
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoredox-catalyzed stereoselective alkylation of enamides with N-hydroxyphthalimide esters via decarboxylative cross-coupling reactions†
Jing-Yu
Guo
a,
Ze-Yu
Zhang
a,
Ting
Guan
a,
Lei-Wen
Mao
a,
Qian
Ban
a,
Kai
Zhao
 *a and
Teck-Peng
Loh
*ab
*a and
Teck-Peng
Loh
*ab
aInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, Nanjing 211816, China. E-mail: ias_kzhao@njtech.edu.cn
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: teckpeng@ntu.edu.sg
First published on 5th August 2019
Abstract
Stereoselective β-C(sp2)–H alkylation of enamides with redox-active N-hydroxyphthalimide esters via a photoredox-catalyzed decarboxylative cross-coupling reaction is demonstrated. This methodology features operational simplicity, broad substrate scopes, and excellent stereoselectivities and functional group tolerance, affording a diverse array of geometrically defined and synthetically valuable enamides bearing primary, secondary or tertiary alkyl groups in satisfactory yields.
Introduction
As a crucial subclass of enamines endowed with a delicate balance of reactivity and stability, enamides have attracted increasing attention among the chemical community as pivotal and versatile building blocks which are of recognized synthetic value in the construction of biologically and pharmaceutically active molecules,1 especially small but complex nitrogen-containing compounds.2 In the past few decades, we have witnessed a booming development in new synthetic strategies for the regio- and stereo-selective functionalization of enamides, especially at their β-C(sp2)–H bond, which are capable of producing enamides bearing a diverse array of functional groups through arylation,3 alkenylation,4 trifluoromethylation,5 difluoroacetylation,6 alkynylation,7 acylation,8 sulfonylation9 and other useful transformations.10 Nevertheless, the coupling of alkyl moieties to enamides has been considered a more challenging task with scarce advances demonstrated.11 One of the existing scenarios for the direct C–H alkylation of enamides was achieved by using electron-deficient bromides as alkylating agents as established by Yu and co-workers through visible-light photoredox-catalysis (Scheme 1a, eqn (1))11a and by our group through a palladium-catalyzed strategy (Scheme 1a, eqn (2))11b. Recently, another elegant methodology for the branch-selective alkylation of enamides with terminal olefins was demonstrated by Dong and co-workers (Scheme 1b).11c However, the success to date has been somewhat restricted with respect to the limited scope of both enamides and alkylating reagents and the relatively strict reaction conditions. Thus, the development of a robust and generally applicable method for the preparation of enamides bearing a diverse range of alkyl groups with versatile functionalities has been considered a remaining challenge.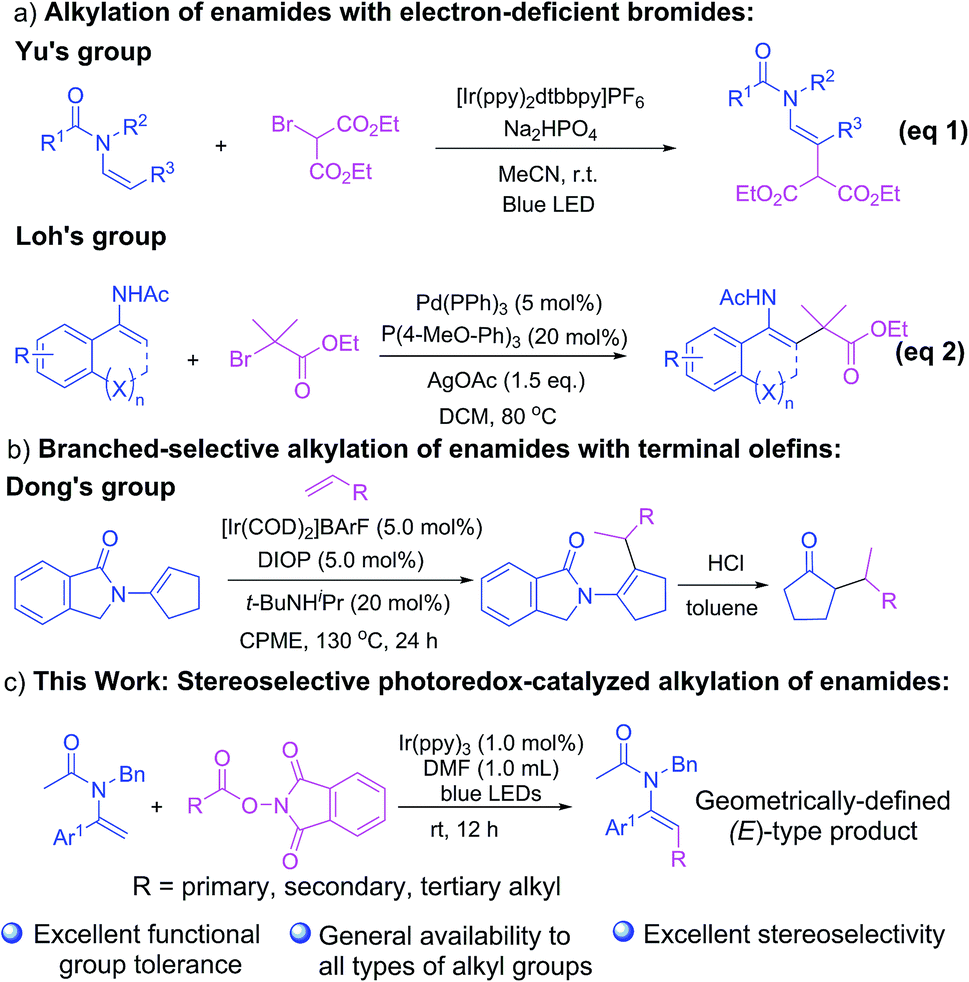 | ||
| Scheme 1 Alkylation of enamides. | ||
The redox-active alkyl N-hydroxyphthalimide esters (NHP) derived from alkanoic acids, as demonstrated for the first time by Okada12 and Overman,13 have entered into an era of “Renaissance” in the past few years in a myriad of cross-electrophile coupling reactions as C(sp3) radical equivalents through single-electron-transfer reduction and decarboxylation. Recent advances in this arena have witnessed a rapid development in a broad range of decarboxylative cross-coupling reactions to forge C(sp3)–C or C(sp3)–X (X = Si, B, Se, etc.) bonds via transition-metal14,15 and photoredox catalysis,16,17 as elegantly established by the groups of Baran,14a–g,o,15b Weix,14h,i Fu,16c–g,17a,b Oestreich,15 Phipps,16a Xiao,16l and many others.18 Very recently, Fu and co-workers demonstrated a brand new catalytic combination of sodium iodide and triphenylphosphine for the cross-coupling of redox-active esters with silyl enol ethers or heteroarenes without resorting to the use of dye or transition-metal based photocatalysts.19 Enlightened by these seminal breakthroughs, we herein demonstrate a robust and practical protocol for stereoselective decarboxylative cross-coupling of NHP esters with enamides, forging a diverse array of geometrically defined alkylated enamides bearing various functional groups under mild conditions (Scheme 1c). Notably, this approach allows the incorporation of various primary, secondary and tertiary alkyl groups into enamides, which represents a significant advance and a crucial complement to existing methods11a,b which only enable the incorporation of electron-deficient secondary alkyl groups.
