
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined high degree of carboxylation and electronic conduction in graphene acid sets new limits for metal free catalysis in alcohol oxidation†
Matías
Blanco
 *a,
Dario
Mosconi
a,
Michal
Otyepka
bc,
Miroslav
Medveď
b,
Aristides
Bakandritsos
b,
Stefano
Agnoli
*a and
Gaetano
Granozzi
a
*a,
Dario
Mosconi
a,
Michal
Otyepka
bc,
Miroslav
Medveď
b,
Aristides
Bakandritsos
b,
Stefano
Agnoli
*a and
Gaetano
Granozzi
a
aDepartment of Chemical Sciences, INSTM Unit, University of Padova, Via F. Marzolo 1, 35131, Padova, Italy. E-mail: matias.blancofernandez@unipd.it; stefano.agnoli@unipd.it
bRegional Centre for Advanced Technologies and Materials, Faculty of Science, Palacký University Olomouc, Šlechtitelů 27, 771 46 Olomouc, Czech Republic
cDepartment of Physical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 771 46 Olomouc, Czech Republic
First published on 6th September 2019
Abstract
Graphene oxide, the most prominent carbocatalyst for several oxidation reactions, has severe limitations due to the overstoichiometric amounts required to achieve practical conversions. Graphene acid, a well-defined graphene derivative selectively and homogeneously covered by carboxylic groups but maintaining the high electronic conductivity of pristine graphene, sets new activity limits in the selective and general oxidation of a large gamut of alcohols, even working at 5 wt% loading for at least 10 reaction cycles without any influence from metal impurities. According to experimental data and first principles calculations, the selective and dense functionalization with carboxyl groups, combined with excellent electron transfer properties, accounts for the unprecedented catalytic activity of this graphene derivative. Moreover, the controlled structure of graphene acid allows shedding light upon the critical steps of the reaction and regulating precisely its selectivity toward different oxidation products.
Introduction
The term carbocatalysis stands for carbon-based catalysis, embracing metal-free nanocarbon catalysis, where the carbon material as a whole or some specific regions of it act as active sites to speed-up chemical processes.1,2 Research interest on heterogeneous carbocatalysis is growing, since it is based on extremely low-cost and abundant materials, without the need for metal centers, therefore complying with the standards of sustainability and environmental protection required by modern society. The limited or abolished use of critical materials, the implementation of “green” methods for their synthesis, and easy recycle procedures are key aspects that make carbocatalysts the most eligible materials for the development of a sustainable circular economy.3–5Despite the rapid progress in this field, there are still many unmet challenges for the development of more efficient and well-defined materials able to work at truly catalytic loadings, and for the fundamental understanding of catalytic mechanisms at the molecular scale. Graphene oxide (GO) is one of the most studied carbocatalysts,1,6 commonly obtained by the chemical oxidation of graphite,7 and its most accepted structure consists of small patches of aromatic domains separated by defective regions.8 Regarding the surface chemistry, hydroxyl and epoxy groups are present on the basal plane, while carboxylic and carbonyl functions are located at the edges and around defects.9 Therefore, this material, since it has plenty of potential active sites that can be employed in chemical transformations, has attracted widespread attention in the catalysis community. Several studies have explored the catalytic role of GO as a multifunctional catalytic platform, e.g., in the oxidation of alcohols10 and olefins,11 as an oxygen activator in the hydration of alkynes,12 as a co-catalyst in the Michael-type Friedel–Crafts alkylations13 and as a solid acid in aldol condensations.14 However, in most of these cases, the activity of GO cannot be truly defined as catalytic, because many clues suggest that GO acts indeed as a stoichiometric reagent, and the effective “catalyst” loadings employed in such reports are in the overstoichiometric range, typically 50–400 wt%.15,16 Among the reactions catalyzed by GO, the selective oxidations of alcohols are extremely appealing, being fundamental processes in industrial processes that lead to key intermediates for the production of polymers, pharmaceutical synthons and fragrances.17 In the alcohol oxidation, the activity can be triggered in the catalytic range by the addition of a promoter, e.g. nitroxyl-based species combined with nitric acid, usually in stoichiometric amounts.18 This synergistic approach is common in the literature, but the combination with inorganic solid acids19 or metal containing carbon materials20 is usually required to obtain good performances, making the process less attractive for applications. A general HNO3-mediated mechanism was suggested21 based on the formation of active N-oxides (NOx) facilitating the oxidation,22 but no first principles calculations or definitive experimental confirmation has been provided.
Here, we propose Graphene acid (GA) as a potent and well-defined metal-free carbocatalyst for the controlled and highly selective alcohol oxidation, and thanks to DFT calculations, we delineate at the molecular scale the reaction steps involved in the catalytic cycle. Interestingly, we demonstrate the catalytic nature of the reaction and that the final oxidant is atmospheric oxygen, which is the most eligible reactant for truly sustainable large scale alcohol oxidation processes. GA is a graphene derivative with the basal plane densely covered by COOH groups (up to 9–13 at%, see Fig. S1†).23 Interestingly, in contrast to GO, GA is electronically conductive,23–25 which is a key property in redox catalytic reactions,26 but at the same time comprises oxygenated groups that are strong oxygen activators. The carbocatalytic activity of GA was tested in benzyl alcohol (BA) oxidation as a model reaction. Both the overstoichiometric (up to GA loading of 20 wt%), and low loading (1–5 wt%) regimes in the presence of HNO3 were investigated to unravel the role played by each component in the catalytic cycle. The influence of metallic impurities was ruled out by recycling experiments coupled with ICP analysis. DFT calculations performed for a finite-size GA model corroborated the reaction mechanism deduced by the experiments.
