrayl)tetrakis(ethyne-2,1-diyl))-tetrabenzoate (BPETB4−). Solvothermal reactions between H4BPETB and Zr6/Hf6 cluster afforded PCN-822(Zr/Hf) with a (4,8)-connected sqc topology. Due to the well-isolated pyrene chromophores in the framework, the MOFs preserve the visible-light absorption of H4BPETB and, simultaneously, show improved efficiency in singlet oxygen generation. PCN-822(Hf) can also be used as a photosensitizer for aerobic photo-oxidation of amines to imines. The heavy Hf6 clusters enhance the intersystem crossing (ISC) in the material, promoting the generation of singlet oxygen. Furthermore, the photo-induced charge separation was also observed in the system, which triggers the O2˙− production. Combining the energy and charge transfer, PCN-822(Hf) exhibits excellent photocatalytic activity in transforming amines to imines.
Results and discussion
The pyrene-based ligand was synthesized in a moderate yield via modified Sonogashira coupling reaction.45 In H4BPETB, the four phenylacetylenic substituents are attached to the 4,5,9,10-positions (K-region) of pyrene with the aim of extending the π conjugation of the pyrene chromophore, more importantly, red-shifting the wavelength of absorption. It has been revealed that the electrophilic substitution of pyrene prefers to occur at 1,3,6,8-positions.46 To tackle the synthetic obstacle, two tBu groups were attached to the 2- and 7-positions, leading to the K-region substitution (Scheme S1†). Yellow single crystals of PCN-822(Zr) were obtained under solvothermal reaction conditions between zirconium(IV) chloride and H4BPETB with trifluoroacetic acid (TFA) as a modulating reagent. Similar reaction with hafnium(IV) chloride as a starting material afforded single crystals of PCN-822(Hf). Powder X-ray diffraction (PXRD) studies demonstrated the crystalline phase purity of both MOFs. As the MOFs share the same framework structure confirmed by single-crystal diffraction studies, the structure of PCN-822(Zr) is discussed as a representative (Fig. 1 and Table S1†). PCN-822(Zr) crystalizes in the tetragonal I41/amd space group. In the Zr6(μ3-O)4(μ3-OH)4 cluster core, six Zr atoms form an idealized octahedron where eight edges are bridged by eight carboxylate groups from BPETB4− ligands and the triangular faces are capped by eight μ3-oxygen atoms. The symmetry of the cluster core is D2d, which is distorted from the ideal cubic symmetry (Fig. 1a). In the BPETB linker, each carboxylate connects with one Zr6 cluster and the peripheral benzene rings have a dihedral angle of 10.99° deviated from the pyrene plane (Fig. 1b). Consequently, PCN-822(Zr) adopts a three-dimensional (3D) structure with a (4,8)-connected sqc topology, which is isostructural to PCN-225.47 The framework contains two types of channels along the a and b-axes, while no apparent pores are observed along c-axis. The small channel with a size of 0.6 × 1.8 nm2 in quadrangle shape is surrounded by two Zr6 clusters and two BPETB4− ligands, whereas the larger one with a size of 0.8 × 3.2 nm2 in pear-like shape is comprised of three Zr6 clusters and three BPETB4− ligands (Fig. 1c). The solvent accessible volume in fully evacuated PCN-822(Zr) is 56.5% calculated by Mercury with a probe of 1.8 Å. Due to the relatively large 1D channels in two of the three dimensions, activation attempts of the MOFs for N2 adsorption isotherm measurements were unsuccessful. Instead, the porosity of the materials was confirmed by dye adsorption in solution, which shows that the materials could adsorb ∼ two methyl orange (MO, ∼1.42 nm × 0.35 nm × 0.19 nm) per formula unit (S6†).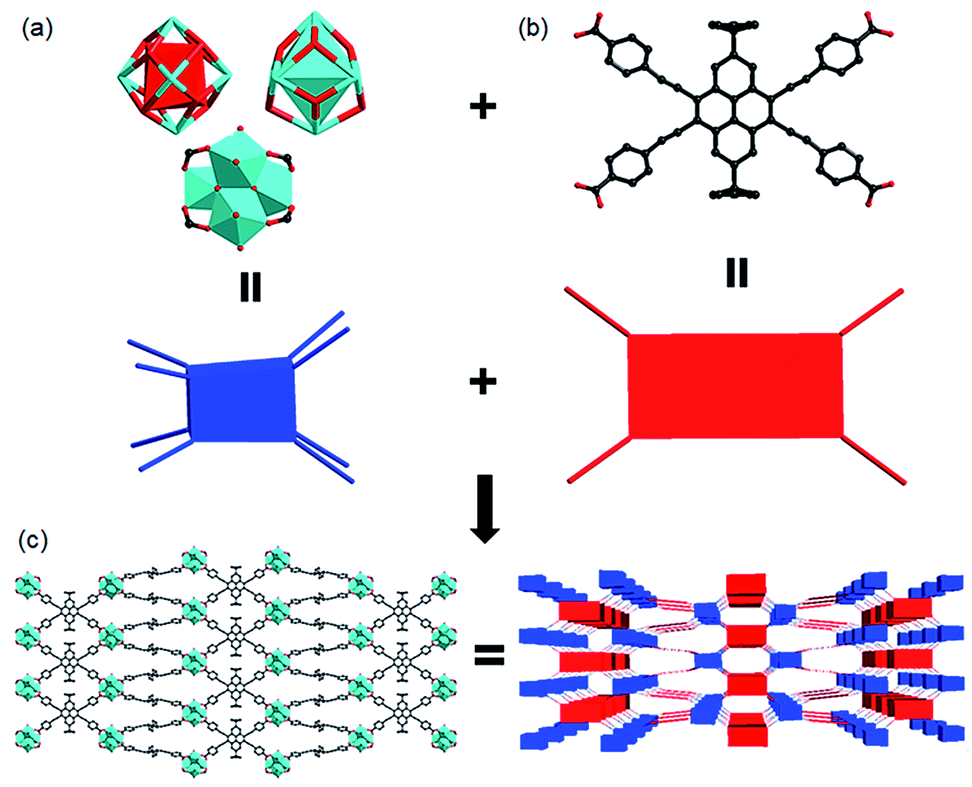 | ||