Results and discussion
At the outset of our investigation, N-benzyl-N-(1-phenylvinyl)acetamide (1a) and 1,3-dioxoisoindolin-2-yl cyclohexanecarboxylate (2a) were selected as model substrates for the screening of optimal reaction conditions (Table 1). Initial screening of common photocatalysts showed that fac-Ir(ppy)3 was superior to Ru(bpy)3Cl2 and Eosin Y (Table 1, entry 1 vs. entries 2 and 3). Further investigation of solvents revealed that DMF was the optimal choice for the transformation (Table 1, entry 1 vs. entries 7–9) and the most appropriate concentration of the enamides was 0.3 M (Table 1, entries 11 and 12 vs. entries 1 and 10). The optimal loading of the photocatalyst proved to be 1.0 mol% with respect to reaction time and efficiency (Table 1, entries 4–6 vs. entry 1). The employment of 1.2 eq. of NHP esters instead of 1.5 eq. led to an increase of the product yield (Table 1, entry 12 vs. entry 11). Control experiments revealed that the photoredox catalyst and light were both of crucial importance for this transformation, and no desired product was formed in the absence of the photocatalyst or without irradiation (Table 1, entries 13 and 14).|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst (mol%) | Solvent | Time (h) | Yieldb |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.45 mmol), and solvent (3.0 mL). b Isolated yields. c 2.0 mL DMF. d 1.0 mL DMF. e 0.36 mmol 2a was used. f The reaction was carried out in darkness. | ||||
| 1 | fac-Ir(ppy)3 (1.0) | DMF | 12 | 63 |
| 2 | Eosin Y (10) | DMF | 12 | 36 |
| 3 | Ru(bpy)3Cl2 (1.0) | DMF | 12 | 52 |
| 4 | fac-Ir(ppy)3 (0.1) | DMF | 24 | 58 |
| 5 | fac-Ir(ppy)3 (0.2) | DMF | 24 | 58 |
| 6 | fac-Ir(ppy)3 (2.0) | DMF | 12 | 49 |
| 7 | fac-Ir(ppy)3 (1.0) | DMAc | 12 | 57 |
| 8 | fac-Ir(ppy)3 (1.0) | CH3CN | 12 | 24 |
| 9 | fac-Ir(ppy)3 (1.0) | DCM | 12 | Trace |
| 10c | fac-Ir(ppy)3 (1.0) | DMF | 12 | 63 |
| 11d | fac-Ir(ppy)3 (1.0) | DMF | 12 | 68 |
| 12d,e | fac-Ir(ppy)3 (1.0) | DMF | 12 | 76 |
| 13 | None | DMF | 12 | 0 |
| 14f | fac-Ir(ppy)3 (1.0) | DMF | 12 | 0 |
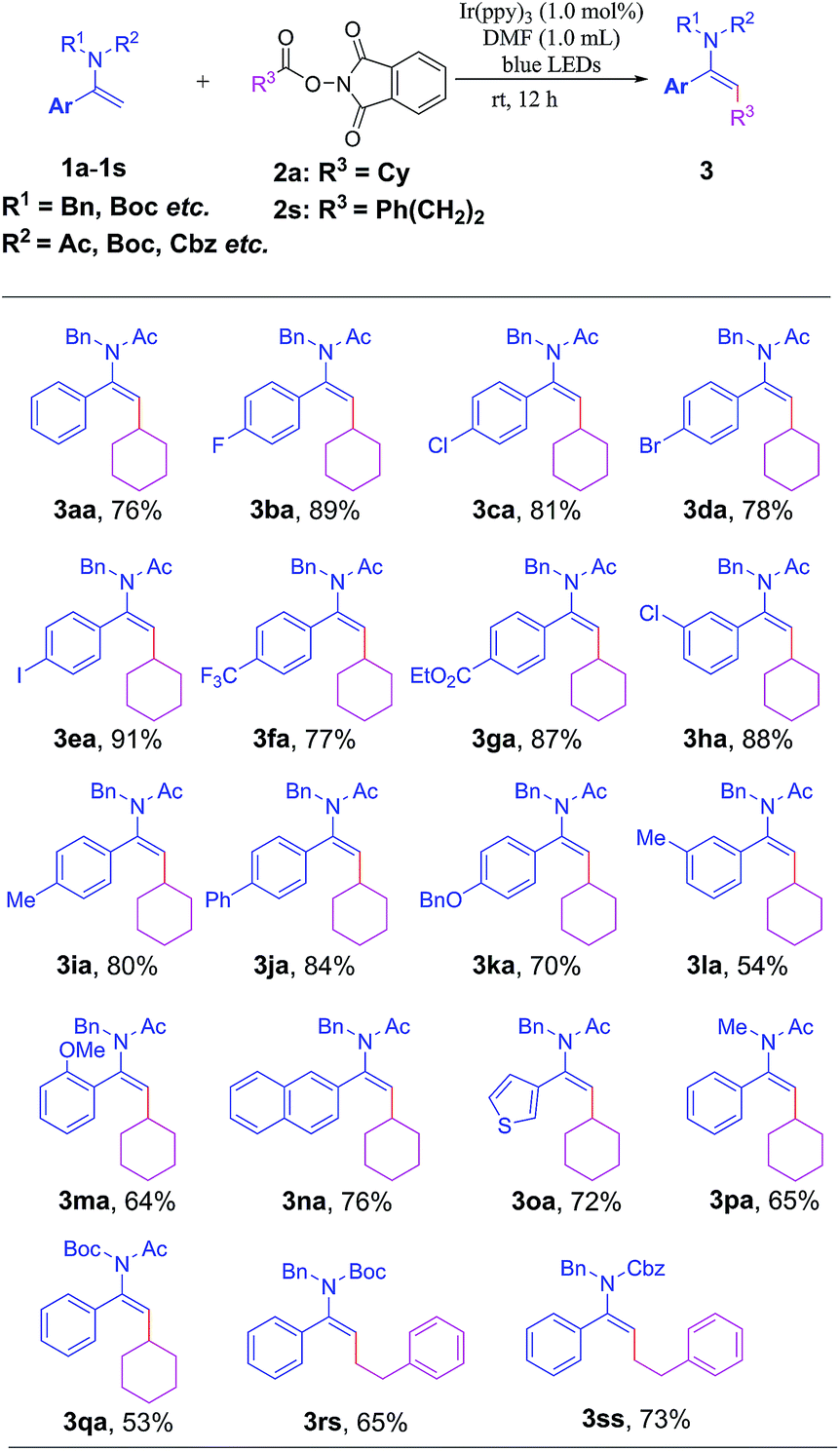