Experimental
General
All chemicals were purchased from Aldrich. Reagent grade or better quality was employed in all experiments. GA was synthetised following the procedure described previously.23 All air-sensitive reactions were carried out in a N2 atmosphere employing Schlenk techniques with N2 degassed solvents. GO was synthesized according to a modified Hummers procedure.27 3.0 g of graphite was mixed with 75 mL of a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2SO4 and H3PO4. Then 9.0 g of KMnO4 was slowly added while cooling the mixture in an ice bath. The mixture was stirred for 3 h at 0 °C and then overnight at room temperature. After that, a volume of 150 mL of water was added heating at 80 °C. After stirring for 1 h, the obtained mixture was sonicated for 1 h at 35 °C. To quench the reaction, 3 mL of 30% wt of H2O2 was added, stirred for additional 2 h, sonicated for 1 h and finally, diluted with additional water. The supernatant was profusely washed with water, dialyzed for 72 h against DI water and 8 h against Milli-Q quality water, and finally lyophilized.
:1 mixture of H2SO4 and H3PO4. Then 9.0 g of KMnO4 was slowly added while cooling the mixture in an ice bath. The mixture was stirred for 3 h at 0 °C and then overnight at room temperature. After that, a volume of 150 mL of water was added heating at 80 °C. After stirring for 1 h, the obtained mixture was sonicated for 1 h at 35 °C. To quench the reaction, 3 mL of 30% wt of H2O2 was added, stirred for additional 2 h, sonicated for 1 h and finally, diluted with additional water. The supernatant was profusely washed with water, dialyzed for 72 h against DI water and 8 h against Milli-Q quality water, and finally lyophilized.
Characterization techniques
The surface chemical characterization of the catalyst has been carried out using X-ray photoelectron spectroscopy (XPS) in a custom-made UHV system working at a base pressure of 10−10 mbar, equipped with an Omicron EA150 electron analyzer and an Omicron DAR 400 X-ray source with a dual Al–Mg anode. Core level photoemission spectra (C 1s, N 1s and O 1s) were collected at rt with a non-monochromated Mg Kα X-ray source (1253.6 eV) and using an energy step of 0.1 eV, 0.5 s of integration time, and a 20 eV pass energy. The TEM images were acquired using an FEI Tecnai 12 microscope with an acceleration voltage of 100 kV. The Raman spectra were collected using a ThermoFisher DXR Raman microscope using a laser with an excitation wavelength of 532 nm (5 mW), focused on the sample with a 50× objective (Olympus). The UV-visible absorption spectroscopy data were acquired using a Cary 50 spectrometer (Varian), in the 200–800 nm range. In this case, powder samples were dispersed in 2-propanol, forming a stable colloidal dispersion. Solid state Fourier Transform Infrared (FT-IR, KBr disk technique) absorption spectra were recorded with a Nicolet Nexus FT-IR spectrometer. Inductive Coupled Plasma Mass Spectrometry (ICP-MS) metal analysis was performed on an Agilent 7700x quadrupole ICP-MS instrument. Samples were digested with a mixture of acids using a microwave digestion unit Milestone MLS 1200 Mega. The Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker Avance 300 MHz (300.1 MHz for 1H, 298 K); chemical shifts (δ) are reported in units of parts per million (ppm) relative to the residual solvent signals and coupling constants (J) are expressed in Hz.Overstoichiometric benzyl alcohol oxidation
The typical catalytic test in the benzyl alcohol oxidation in the overstoichiometric regime was performed, if not otherwise stated, as follows: 1 mmol (0.108 g) of benzyl alcohol was mixed, in a Teflon-capped vial, with an adequate amount of GA powder to generate the corresponding 1–20 wt% loading of catalyst, and the mixture was heated at the target temperature, typically 150 °C, for 24 h. Then, it was allowed to cool to room temperature, the solid was filtered, CDCl3 was added and immediately submitted to NMR analysis. If a solvent was employed, 1 M concentration of benzyl alcohol was used.Alcohol oxidation with HNO3
The standard catalytic test in the alcohol oxidation with nitric acid was performed, if not otherwise stated, as follows: 1 mmol of alcohol was mixed, in a 20 mL volume Teflon-capped vial (see Fig. S4†), with a certain amount of graphenic material powder (typically GA, 5 mg), and the mixture was suspended in 2 mL of solvent, normally 1,4-dioxane. Then, 2 mmol of the oxidant, typically HNO3 (concentrated, >65%) was added, the vial was closed tightly, and heated at the target temperature, usually 90 °C, for the desired time. Then, it was allowed to cool to room temperature, the solid was filtered, CDCl3 was added and immediately submitted to NMR analysis.The study of the catalytic cycle required the systematic modification of the standard reaction parameters, such as the catalyst (GA, GO or no-catalyst), solvent, temperature (from rt to 90 °C), oxidant (various acids, different amounts of HNO3, different amounts of NaNO2 combined with different acids or even NOBF4), reactor (pressured autoclave or open-to-air vial) and atmosphere (air or inert). Given this set of modified conditions, the changes in the performance of the reaction are routinely compared with the standard catalytic run in the Results and discussion section.