| Fig. 1 Representative structure and topology of (a) the 8-connected Zr6/Hf6 clusters and (b) the tetratopic BPETB4− that constitute (c) the (4,8)-connected PCN-822. | ||
Both MOFs possess excellent thermal and chemical stability. Thermal stability of PCN-822(Zr/Hf) was evaluated by thermogravimetric (TG) analyses under air stream. It shows that the decomposition temperature of the two MOFs is around 450 °C and the weight loss before 320 °C corresponds to the loss of isolated N,N-dimethylformamide (DMF) molecules and coordinated hydroxide groups/water molecules (Fig. S4†). Water treatment tests were also performed to assess chemical stability of the MOFs. The as-synthesized samples were immersed into aqueous solutions with various pH values and allowed to stay overnight. The PXRD patterns of the samples after water treatment remain unchanged, indicating the crystallinity of the MOFs in the aqueous solutions with pH values ranging from 2 to 11 was preserved (Fig. 2).
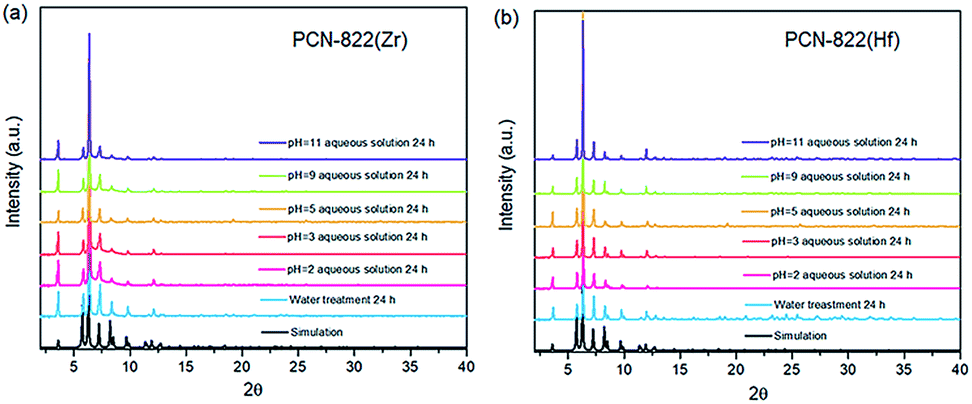 | ||
| Fig. 2 PXRD patterns of (a) PCN-822(Zr) and (b) PCN-822(Hf) after water treatment for 24 hours. Both MOFs retain their crystallinity in aqueous solutions with pH values from 2–11. | ||
Photophysical properties
Given a good photosensitizer is supposed to have efficient absorption of light, especially in the visible range, we initially collected the solid-state absorption of PCN-822(Zr), PCN-822(Hf) and H4BPETB.48 According to the UV-vis spectra, H4BPETB exhibits a broad absorption band ranging from 225 to 700 nm, while PCN-822 (Zr/Hf) have slightly narrowed absorption bands from 225 to 650 nm (Fig. 3a, dashed lines). Obviously, the electrons of the MOFs could be sufficiently promoted to an excited state upon blue-green light irradiation. Density functional theory (DFT) calculations were conducted to study the optical band gap of the materials (Fig. S15†). It has been shown that the highest occupied molecular orbital (HOMO) of H4BPETB in free state is mostly delocalized over the central π-conjugated pyrene core, and the lowest unoccupied molecular orbital (LUMO) is formally delocalized over both the central π-conjugated pyrene core and the exterior phenylacetylenic carboxylate units. In comparison with free-state H4BPETB, BPETB4− shows similar frontier molecular orbital distributions in the MOFs, which explains the almost unchanged absorption of the MOF fragments.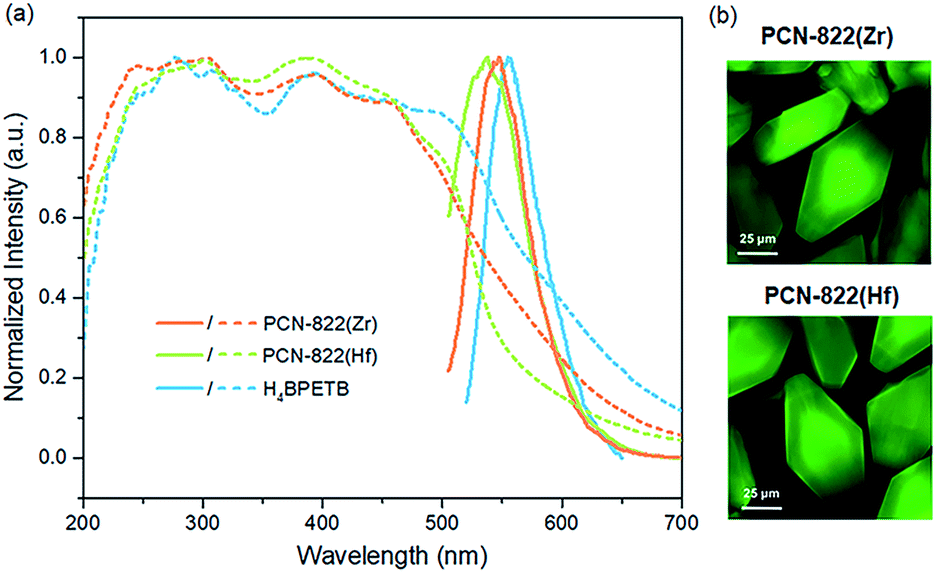 | ||
| Fig. 3 (a) UV-vis spectra of PCN-822(Zr/Hf), H4BPETB (dashed line) and solid-state photoluminescent spectra of PCN-822(Zr/Hf) and H4BPETB (solid line). (b) Fluorescent microscopic images of PCN-822(Zr/Hf) upon excitation at 488 nm. | ||