With the optimal reaction conditions in hand, we next examined the substrate scope with regard to different enamides or enecarbamates 1a–1s with NHP esters 2a or 2s; the results are summarized in Table 2. It was found that substrates bearing either electron-withdrawing (1b–1h) or electron-donating groups (1i–1n) were viable in this transformation to furnish the desired products 3ba–3na in considerable yields. The substrates with ortho- or meta-substituents were also well tolerated to give 3ma, 3ha and 3la in synthetically applicable yields, respectively. Substrates bearing halogen atoms (–Cl, –Br, or –I) also afford 3ca–3ea in excellent yields, enabling them to be amenable for further functionalization through cross-coupling reactions. A range of useful functional groups such as CF3 and CO2Et were also applicable to this reaction to give 3fa and 3ga in 77% and 87% yields, respectively. Notably, a heterocyclic skeleton such as a 3-thienyl moiety was also well tolerated to give the target product 3oa in 72% yield. Replacing the N-protecting benzyl group with methyl and Boc did not attenuate the reaction efficiency, affording 3pa and 3qa in synthetically useful yields. Gratifyingly, a handful of enecarbamates with N-Boc (1r) or N-Cbz (1s) substituents could smoothly react with redox-active ester 2s, giving rise to alkylated enamides 3rs and 3ss in 65% and 73% yields, respectively. Notably, in all cases, this transformation proceeded smoothly in a stereoselective manner to afford geometrically defined E-type alkylated enamides (see the ESI† for details); the stereochemistry has been unambiguously confirmed through X-ray crystallography of 3ce as shown in Table 3.20
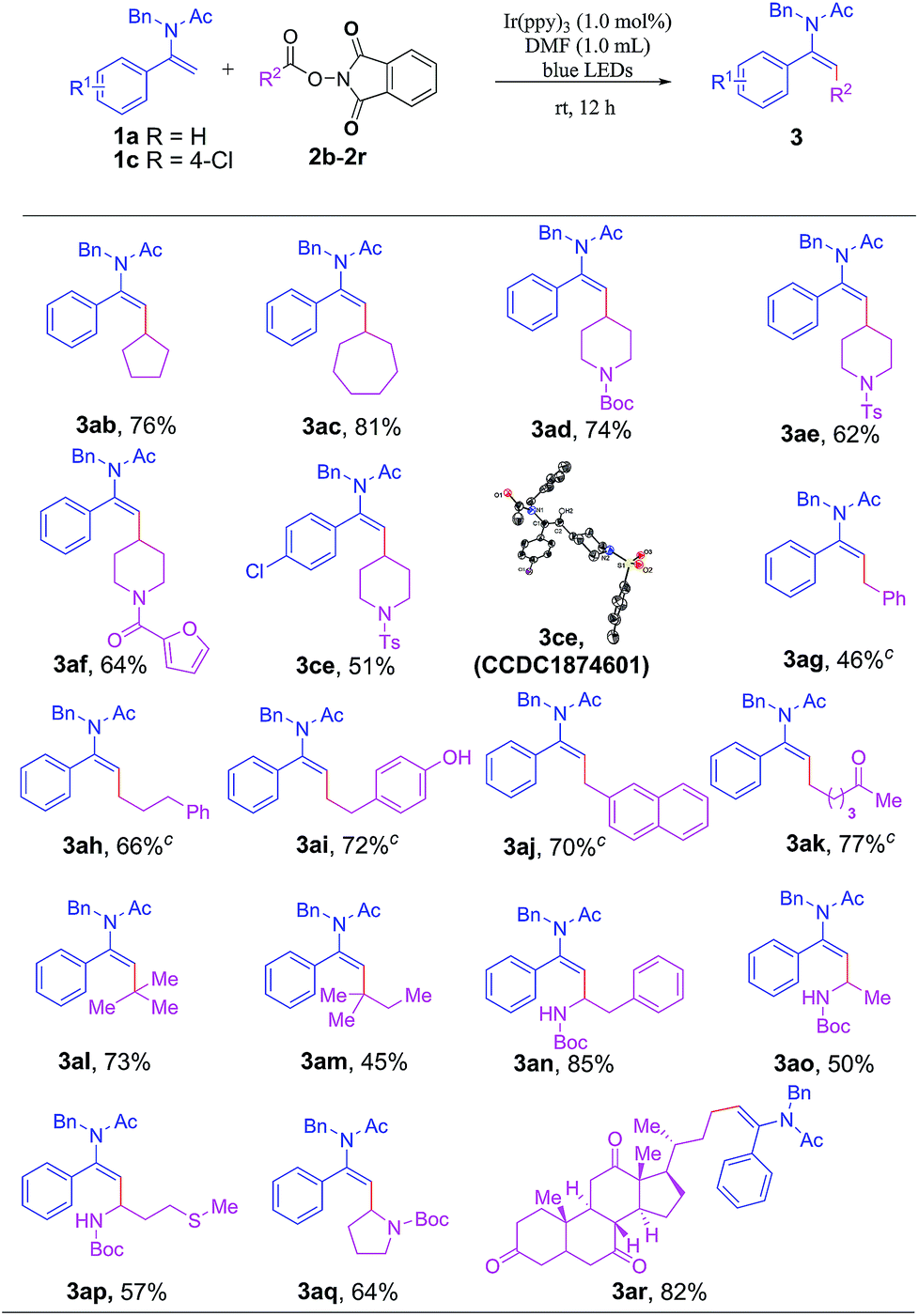
Next, we investigated the generality of this reaction with respect to the scope of various NHP esters (Table 3). A broad range of NHP esters with different cyclic moieties were amenable to this transformation to give 3ab–3af and 3ce in moderate to good yields. It is worth noting that the protecting groups on piperidine such as tert-butyloxycarbonyl (Boc), p-toluenesulfonyl (Ts) or even heterocyclic 2-furancarbonyl were well tolerated. A plethora of NHP esters with primary alkyl groups were also readily applicable to this reaction to forge 3ag–3ak smoothly. Several useful functional groups such as phenol and ketone were also compatible with this transformation to give 3ai and 3ak in good yields, respectively. Especially noteworthy was the excellent compatibility of tertiary alkyl groups for this transformation, enabling the