Recovery experiments were conducted after washing the catalyst by centrifugation for 3 cycles with water and 3 additional cycles with acetone, sonicating for 5 min between each cycle, and vacuum-dried. After that, the solid was submitted to a new catalytic cycle without adding in any case a new catalyst precursor. The procedure was repeated 10 times.
Computational details
All calculations were performed with the Gaussian 09 program28 employing the ωB97X-D/6-31+G(d) level of theory.29,30 GA was modelled using a finite-size model (ovalene) functionalized by carboxyl and hydroxyl groups (Fig. S7†) according to the composition of the catalyst as revealed by the experimental C 1s photoemission line deconvolution data reported in Table S1.† To understand the different performances of the catalyst in various solvents, two representative solvents (toluene and dimethyl sulfoxide (DMSO)) were considered in theoretical models. The solvent effects were included by using the universal continuum solvation model based on solute electron density (SMD).31 Whereas the structures of molecular species (such as HNO3, NO2, H2O, benzyl nitrite, and benzaldehyde) were fully relaxed in the geometry optimization, to mimic the semilocal rigidity of graphene sheets, the GA model was obtained by constrained geometry optimizations keeping the edge carbon atoms frozen.Results and discussion
GA was obtained by the controlled hydrolysis of cyanographene, which was prepared by the substitution and defluorination of fluorographite.23 GO, used as a benchmark, was synthesized according to a modified Hummers protocol.32 The X-ray photoemission spectroscopy (XPS) investigation of the C 1s spectrum of GA (Fig. 1) showed the presence of large aromatic domains (61.5% of C–C sp2 bonds, Table S1†)33 together with a large fraction of COOH groups, as deduced by the component centered at 289 eV (9.7% of the total C 1s spectrum area, i.e. 11% functionalization degree). No traces of metals were observed by XPS (Fig. S2 and Table S2†), as expected from the synthesis protocol. Nevertheless, since our goal is to employ GA as the carbocatalyst, ICP-MS analysis was also performed to quantify the metallic content below the XPS detection limit, revealing ppb levels of metals such as Cr, Mn, Fe, Ni or Cu at the GA surface; however, our graphene derivative does not contain Co, Pt or Pd (Table S5†). On the other hand, the GO sample presented the standard complex distribution of the oxygen species, with smaller aromatic domains (53.4%) and a double amount of hydroxyl and epoxy groups compared to the GA (Table S1†). Moreover, COOH groups in GO accounted only 7% of the total C 1s intensity. Metallic impurities are higher on this sample, in particular Mn and Pt, as demonstrated by ICP-MS analysis (Table S5†). The Raman spectra of both samples agreed with the prototypical pattern of an oxidized carbon material, having the most prominent bands centered at 1350 cm−1 (D band) and 1580 cm−1 (G band),34 yielding a ID/IG ratio of 1.05 and 0.92 for GA and GO, respectively (see Fig. S3†). Both materials presented the typical flake-like morphology of graphene derivatives in the monolayer regime, as depicted by TEM analysis (see Fig. S3†). The lateral sizes ranged from hundreds of nm on GA to tens of μm in GO, in excellent agreement with previous reports.23,26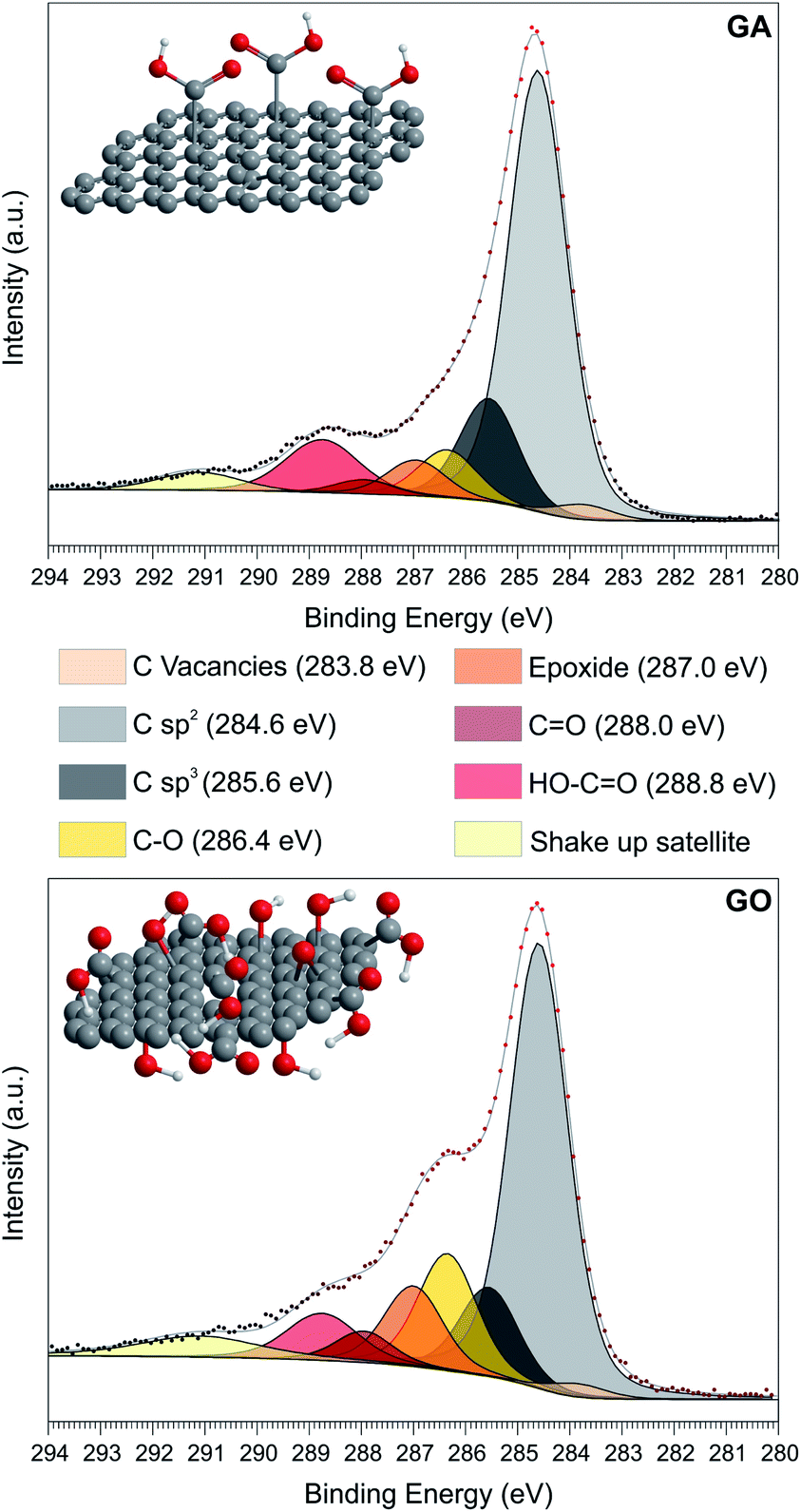 | ||
| Fig. 1 XPS C 1s core level spectra with separation into chemically shifted components, of GA (upper) and GO (down) with the corresponding structural models. | ||