As pyrene is a conventional fluorophore, the photo-luminescence (PL) properties of H4BPETB and the MOFs were also studied. H4BPETB emits green light centred at 554 nm, while the MOFs show emission maxima at 547 nm and 538 nm for PCN-822(Zr) and PCN-822(Hf), respectively (Fig. 3a, solid lines). The small blue-shifts as well as the similar emission profiles between the ligand and the MOFs indicate that the emission is ligand-based. The green fluorescence of MOFs was further confirmed by the confocal scanning microscopic images shown in Fig. 3b.
Photooxidation of 1,5-dihydroxynaphthalene (DHN)
The ability of PCN-822(Zr/Hf) in singlet oxygen generation was evaluated by photooxidation of 1,5-dihydroxynaphthalene (DHN), where DHN acts as an efficient 1O2 scavenger to produce 5-hydroxy-1,4-naphthalenedione (juglone) (Fig. 4a).49 The reactions were carried out with 0.036 mmol of DHN and 1% PCN-822(Zr/Hf) in acetonitrile under the irradiation of visible light and the UV-vis absorption of the mixture was recorded at an interval of one hour. As the reaction occurred, the absorption peak of DHN at 301 nm decreased, whereas the characteristic peak of juglone at 410 nm increased gradually. With PCN-822(Hf) as a photosensitizer, the absorption peak of DHN disappeared completely in 6 hours, indicating the consumption of the starting material (Fig. 4b). However, when PCN-822(Zr) was used as a photosensitizer, it took longer time for the DHN absorption band to decay, meanwhile, the new absorption band of juglone developed more slowly than that sensitized by PCN-822(Hf) (Fig. 4c and S16†). Clearly, PCN-822(Hf) exhibits higher capability in 1O2 generation. The better performance might be ascribed to the heavy-metal effect of hafnium, which enhances the intersystem crossing of the materials. The reliability of the measurements was further tested by a control experiment with no addition of photosensitizer. The absorption spectra show slightly increase at 410 nm and negligible changes at 301 nm, revealing the indispensable role of photosensitisers (Fig. S17†).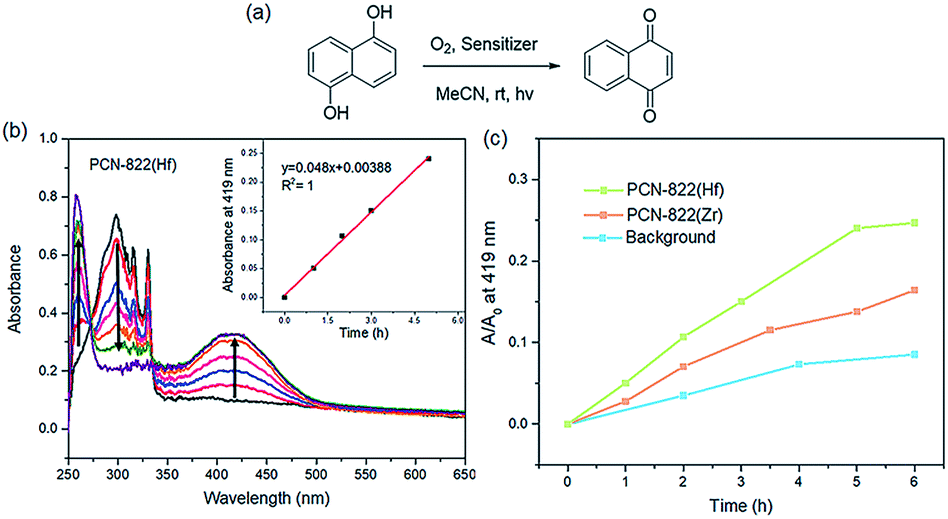 | ||
| Fig. 4 (a) Photooxidation of DHN sensitized by PCN-822 and H4BPETB in the present of oxygen and light irradiation. (b) Changes of UV-vis spectra for oxidation of DHN sensitized by PCN-822(Hf). Insert: changes of juglone absorbance (at 419 nm) with reaction time. (c) Comparison of PCN-822(Hf) and PCN-822(Zr) in juglone development. | ||
Oxidation of various amines
With the excellent performance in 1O2 generation, PCN-822(Hf) was chosen as photosensitizer for aerobic oxidation of amines to imines. Imines and derivatives are versatile intermediates in organic transformations and they are widely used in industry for the synthesis of fine chemicals and pharmaceuticals. Traditional synthetic protocols of imines generally involve unstable aldehydes, dehydrating agents as well as Lewis acid catalysts. Hence, significant efforts have been dedicated to develop mild and practical methodologies for imine formation.50,51 Among all the methodologies, photoinduced aerobic oxidation of amines turns out as an effective and sustainable approach from the view of green chemistry, as it can efficiently utilize sunlight, requiring relative