formation of enamides 3al and 3am bearing a quaternary carbon centre which were relatively difficult to be produced through other synthetic methods. In addition, various natural amino acid-derived NHP esters were viable substrates, affording synthetically valuable products 3an–3aq in moderate to good yields. Gratifyingly, an NHP ester bearing a naturally occurring dehydrocholic acid fragment containing three base-sensitive ketone groups was readily amenable to the transformation to afford 3ar in 82% yield.
To showcase the synthetic utility and practicality of this transformation. We have conducted a range of further transformations of the alkylated enamides. A gram-scale reaction of 1a with 2a proceeded smoothly, affording 3aa in good yield and stereoselectivity (Scheme 2a). Notably, upon treatment with trifluoroacetic acid at 110 °C, the E-configured enamides 3aa, 3fa and 3ae could be converted to their Z-isomers in moderate yields (which might be attributed to decomposition), allowing us to easily control the stereochemistry of the alkylated enamides (Scheme 2b).21 The alkylation of enamide 4 with NHP ester 5 proceeded smoothly under standard reaction conditions to give the desired product 6 in 65% yield, which underwent a subsequent palladium-catalyzed intramolecular Heck coupling to furnish a synthetically and pharmaceutically crucial isoquinoline derivative 7 in 76% yield (Scheme 2c). Next, Pd/C-catalyzed hydrogenation of enamide 3aa was successfully conducted under mild conditions to give benzylamine 8 in 69% yield (Scheme 2d). To our delight, the hydrolysis of alkylated enamides in the presence of concentrated HCl (aq.) afforded a broad range of α-alkylated ketones in excellent yields (Scheme 2e). Interestingly, when 3ak was applied under the hydrolysis condition, a cascade hydrolysis-intramolecular cyclization reaction occurred to give 9e in 65% yield (Scheme 2f). Gratifyingly, when alkylated enamide 3aa was treated with m-chloroperoxybenzoic acid (m-CPBA), α-acyloxyketone 10 was obtained in 75% yield after a tandem epoxidation-intramolecular nucleophilic addition–elimination–hydrolysis process (Scheme 2g). It is worth noting that the N-Boc protecting group of 3qa could be removed efficiently by treatment with zinc bromide to give the desired product 11 under mild reaction conditions (Scheme 2h).