Motivated by the overstoichiometric strategy of Bielawski et al.,10 we tested the catalytic activity of the selectively –COOH covered and metal-free surface of GA without any promoter in the BA oxidation, employing different GA loadings (from 1 wt% to 20 wt% with respect to BA). Under the standard catalytic conditions reported in Table 1, no conversion was detected with 1 wt% GA loading, while the reaction progressively yielded the intended products until 27% conversion was reached when the amount of the added catalyst was increased to 20 wt% loading. In all positive tests (i.e. from 5 wt% to 20 wt% loading), NMR analysis of the reaction crude confirmed 100% selectivity for benzaldehyde, i.e., without detection of any trace of benzoic acid. The obtained conversion value in the GA-catalyzed test was very similar (Table 1) to the results obtained with GO (25% of conversion) and also to that reported by Bielawski et al. (27%).10 By a linear extrapolation of our “overstoichiometric” catalytic data, we deduced a behaviour identical to that reported in the literature,10i.e. full conversion with high selectivity for benzaldehyde, at 200 wt% loading of GA. However, we did not pursue this route further because we were interested to use only catalytic amounts of active material. As a matter of fact, it has been decisively reported that, when working under overstoichiometric conditions, GO acts as a reagent, undergoing important changes in its C:O ratio, rather than being a true catalyst that remains essentially unaltered at the end of the chemical conversion.16 Here, we are interested only in the study of the true catalytic activity of GA, and not in the possible stoichiometric reactions that may be happening when it is used in an extremely large excess.
|
|
||||
|---|---|---|---|---|
| Catalyst | Loadingb | Solvent | T | Yieldd |
| a 1 mmol of BA and the amount of catalyst to reach the desired loading. b wt% vs. BA. c °C. d %, determined by 1H-NMR spectroscopy. e Reported by Bielawski et al.10 | ||||
| GA | 10 | DMF | Rt | — |
| GA | 10 | DMF | 150 | — |
| GA | 1 | Neat | 150 | — |
| GA | 5 | Neat | 150 | 6 |
| GA | 10 | Neat | 150 | 15 |
| GA | 20 | Neat | 150 | 27 |
| GO | 20 | Neat | 150 | 25 |
| GOe | 20 | Neat | 150 | 27 |
No conversion was detected when the reaction was performed in a compatible solvent such as dimethylformamide (DMF) (Table 1).
However, this scenario completely changed when nitric acid was added to the reaction mixture. When 2 equivalents of HNO3 as the promoter were introduced in the reaction medium, GA (5 wt% loading) oxidized BA quantitatively in 75 minutes in an air atmosphere (Fig. 2), and the formation of brown NOx fumes demonstrated an outstanding performance compared to other metal- and carbo-catalysts reported in the literature for this reaction (Fig. S3 and Table S3†). The selectivity toward benzaldehyde was 70%, because the excess of HNO3 could lead to the competing formation of benzoic acid (Fig. 2), which is progressively formed when the reaction time is increased. Interestingly, in the corresponding catalytic test with benchmark GO, it was not possible to achieve this activity: in this case, no NOx evolution was observed from the reaction vessel (Fig. S4†), probably because of GO's inherent insulating nature35 that blocks its activity as a redox mediator, which might prevent the decomposition of HNO3 at the GO surface. Indeed, Larsen et al.36 proposed a mechanism for redox reactions involving organic molecules and carbon-based catalysts, where the carbon material acts as an electron reservoir that is able to store and successively inject electrons back to the reactants. This mechanism was later confirmed by theory and experiments.37 Obviously, this mechanism cannot be applied with an insulating material acting as a catalyst, thus justifying the negligible activity shown by GO. Therefore, the key to the superior catalytic activity of GA is not simply related to the presence of carboxyl groups, which are known to be oxygen activators,38,39 but to the fact that these catalytically active centers are electronically connected to a reduced carbon scaffold that can be used as an electron reservoir/sink. GO bears 23% less carboxyl groups than GA and is located only at the nanosheet edges on very defective areas, whereas on GA they are directly placed on the basal plane, maximizing the possibility of an easy and fast electron transfer.