mild and neat conditions.52–55 At the outset, dibenzylamine was chosen as a model substrate to optimize the reaction conditions (Table S2†). The reaction was generally performed under O2 atmosphere upon excitation of visible light at room temperature. It turned out that solvents exerted remarkable influence on oxidation of dibenzylamine, where acetonitrile was proved to be the most suitable medium compared with other types of solvents (entries 1–5). It was also noted that prolonged reaction deteriorated the formation of target product, leading to the dominating aldehyde byproducts (entry 4). Finally, with 1.0 mol% of PCN-822(Hf) photosensitizer, the starting material could be efficiently converted to the imine-based product with the highest yield in 6 h (entry 6). Furthermore, control experiments were conducted without light irradiation or O2 atmosphere (entries 7 and 8). As expected, no product was observed under these reaction conditions. The necessity of photosensitizer was also confirmed by entry 9 with no addition of PCN-822(Hf) and it gave no yield of imine. Interestingly, when the esterified ligand, Me4BPETB, was used as photosensitizer, only 22.2% yield of product was achieved. The dramatic decrease in the formation of imine might be partially ascribed to the aggregation-caused quenching (ACQ) effect of the pyrene-based ligand in the solution (entry 10).PCN-822(Hf) also shows excellent chemical stability and recyclability. The MOFs separated from the reaction mixture could be reused two more times without loss of catalytic activity. The PXRD patterns of the samples recovered from each recycle showed no essential change in the profiles from the pristine ones, suggesting the intact crystallinity of the MOFs under the catalytic conditions (Fig. S18 and S19†).
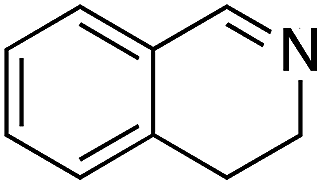
Under the optimized conditions, the oxidation of various amines was further performed to evaluate the general capability of PCN-822(Hf). As shown in Table 1, both primary and secondary amines could be oxidized to the corresponding imines with satisfactory yields. Primary amines underwent oxidative coupling reaction photocatalyzed by PCN-822(Hf). It was noted that para-substituted benzylamines with electron-donating groups (CH3 and OCH3) afforded higher yields of imines than those with electron-withdrawing groups (Cl and CF3), which indicates the remarkable electronic effect on the oxidation reactions (entries 1–5). In addition, no product was observed when catalysing aniline, suggesting that the coupling reaction experiences an abstraction of α-hydrogen of the substrates (entry 6). Compare to primary amines, secondary amines gave imines by oxidative dehydrogenation. Due to the possible oxidative cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, they presented relatively lower yields and selectivity in imine formation (entries 7–10). Substrates with bulky substitutes also reacted smoothly, which might be facilitated by the large channels of the MOF (entries 8–10). Besides aromatic amines, heterocyclic amines such as 1,2,3,4-tetrahydroisoquinoline could also be oxidized to dehydrogenated imine with good yield and high selectivity (entry 11).