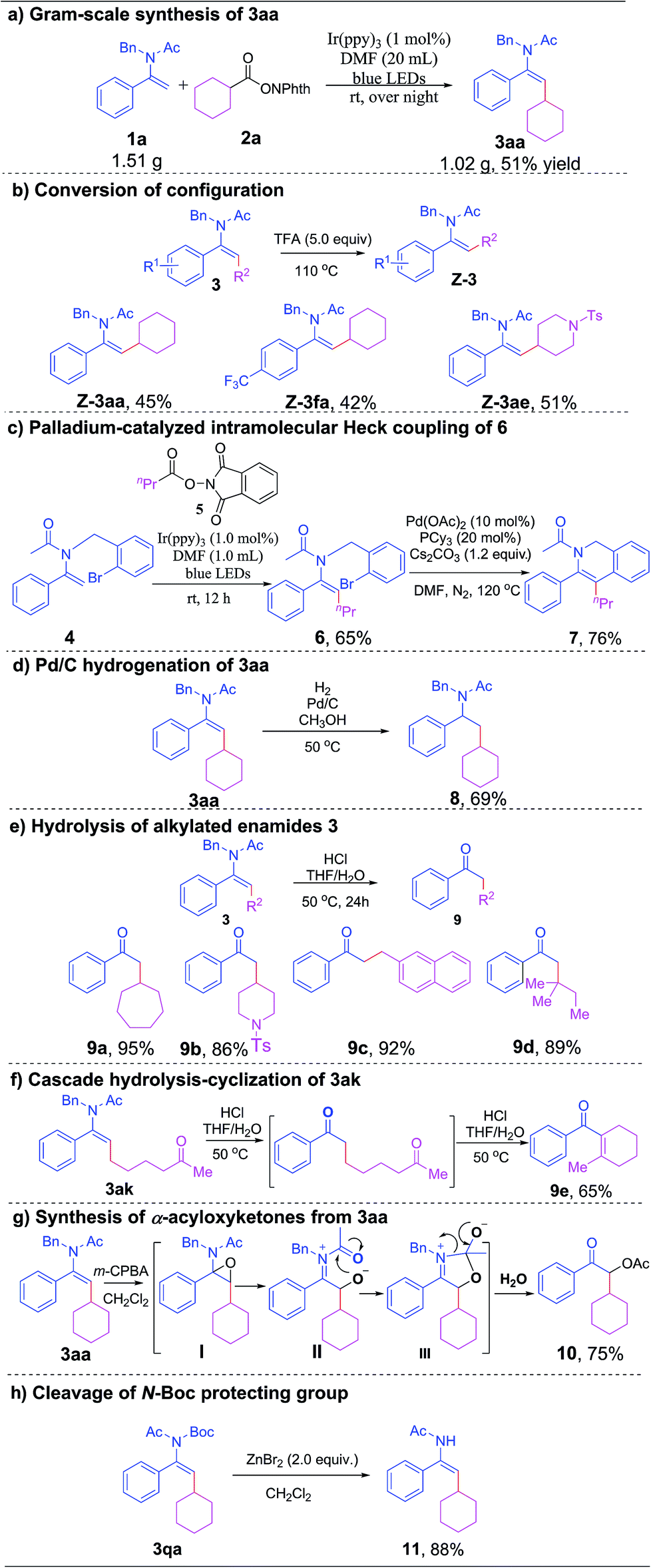 | ||
| Scheme 2 Synthetic applications of alkylated enamides. | ||
A number of preliminary mechanistic studies were conducted to shed more light on the reaction pathway. Initially, a radical-trapping experiment in the presence of a radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was performed. A complete inhibition of the reaction was observed and the alkyl radical could be intercepted by TEMPO to generate intermediate 12 as detected by GC-MS, which suggested that the reaction went through a plausible radical mechanism (Scheme 3a). Secondly, the coupling of a radical-clock-containing NHP ester 13 with enamide 1a afforded the ring-opening product 14, which strongly supported the participation of radical intermediates (Scheme 3b). In addition, we have determined a quantum yield of Φ = 0.71 for the model reaction of 1a with 2a (see the ESI† for details),22 implying that it is highly possible for the reaction to proceed through a photoredox catalytic pathway rather than a radical-chain mechanism.
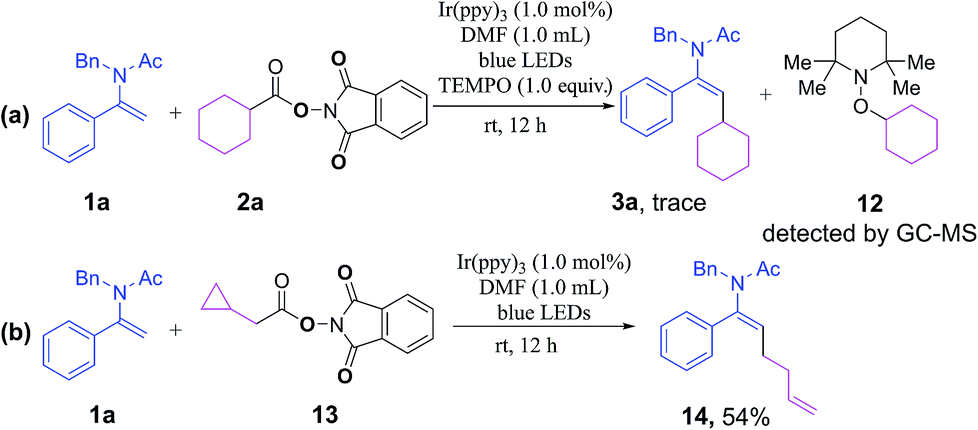 | ||
| Scheme 3 Preliminary mechanistic studies. | ||
Based on the above observations, we have proposed a plausible mechanism for the photoredox-catalyzed decarboxylative alkylation of enamides with NHP esters. Initially, the iridium photocatalyst [fac-Ir(ppy)3] is excited to [fac-Ir(ppy)3]* via the absorption of a photon under blue LED irradiation. Secondly, the single electron transfer (SET) between [fac-Ir(ppy)3]* and NHP ester 2 generates a radical anion A which is readily able to produce an alkyl radical species Bvia decarboxylation. Thirdly, the alkyl radical is intercepted by enamides to furnish a radical intermediate C which is subsequently oxidized by the oxidative photocatalyst [fac-Ir(ppy)3]+ through SET to forge a cationic intermediate D, which is in equilibrium with iminium ion E, along with the regeneration of [fac-Ir(ppy)3]. Finally, the deprotonation of D or E gives alkylated enamides. The stereoselectivity for this transformation could be rationalized through the conformational analysis of iminium ion E:23 conformer 1 is sterically favorable in contrast to conformer 2 in view of minimized allylic strain between the benzyl group and alkyl group, leading to the formation of the E-configured alkylated enamides (Scheme 4).