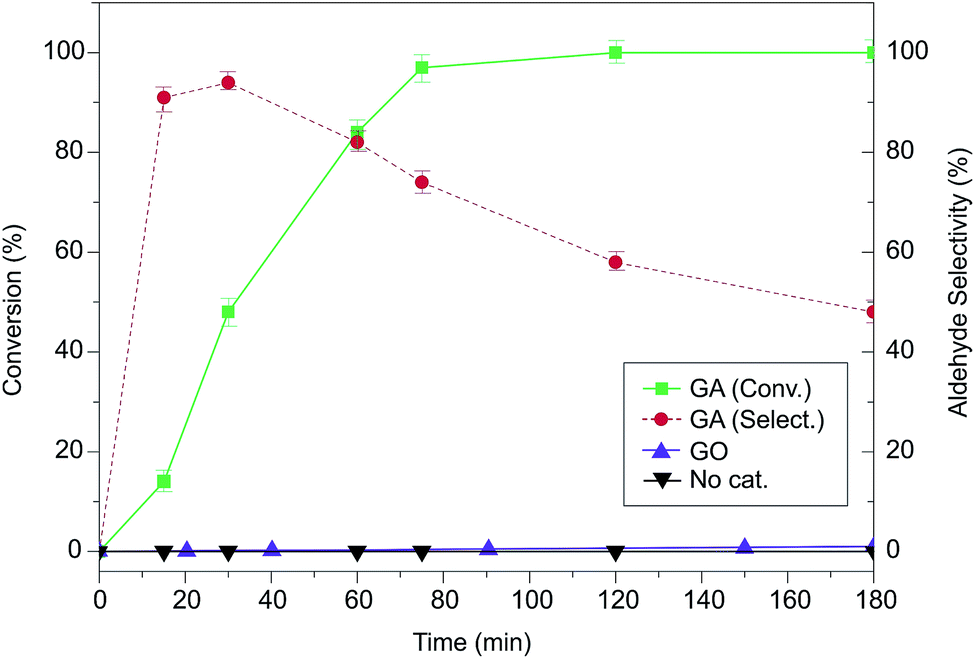 | ||
| Fig. 2 Time evolution of benzyl alcohol oxidation catalysed by graphene materials. | ||
Metallic impurities do not seem to have any effect on the catalytic activity for both samples. Indeed, the GO control sample carries higher metal amounts than GA as demonstrated by ICP analysis (Table S5†), but its activity is almost negligible in this oxidation, confirming the outstanding carbocatalytic nature of GA. Finally, the reagents used in the catalytic reaction (HNO3 and 1,3-dioxane), despite the significant content of Cr, and Fe (Table S5†), also displayed undetectable conversion (Fig. 2) suggesting that these impurities cannot be considered as the source of the catalytic activity.
To gain a deeper insight into the reaction mechanism and understand the excellent behavior of GA as a carbocatalyst, we systematically changed several reaction parameters (Fig. 3). We found that a minimum GA loading of 5 wt% was necessary to start the conversion, while increasing the loading to 10 wt% or 20 wt% did not significantly affect the obtained mixture of products (Fig. S4†). On the other hand, at least 2 equivalents of HNO3 employing a conventional vessel in an air atmosphere were required to achieve a good oxidation performance, while when the acid content was reduced (0.25–1.5 eq.), the activity was largely quenched (Fig. 3a). Adding an excess of HNO3 (3 or 4 eq.), however, led to a slight increase in the benzoic acid production as the oxidizing strength of the medium is progressively increased. Notably, the nature of the acid played a very important role: 2 equivalents of HCl, H2SO4 or acetic acid only yielded traces of products. Thus, the decomposition of the nitric acid into NOx derivatives seems to be a key step to initiate the BA oxidation. In addition, if the reaction was performed at a temperature lower than 90 °C (from rt to 70 °C) no conversion was observed (Fig. 3b). Moreover, the reaction was also solvent dependent, because full conversion was achieved only with 1,4-dioxane and dimethylsulfoxide (DMSO). In contrast, in toluene, DMF and water, no conversion could be achieved even after overnight reactions (Fig. S5†).