N bond, they presented relatively lower yields and selectivity in imine formation (entries 7–10). Substrates with bulky substitutes also reacted smoothly, which might be facilitated by the large channels of the MOF (entries 8–10). Besides aromatic amines, heterocyclic amines such as 1,2,3,4-tetrahydroisoquinoline could also be oxidized to dehydrogenated imine with good yield and high selectivity (entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%) | Sel. (%) |
| a Reaction conditions: amine (0.1 mmol), PCN-822(Hf) (1.0 mol%), acetonitrile (2.0 mL), 1 atm O2, rt., LED light (λ = 450 nm), 100 mW cm−2. Yields were determined by 1H NMR with mesitylene as internal standard. | ||||
| 1 |
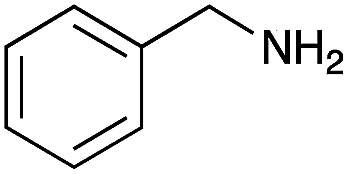
|
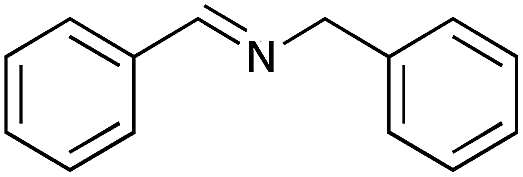
|
93 | 98 |
| 2 |
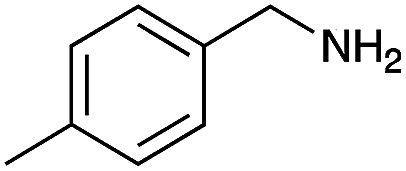
|
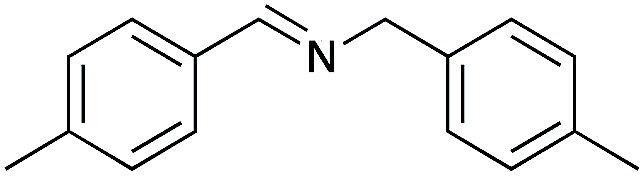
|
90 | 98 |
| 3 |
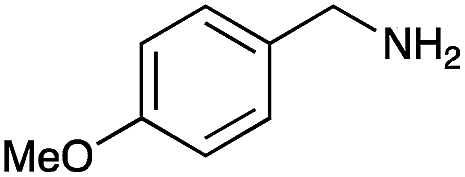
|
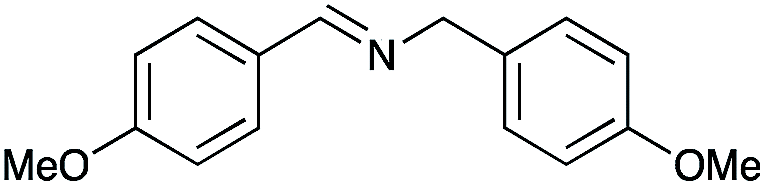
|
91 | 98 |
| 4 |
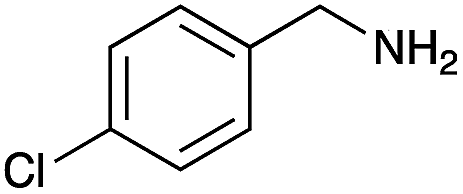
|
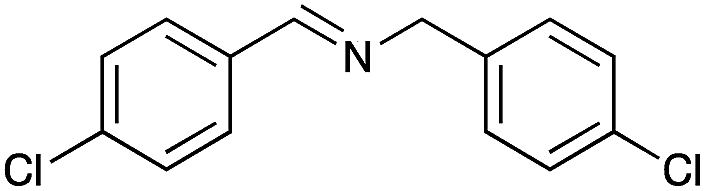
|
81 | 90 |
| 5 |
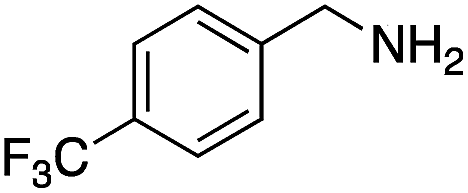
|
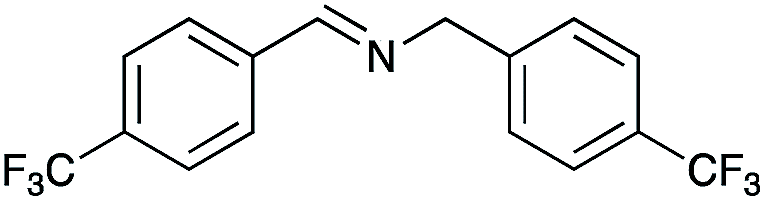
|
83 | 96 |
| 6 |
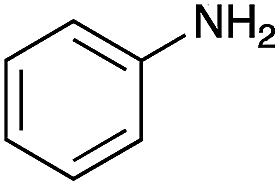
|
— | — | — |
| 7 |
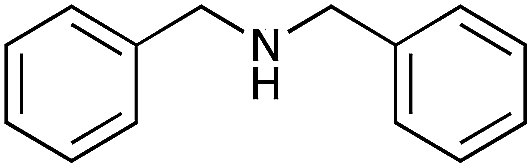
|
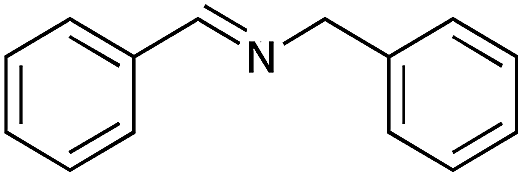
|
79 | 84 |
| 8 |
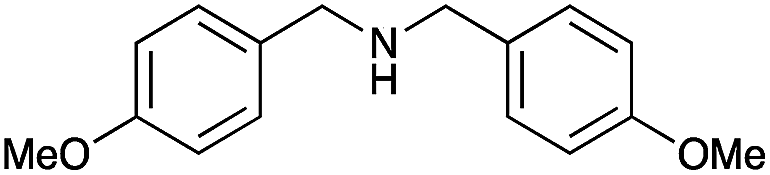
|
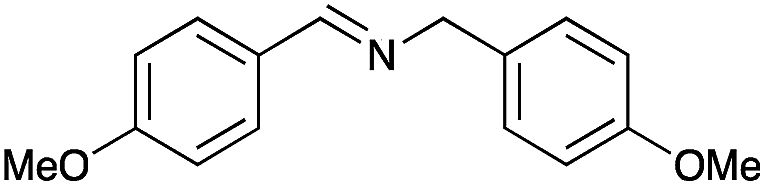
|
76 | 83 |
| 9 |
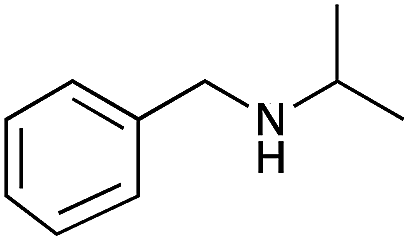
|
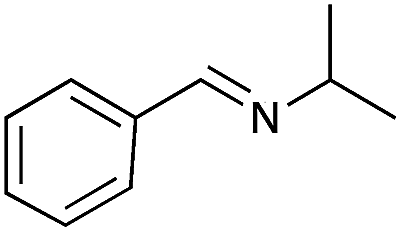
|
82 | 86 |
| 10 |
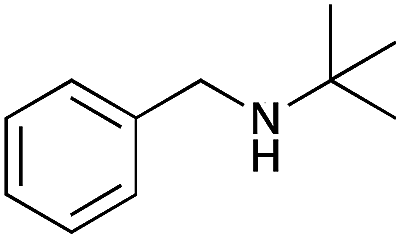
|
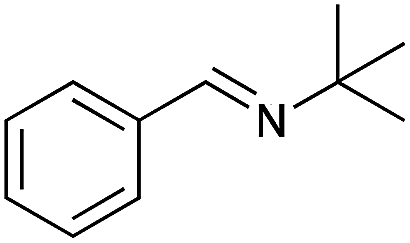
|
79 | 85 |
| 11 |
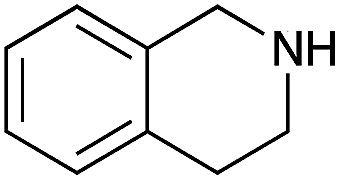
|
|
92 | 95 |
Mechanism analysis
Although the mechanism underlying the oxidation reaction remains disputable, two possible pathways are proposed to explain the excellent photocatalytic performance of PCN-822(Hf) (Fig. 5). Firstly, as PCN-822(Hf) has been proved to be an excellent singlet oxygen generator, a mechanism is proposed where the material produces 1O2 species to oxidize amine via energy transfer upon the light irradiation. The