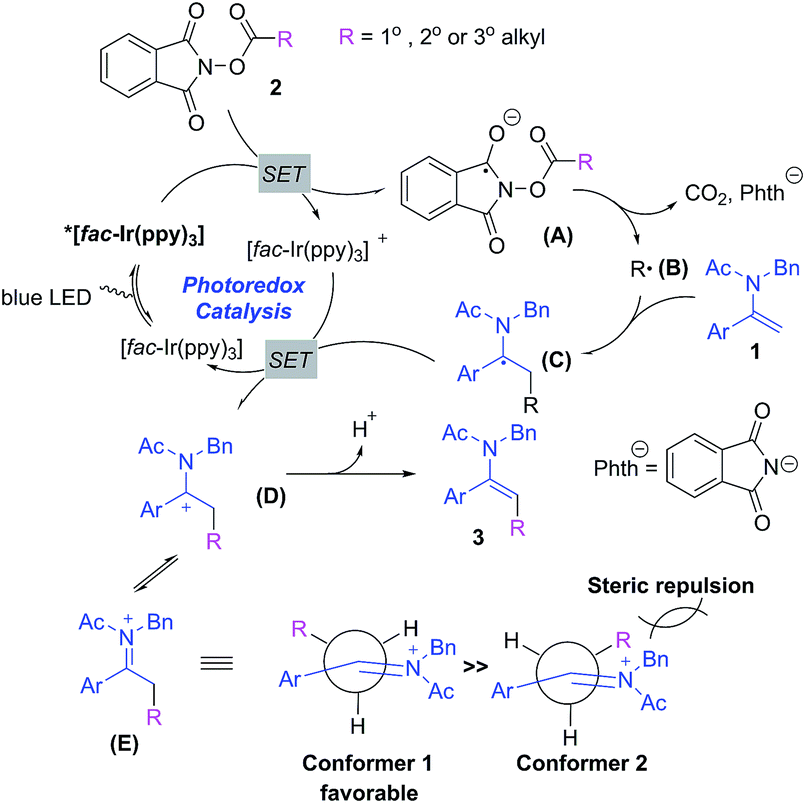 | ||
| Scheme 4 Plausible mechanism. | ||
Conclusions
We have developed a novel, efficient and generally applicable approach for the chemo- and stereo-selective alkylation of enamides with NHP esters. A wide array of enamides and NHP esters bearing various functional groups were viable for this protocol to afford synthetically important and geometrically defined enamides bearing primary secondary or tertiary alkyl groups in moderate to good yields and excellent stereoselectivities. A plethora of further transformations were applied to showcase the synthetic value of this transformation. A radical reaction pathway was proposed through mechanistic investigation. The simple operation and the easy availability of the starting materials also allowed this method to pave a new way for the preparation of synthetically crucial alkylated enamides.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from Nanjing Tech University (Start-up Grant No. 39837137, 39837101 and 3827401739), the National Natural Science Foundation of China (21372210, 21672198, and 21801129), the State Key Program of National Natural Science Foundation of China (21432009), and the Natural Science Research Projects in Jiangsu Higher Education Institutions (18KJB150018).References
- (a) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed; (b) C. Curti, M. Laget, A. O. Carle, A. Gellis and P. Vanelle, Eur. J. Med. Chem., 2007, 42, 880 CrossRef CAS PubMed; (c) H. Yang, R. G. Carter and L. N. Zakharov, J. Am. Chem. Soc., 2008, 130, 9238 CrossRef CAS PubMed; (d) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (e) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS PubMed.
- (a) S. Tong, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, Angew. Chem., Int. Ed., 2012, 51, 4417 CrossRef CAS PubMed; (b) M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Chem. Commun., 2012, 48, 8105 RSC; (c) G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235 CrossRef CAS PubMed; (d) C.-H. Lei, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 4708 CrossRef CAS PubMed; (e) M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 608 CrossRef CAS PubMed; (f) C.-H. Lei, L. Zhao, D.-X. Wang, J. Zhu and M.-X. Wang, Org. Chem. Front., 2014, 1, 909 RSC; (g) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746 CrossRef CAS PubMed; (h) P. Song, P. Yu, J.-S. Lin, Y. Li, N.-Y. Yang and X.-Y. Liu, Org. Lett., 2017, 19, 1330 CrossRef CAS PubMed.
- (a) S. Pankajakshan, Y.-H. Xu, J. K. Cheng, M. T. Low and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 5701 CrossRef CAS PubMed; (b) H.-L. Wang, R.-B. Hu, H. Zhang, A.-X. Zhou and S.-D. Yang, Org. Lett., 2013, 15, 5302 CrossRef CAS PubMed; (c) F. Bartoccini, D. M. Cannas, F. Fini and G. Piersanti, Org. Lett., 2016, 18, 2762 CrossRef CAS PubMed; (d) N. Gigant, L. Chausset-Boissarie, M. Belhomme, T. Poisson, X. Pannecoucke and I. Gillaizeau, Org. Lett., 2013, 15, 278 CrossRef CAS PubMed.