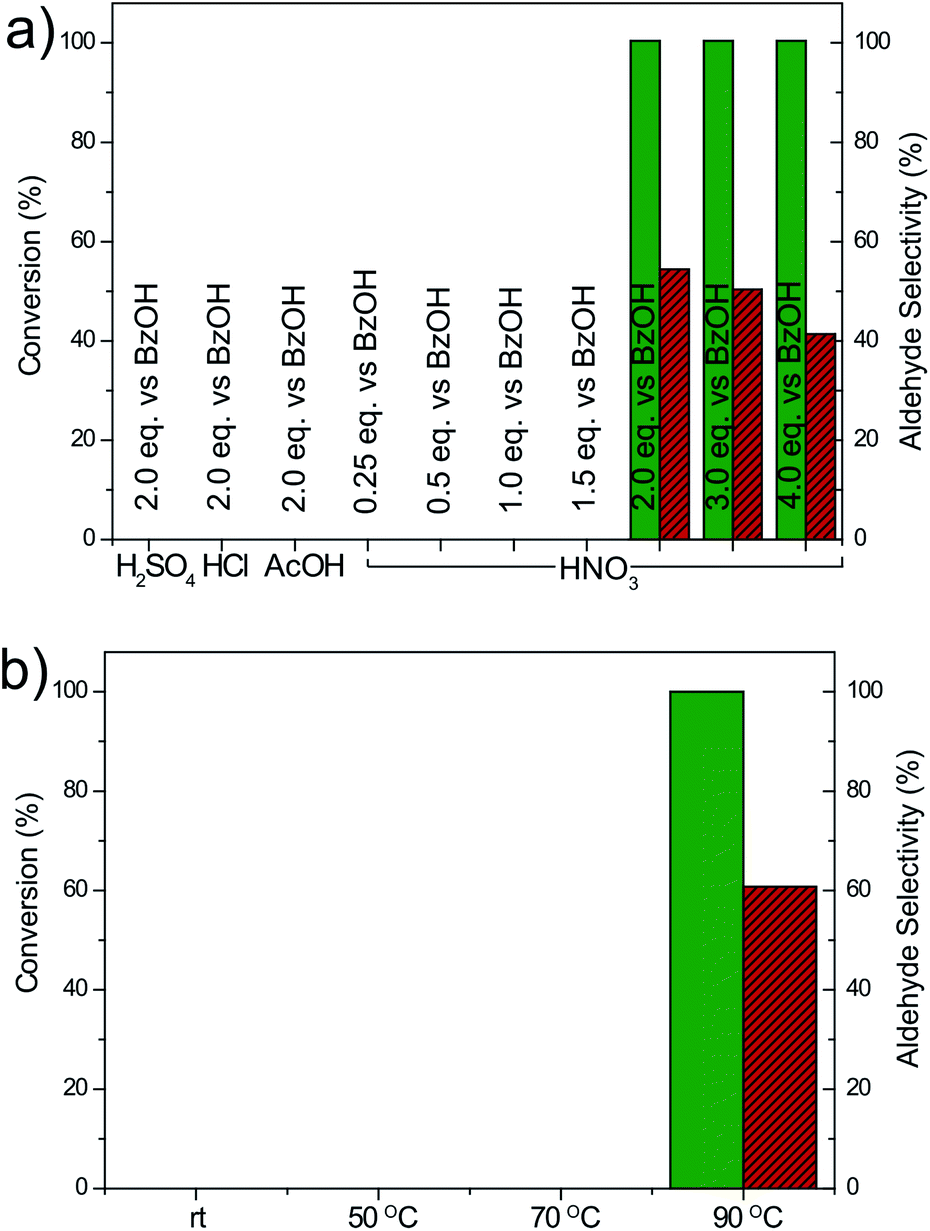 | ||
| Fig. 3 Oxidation conversion (%) of benzyl alcohol under standard conditions with 5 wt% GA with nitric acid promoter changing (a) the nature and amount of the acid and (b) the temperature. | ||
Given these first experimental proofs, we carried out density functional theory (DFT) calculations to unravel the reaction mechanism. We assumed that the high temperature needed to yield the products (Fig. 3b) was likely related to the decomposition of the HNO3 on the GA surface, which might be the initiating step of the catalytic cycle. This was firstly addressed by investigating the possible binding modes of HNO3 on GA (Fig. S6†). As expected, the most favorable binding mode corresponded to the formation of two hydrogen bonds (2HB) between the HNO3 and GA (Fig. S6a†). In the gas phase and an almost non-polar solvent such as toluene, a binding mode involving one hydrogen bond (1HB) was also found to be favorable (Fig. S6b†). Interestingly, in the case of DMSO, a “parallel” (P) binding mode was identified, in which the orientation of the HNO3 molecule was highly convenient for the formation of NO2 and H2O (Fig. S6c†). For all three binding modes, a decrease of the partial charge on N (which is a predisposition for reduction), compared to isolated HNO3 (qN = +0.12e; in DMSO) was observed. The energy differences between the identified local minima indicated that, under the experimental conditions (T = ∼90 °C), the HNO3 molecule was not fixed in one particular position and could also adopt orientations convenient for the reaction, such as, e.g., the P binding mode with the most pronounced decrease of qN (−0.22e). In the next step, the reaction energies for the GA assisted transformation of HNO3 to NO2 were calculated (Table 2). In general, all reactions were thermodynamically favorable; nevertheless, the most negative reaction energies were obtained for the DMSO solvent, in line with the experimental observations. Moreover, the HNO3 reduction was accompanied by a partial decarboxylation of GA and production of NO2 that hydrolysed to give nitric and nitrous acids.21 To confirm this theoretical prediction, we performed a spectroscopic analysis of the GA catalyst after reaction. The XPS, Raman and FTIR measurements on the sample after BA oxidation suggested a small decrease of carboxyl groups accompanied by a parasitic oxidation of the material (Fig. S8 and S9, Table S2†). It should also be noted that due to the complexity of the simultaneous electron transfer, hydrogen transfer and decomposition process, the kinetics of the reduction was not addressed computationally.