generation of the singlet oxygen was further confirmed by electron paramagnetic resonance (EPR) spectra, which displayed featured signals in the (Fig. S20†). Regarding to the semiconductor nature of MOFs, an alternative mechanism involving photoinduced electron-transfer process is also operable. It is assumed that the reaction is initiated by the excitation of PCN-822(Hf) upon visible-light irradiation, which gives rise to electron (e−) and hole (h+) pairs separated in the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the material. The photogenerated electron reduces molecular oxygen to its activated superoxide radical species, O2˙−, while the hole oxidizes amine to a cationic amine radical. The amine radical and O2˙− then react with each other to produce imine via proton and hydrogen transfer. For primary amine, the formed imine serves as an intermediate, which is further attacked by another amine to give the final coupled product. EPR spectra showed an enhanced signal upon light irradiation relative to that taken under darkness, indicating the photogenerated radicals, like electron–hole pairs (Fig. S21†). To further ascertain the photoinduced electron-transfer mechanism, the energy diagram of PCN-822(Hf) was estimated by UV-vis spectra combined with cyclic voltammetry (S10†). According to cyclic voltammogram, the reduction potential of PCN-822(Hf) was −0.55 V vs. standard hydrogen electrode (SHE), corresponding to LUMO level (Fig. S22†). The relatively more negative potential theoretically enables the material to reduce molecular oxygen to O2˙− (E(O2/O2˙−) = −0.33 V vs. SHE). The optical band gap of PCN-822(Hf) was calculated to be 2.17 eV on the basis of Tauc plot, from which HOMO level was determined to be 1.62 V vs. SHE (Fig. S12†). As the E1/2 (M+/M) of amines is around +1.47 V vs. SHE, the more positive HOMO level of PCN-822(Hf) should be sufficient for the amine oxidation.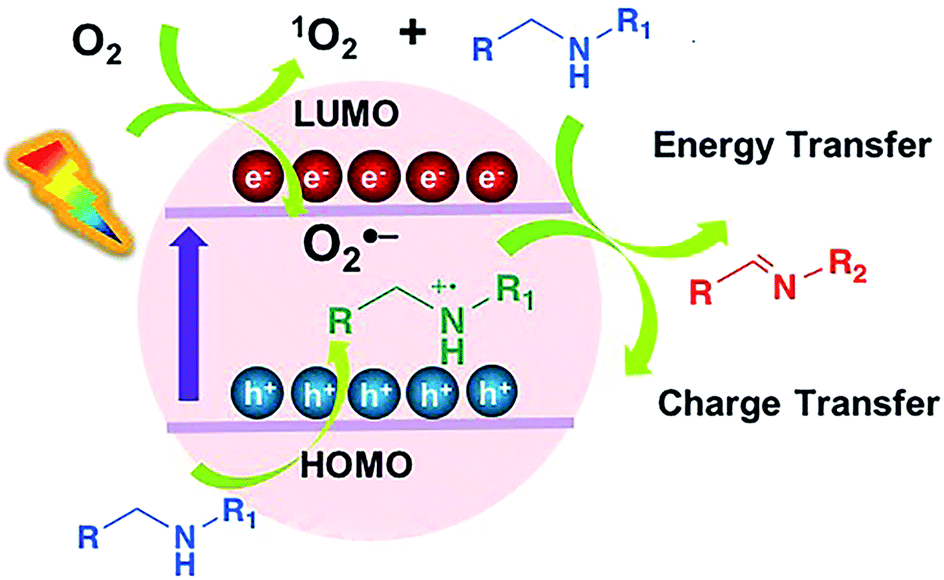 | ||
| Fig. 5 Proposed mechanisms of oxidation of amines. | ||
Conclusions