- (a) T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem.–Eur. J., 2011, 17, 7167 CrossRef CAS PubMed; (b) Y.-H. Xu, Y. K. Chok and T.-P. Loh, Chem. Sci., 2011, 2, 1822 RSC; (c) N. Gigant and I. Gillaizeau, Org. Lett., 2012, 14, 3304 CrossRef CAS PubMed; (d) W. Zhu, L. Zhao and M.-X. Wang, J. Org. Chem., 2015, 80, 12047 CrossRef CAS PubMed.
- (a) C. Feng and T.-P. Loh, Chem. Sci., 2012, 3, 3458 RSC; (b) R. Rey-Rodriguez, P. Retailleau, P. Bonnet and I. Gillaizeau, Chem.–Eur. J., 2015, 21, 3572 CrossRef CAS PubMed; (c) H. Wang, Y. Cheng and S. Yu, Sci. China: Chem., 2016, 59, 195 CrossRef CAS.
- G. Caillot, J. Dufour, M.-C. Belhomme, T. Poisson, L. Grimaud, X. Pannecoucke and I. Gillaizeau, Chem. Commun., 2014, 50, 5887 RSC.
- C. Feng, D. Feng and T.-P. Loh, Chem. Commun., 2014, 50, 9865 RSC.
- (a) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430 CrossRef CAS PubMed; (b) H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2012, 14, 4358 CrossRef CAS PubMed; (c) R. Ding, Q.-C. Zhang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2014, 50, 11661 RSC; (d) Z. Xiong, D. Liang and S. Luo, Org. Chem. Front., 2017, 4, 1103 RSC.
- (a) Y. Li, K. Cheng, X. Lu and J. Sun, Adv. Synth. Catal., 2010, 352, 1876 CrossRef CAS; (b) D. Sun and R. Zhang, Org. Chem. Front., 2018, 5, 92 RSC; (c) T.-H. Zhu, X.-C. Zhang, K. Zhao and T.-P. Loh, Org. Chem. Front., 2019, 6, 94 RSC; (d) T.-H. Zhu, X.-C. Zhang, X.-L. Cui, Z.-Y. Zhang, H. Jiang, S.-S. Sun, L.-L. Zhao, K. Zhao and T.-P. Loh, Adv. Synth. Catal., 2019, 361, 3593 CrossRef CAS.
- W. Yu, J. Chen, K. Gao, Z. Liu and Y. Zhang, Org. Lett., 2014, 16, 4870 CrossRef CAS PubMed.
- (a) H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem.–Eur. J., 2012, 18, 15158 CrossRef CAS PubMed; (b) R. Ding, Z.-D. Huang, Z.-L. Liu, T.-X. Wang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2016, 52, 5617 RSC; (c) D. Xing and G. Dong, J. Am. Chem. Soc., 2017, 139, 13664 CrossRef CAS PubMed; (d) C. Baudequin, A. Zamfir and S. B. Tsogoeva, Chem. Commun., 2008, 4637 RSC; (e) B. Maji, S. Lakhdar and H. Mayr, Chem.–Eur. J., 2012, 18, 5732 CrossRef CAS PubMed; (f) M.-N. Zhao, W. Du, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Eur. J. Org. Chem., 2013, 7989 CrossRef CAS; (g) P. Li, J. Zhao, C. Xia and F. Li, Org. Lett., 2014, 16, 5992 CrossRef CAS PubMed; (h) M. Giedyk, K. Goliszewska, K. ó. Proinsias and D. Gryko, Chem. Commun., 2016, 52, 1389 RSC.
- (a) K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736 CrossRef CAS; (b) K. Okada, K. Okamoto and M. Oda, J. Chem. Soc., Chem. Commun., 1989, 1636 RSC; (c) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401 CrossRef CAS; (d) K. Okada, K. Okubo, N. Morita and M. Oda, Tetrahedron Lett., 1992, 33, 7377 CrossRef CAS; (e) K. Okada, K. Okubo, N. Morita and M. Oda, Chem. Lett., 1993, 22, 2021 CrossRef.
- (a) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578 CrossRef CAS PubMed; (b) D. J. Tao, Y. Slutskyy, M. Muuronen, A. Le, P. Kohler and L. E. Overman, J. Am. Chem. Soc., 2018, 140, 3091 CrossRef CAS PubMed; (c) M. R. Garnsey, Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee and L. E. Overman, J. Org. Chem., 2018, 83, 6958 CrossRef CAS PubMed.
- For selected examples of transition metal-catalyzed C(sp3)–C formation, see: (a) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174 CrossRef CAS PubMed; (b) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (c) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS PubMed; (d) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260 CrossRef CAS PubMed; (e) J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906 CrossRef CAS PubMed; (f) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (g) S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 14560 CrossRef CAS PubMed; (h) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016 CrossRef CAS PubMed; (i) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901 CrossRef CAS PubMed; (j) X. Lu, B. Xiao, L. Liu and Y. Fu, Chem.–Eur. J., 2016, 22, 11161 CrossRef CAS PubMed; (k) N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150 CrossRef CAS PubMed; (l) S. S. R. Bernhard, G. M. Locke, S. Plunkett, A. Meindl, K. J. Flanagan and M. O. Senge, Chem.–Eur. J., 2018, 24, 1026 CrossRef CAS PubMed; (m) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809 RSC; (n) T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, Org. Lett., 2019, 21, 816 CrossRef CAS PubMed; (o) T.-G. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2019, 58, 2454 CrossRef CAS PubMed; (p) M. Montesinos-Magraner, M. Costantini, R. Ramírez-Contreras, M. E. Muratore, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2019, 58, 5930 CrossRef CAS PubMed.
- For selected examples of transition metal-catalyzed C(sp3)–X formation, see (a) W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 11649 CrossRef CAS PubMed; (b) J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537 CrossRef CAS PubMed.