| Reaction | Gas phase | Toluene | DMSO |
|---|---|---|---|
| a Parentheses: energies corresponding to the formation of GA-COO˙ after releasing ˙NO2 and H2O. | |||
| 2HB mode | |||
| GA-COOH⋯HNO3 → GA˙ + ˙NO2 + H2O + CO2 | −16.1 (41.5) | −18.4 (28.4) | −19.8 (19.5) |
|
|||
| 1HB mode | |||
| GA-COOH⋯HNO3 → GA˙ + ˙NO2 + H2O + CO2 | −19.5 (38.0) | −20.6 (26.2) | — |
|
|||
| P mode | |||
| GA-COOH⋯HNO3 → GA˙ + ˙NO2 + H2O + CO2 | — | — | −28.1 (11.2) |
Based on the experimental and theoretical validation discussed above, we can propose the following catalytic cycle (Fig. 4): HNO3 decomposes on the material and forms NO2 (ref. 19) (step 1). This brown gas dissolved in the reaction medium must hydrolyze to HNO2 as a consequence of the aqueous environment in which the reaction takes place (step 2).21 The in situ formed HNO2 can react with benzyl alcohol to give benzyl nitrite (step 3), which eventually yields the final products through a GA mediated reaction involving the adsorption of the benzyl nitrite on the GA surface and its subsequent oxidation (steps 4 and possibly 5).22 As a result, a NO molecule is released (step 6),19,20 which is then re-oxidized to NO2 by molecular oxygen (step 7), closing the catalytic cycle.18 It is noteworthy that this reaction, especially steps 1 and 2, require stoichiometric amounts of nitric acid to achieve appreciable conversion of the oxidation products, in accordance with our theoretical and experimental validation, and also with previous studies (see Table S3† for more examples). Nevertheless, the proposed cycle in Fig. 4 should be sustained even with catalytic quantities of HNO3 or NOx since they do not seem to be consumed after closing the mass balances. However, key catalytic species such as NO2, due to their gaseous nature, are prone to be quickly released from the reaction environment when operating under standard conditions. Therefore, to confirm the true catalytic nature of the reaction, we ran a test employing 10 mol% of HNO3 in an autoclave to ensure that all the gases remained in the reaction vessel. Operating in such a way, excellent performances were obtained (95% conversion with 98% selectivity) (see Fig. S10†). Therefore, the NOx derivatives or even the nitric acid would truly act as a co-catalyst in the benzyl alcohol oxidation and not simply as a promoter. On the other hand, HNO2 can be mildly generated if NO2− ions are externally added. Thus, reactions with NaNO2 were performed under standard conditions and in an air atmosphere. We observed that when 1 equivalent of NaNO2 was mixed with 1 equivalent of HNO3 (or any other strong acid to provide HNO2 under standard conditions, Fig. S10†), high conversion with excellent selectivity to the aldehyde was achieved. Therefore, these tests additionally confirmed that steps 1 and 2 of the reaction cycle (Fig. 4) are actually taking place as the initiating processes of this catalytic oxidation. In addition, the selectivity to the aldehyde was higher compared to the standard run (2 eq. of HNO3) due to the milder generation of the co-catalyst, allowing a better control on the selectivity. Moreover, if the reaction intermediates corresponding to step 3 are carefully analyzed during the BA oxidation, the –CH2–(ONO) group belonging to benzyl nitrite is detected as a small singlet at δ ≈ 6.0 ppm in the NMR spectra (Fig. S11†), confirming the set of experimental data discussed above.