In summary, a tetratopic pyrene-based ligand (H4BPETB) was rationally designed to construct two novel porous MOFs, PCN-822(Zr/Hf). The MOFs have a broad adsorption of visible light up to 650 nm. The desirable optical adsorption of the materials is mainly ascribed to the 4,5,9,10-positions (K-region) substitution of the pyrene-derived ligand, which leads to an extended π-conjugated moiety. Merging the merits of pyrene in singlet oxygen generation with visible-light harvesting, PCN-822(Zr/Hf) exhibit an efficient capability in activating O2 to 1O2 under the excitation of visible light. Furthermore, PCN-822(Hf) is demonstrated to be highly active in oxidation of various amines to imines. Possible dual pathways are proposed for the light-driven transformation, involving the energy transfer-based 1O2 generation and photo-induced charge separation. The present work not only exemplifies novel pyrene-based MOFs as inherent visible-light harvesting photosensitizers without the aid of other chromophores, but also illustrates the necessity of the structural design at the molecular level in the MOF-based photosynthesizing system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Centre for Gas Separations, an Energy Frontier Research Centre funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (DE-SC0001015), U.S. Department of Energy, Office of Fossil Energy, National Energy Technology Laboratory (DE-FE0026472) and Robert A. Welch Foundation through a Welch Endowed Chair to H.-C. Z. (A-0030). The authors also acknowledge the financial support of NPRP award NPRP9-377-1-080 from the Qatar National Research Fund. Use of the TAMU Materials Characterization Facility is also acknowledged.Notes and references
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- B. Enrico, D. G. Tiziana, L. Osvaldo and L. Andrea, J. Org. Chem., 2007, 72, 9582–9589 CrossRef PubMed.
- C. Tanielian, C. Wolff and M. Esch, J. Phys. Chem., 1996, 100, 6555–6560 CrossRef CAS.
- M. C. Derosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- W. Zhang, B. Li, H. Ma, L. Zhang, Y. Guan, Y. Zhang, X. Zhang, P. Jing and S. Yue, ACS Appl. Mater. Interfaces, 2016, 8, 21465–21471 CrossRef CAS PubMed.
- X. Ding and B. H. Han, Angew. Chem., Int. Ed., 2015, 54, 6536–6539 CrossRef CAS PubMed.