- For selected examples of photoredox-catalyzed C(sp3)–C formation, see: (a) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef CAS PubMed; (b) J. Schwarz and B. König, Green Chem., 2016, 18, 4743 RSC; (c) Y. Jin, H. Yang and H. Fu, Org. Lett., 2016, 18, 6400 CrossRef CAS PubMed; (d) Y. Jin, M. Jiang, H. Wang and H. Fu, Sci. Rep., 2016, 6, 20068 CrossRef PubMed; (e) J. Li, H. Tian, M. Jiang, H. Yang, Y. Zhao and H. Fu, Chem. Commun., 2016, 52, 8862 RSC; (f) C. Gao, J. Li, J. Yu, H. Yang and H. Fu, Chem. Commun., 2016, 52, 7292 RSC; (g) H. Zhang, P. Zhang, M. Jiang, H. Yang and H. Fu, Org. Lett., 2017, 19, 1016 CrossRef CAS PubMed; (h) A. Tlahuext-Aca, R. A. Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708 CrossRef CAS PubMed; (i) A. F. Garrido-Castro, H. Choubane, M. Daaou, M. C. Maestro and J. Alemán, Chem. Commun., 2017, 53, 7764 RSC; (j) W. Sha, S. Ni, J. Han and Y. Pan, Org. Lett., 2017, 19, 5900 CrossRef CAS PubMed; (k) J.-C. Yang, J.-Y. Zhang, J.-J. Zhang, X.-H. Duan and L.-N. Guo, J. Org. Chem., 2018, 83, 1598 CrossRef CAS PubMed; (l) Y. Zhao, J.-R. Chen and W.-J. Xiao, Org. Lett., 2018, 20, 224 CrossRef CAS PubMed; (m) W. Kong, C. Yu, H. An and Q. Song, Org. Lett., 2018, 20, 349 CrossRef CAS PubMed; (n) A. Tlahuext-Aca, R. A. Garza-Sanchez, M. Schäfer and F. Glorius, Org. Lett., 2018, 20, 1546 CrossRef CAS PubMed; (o) L. Ren and H. Cong, Org. Lett., 2018, 20, 3225 CrossRef CAS PubMed; (p) X.-H. Ouyang, Y. Li, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 6659 CrossRef CAS PubMed; (q) W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907 CrossRef CAS; (r) W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem.–Eur. J., 2017, 23, 2537 CrossRef CAS PubMed; (s) J.-J. Zhang, J.-C. Yang, L.-N. Guo and X.-H. Duan, Chem.–Eur. J., 2017, 23, 10259 CrossRef CAS PubMed; (t) K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719 RSC; (u) Z.-H. Xia, C.-L. Zhang, Z.-H. Gao and S. Ye, Org. Lett., 2018, 20, 3496 CrossRef CAS PubMed; (v) Q. Tang, X. Liu, S. Liu, H. Xie, W. Liu, J. Zeng and P. Cheng, RSC Adv., 2015, 5, 89009 RSC; (w) J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275 RSC; (x) C. Jin, Z. Yan, B. Sun and J. Yang, Org. Lett., 2019, 21, 2064 CrossRef CAS PubMed; (y) G.-L. Dai, S.-Z. Lai, Z. Luo and Z.-Y. Tang, Org. Lett., 2019, 21, 2269 CrossRef CAS PubMed; (z) L. Liu, J. Dong, Y. Yan, S.-F. Yin, L.-B. Han and Y. Zhou, Chem. Commun., 2019, 55, 233 RSC.
- For selected examples of photoredox-catalyzed C(sp3)–X formation, see: (a) M. Jiang, H. Yang and H. Fu, Org. Lett., 2016, 18, 1968 CrossRef CAS PubMed; (b) Y. Jin, H. Yang and H. Fu, Chem. Commun., 2016, 52, 12909 RSC; (c) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440 CrossRef CAS PubMed; (d) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS PubMed; (e) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770 CrossRef CAS PubMed; (f) R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 9501 CrossRef CAS PubMed; (g) C. Zheng, Y. Wang, Y. Xu, Z. Chen, G. Chen and S. H. Liang, Org. Lett., 2018, 20, 4824 CrossRef CAS PubMed.
- (a) D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632 CrossRef CAS PubMed; (b) G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888 CrossRef CAS PubMed; (c) L. Yu, M.-L. Tang, C.-M. Si, Z. Meng, Y. Liang, J. Han and X. Sun, Org. Lett., 2018, 20, 4579 CrossRef CAS PubMed; (d) L. Gao, G. Wang, J. Cao, D. Yuan, C. Xu, X. Guo and S. Li, Chem. Commun., 2018, 54, 11534 RSC.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed.
- Product 3ce in Table 3 (CCDC 1874601) contains the crystallographic data for this paper. All other E-adducts 3aa–3ss (Table 2) and 3ab–3ar (Table 3) exhibit signals similar to those of 3ce for their alkene and benzylic hydrogens in 1H NMR. In addition, NOESY experiments for E-3aa and Z-3aa have been performed to further verify the assignment of stereochemistry.
- Z-3aa, Z-3fa and Z-3ae show 1H NMR signals similar to each other, which are different from those of their E-isomers, especially for the chemical shift of olefinic hydrogens and the spin splitting of benzylic hydrogens.
- M. A. Cismesiaa and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- In iminium ion E, the acetyl group is positioned cis to the Ar group for simplicity; the plausible conformer with the benzyl group cis to the Ar group would also give rise to E-alkylated enamides in order to minimize the allylic strain between the acetyl group on nitrogen and the alkyl group.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1874601. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03070k |
| This journal is © The Royal Society of Chemistry 2019 |