- R. Kumar, E. H. Gleissner, E. G. Tiu and Y. Yamakoshi, Org. Lett., 2016, 18, 184–187 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- W.-P. To, G. S.-M. Tong, W. Lu, C. Ma, J. Liu, A. L.-F. Chow and C.-M. Che, Angew. Chem., 2012, 124, 2708–2711 CrossRef.
- I. Pibiri, S. Buscemi, A. Palumbo Piccionello and A. Pace, ChemPhotoChem, 2018, 2, 535–547 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- J. R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- N. L. Rosi, E. Juergen, E. Mohamed, D. T. Vodak, K. Jaheon, O. K. Michael and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- J. A. Mason, O. Julia, M. K. Taylor, M. R. Hudson, R. Julien, J. E. Bachman, M. I. Gonzalez, C. Antonio, G. Antonietta and C. M. Brown, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- Y. B. Huang, J. Liang, X. S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- G. M. Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
- H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729–4756 RSC.
- M. Rimoldi, A. J. Howarth, M. R. DeStefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, ACS Catal., 2016, 7, 997–1014 CrossRef.
- P. Ramaswamy, N. E. Wong and G. K. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- S. Yuan, J. S. Qin, H. Q. Xu, J. Su, D. Rossi, Y. Chen, L. Zhang, C. Lollar, Q. Wang, H. L. Jiang, D. H. Son, H. Xu, Z. Huang, X. Zou and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 105–111 CrossRef CAS PubMed.
- D. Chen, H. Xing, C. Wang and Z. Su, J. Mater. Chem. A, 2016, 4, 2657–2662 RSC.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS.
- H. Liu, C. Xu, D. Li and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., 2015, 127, 9129–9133 CrossRef.
- J. Park, Q. Jiang, D. Feng and H.-C. Zhou, Angew. Chem., 2016, 128, 7304–7309 CrossRef.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 12502–12510 CrossRef CAS PubMed.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS PubMed.
- J. Park, D. Feng, S. Yuan and H.-C. Zhou, Angew. Chem., 2015, 127, 440–445 CrossRef.
- C. T. Buru, M. B. Majewski, A. J. Howarth, R. H. Lavroff, C. W. Kung, A. W. Peters, S. Goswami and O. K. Farha, ACS Appl. Mater. Interfaces, 2018, 10, 23802–23806 CrossRef CAS PubMed.
- A. J. Howarth, M. B. Majewski and M. O. Wolf, Coord. Chem. Rev., 2015, 282–283, 139–149 CrossRef CAS.
- T. GeorgeáTruscott and J. Edward, J. Chem. Soc., Faraday Trans., 1990, 86, 3075–3080 RSC.
- M. Hissler, A. Harriman, A. Khatyr and R. Ziessel, Chem.–Eur. J., 1999, 5, 3366–3381 CrossRef CAS.
- M. Okamoto and F. Tanaka, J. Phys. Chem. A, 2002, 106, 3982–3990 CrossRef CAS.
- Y. Liu, C. T. Buru, A. J. Howarth, J. J. Mahle, J. H. Buchanan, J. B. DeCoste, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 13809–13813 RSC.
- A. Atilgan, T. Islamoglu, A. J. Howarth, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 24555–24560 CrossRef CAS PubMed.
- A. J. Howarth, C. T. Buru, Y. Liu, A. M. Ploskonka, K. J. Hartlieb, M. McEntee, J. J. Mahle, J. H. Buchanan, E. M. Durke, S. S. Al-Juaid, J. F. Stoddart, J. B. DeCoste, J. T. Hupp and O. K. Farha, Chem.–Eur. J., 2017, 23, 214–218 CrossRef CAS PubMed.
- J.-y. Hu, M. Era, M. R. J. Elsegood and T. Yamato, Eur. J. Org. Chem., 2010, 2010, 72–79 CrossRef.
- Z.-H. Wu, Z.-T. Huang, R.-X. Guo, C.-L. Sun, L.-C. Chen, B. Sun, Z.-F. Shi, X. Shao, H. Li and H.-L. Zhang, Angew. Chem., 2017, 129, 13211–13215 CrossRef.
- H. L. Jiang, D. Feng, K. Wang, Z. Y. Gu, Z. Wei, Y. P. Chen and H. C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- J. Sun, J. Zhao, H. Guo and W. Wu, Chem. Commun., 2012, 48, 4169–4171 RSC.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, Angew. Chem., Int. Ed., 2003, 42, 4077–4082 CrossRef CAS PubMed.
- G. Jiang, J. Chen, J.-S. Huang and C.-M. Che, Org. Lett., 2009, 11, 4568–4571 CrossRef CAS PubMed.
- F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., 2011, 123, 683–686 CrossRef.
- D. Sun, L. Ye and Z. Li, Appl. Catal., B, 2015, 164, 428–432 CrossRef CAS.
- C. Xu, H. Liu, D. Li, J. H. Su and H. L. Jiang, Chem. Sci., 2018, 9, 3152–3158 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1908132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03080h |
| This journal is © The Royal Society of Chemistry 2019 |