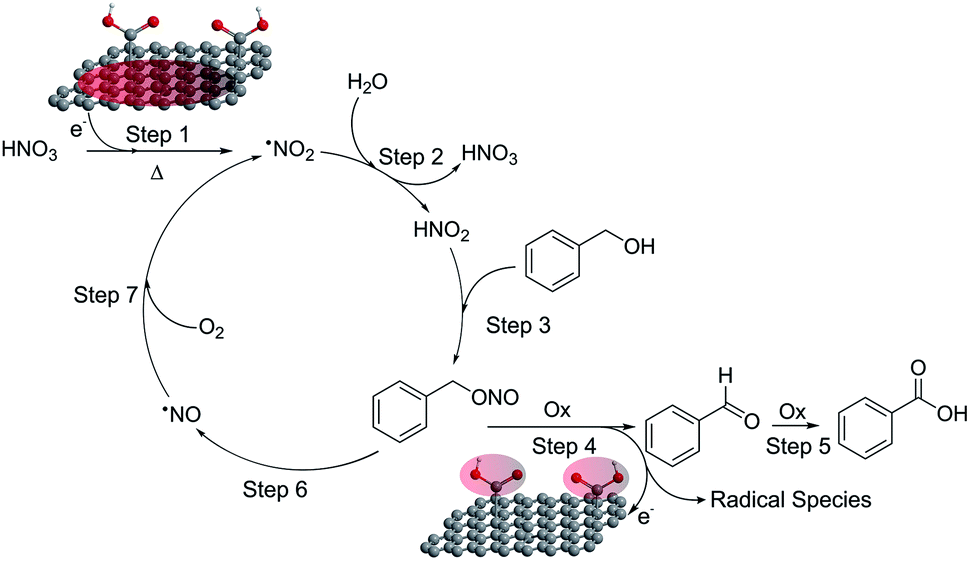 | ||
| Fig. 4 Catalytic cycle involving GA. | ||
Step 4 of the catalytic cycle was investigated by DFT calculations. As for the HNO3 reduction, firstly we identified the binding modes of benzyl nitrite on GA (Fig. 5). In solvents, the mode with a hydrogen bond between the oxygen bound to the benzyl moiety and the hydrogen atom from the GA's COOH was the most favorable. Obviously, this structure was convenient for a subsequent release of NO. Although the cleavage of the N–O bond required ca. 40 kcal mol−1 according to our calculations, it could be compensated by the energetically favorable stabilization of the benzyloxy radical by its reaction with O2 or NO2 (Table S4 and Fig. S7†) to form benzaldehyde. Moreover, the relatively high energy of the N–O bond dissociation can be overcome at moderate temperature, which in fact represents a limiting factor for BA oxidation. We also explored the experimental conditions that allowed oxidation of the aldehyde to the corresponding carboxyl acid (step 5). When BA oxidation was performed with stoichiometric amounts of HNO3 in an open vessel, where gases can be easily released and oxygen constantly provided, full conversion was reached and the reaction was also fully selective toward the acid (Fig. 5d). Thus, the release of gases accelerated the oxidations reaching the level when two oxidations (1 for the alcohol and 1 for the aldehyde) occurred successively.
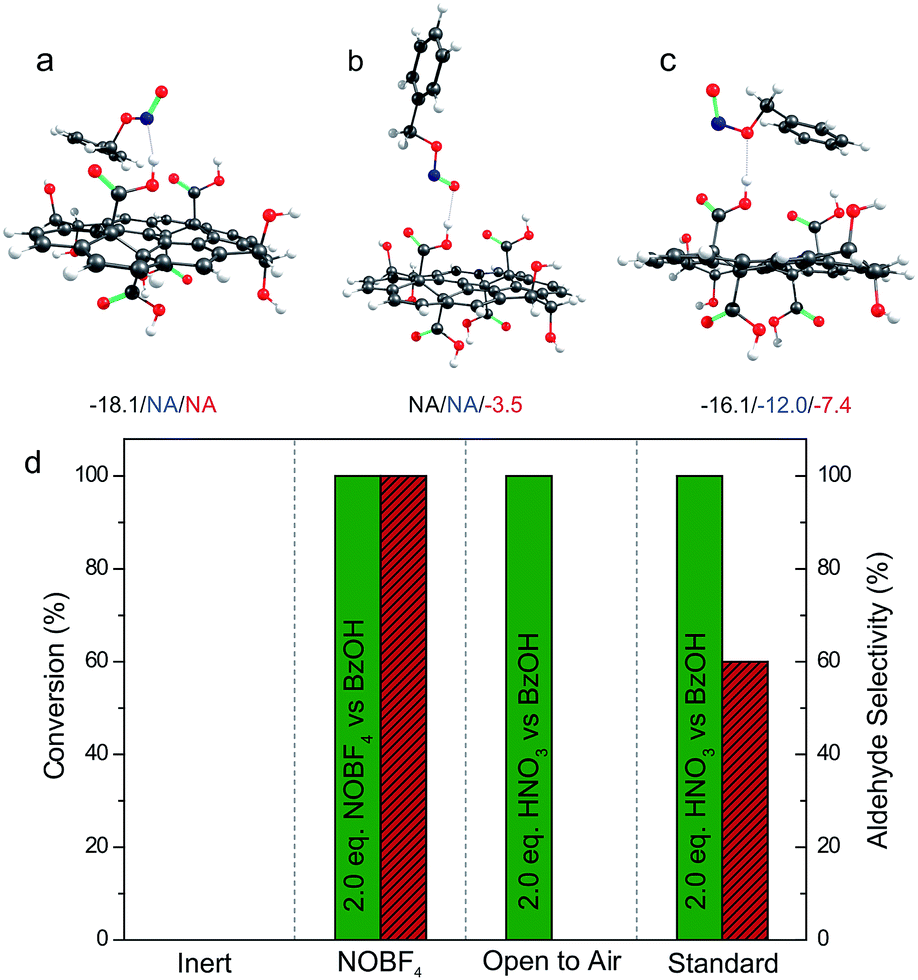 | ||
| Fig. 5 (a–c) Binding modes of benzyl nitrite on GA. The binding energies (in kcal mol−1) were calculated at the ωB97X-D/6-31+G(d) level of theory; the black/blue/red values correspond to the gas phase/toluene/DMSO, respectively. (d) Analysis of cycle terminating steps. | ||
The terminating step of the cycle (step 6) was explored (Fig. 5d) performing new catalytic tests changing a variety of parameters. On the one hand, the presence of NO in the catalytic cycle after the BA oxidation (step 6) was confirmed by running a catalytic test employing the [NO:BF4] complex as the oxidant (standard vessel, air atmosphere, stoichiometric in the oxidant). In this particular case (Fig. 5d), a complete conversion with full selectivity to the aldehyde was obtained as well. This simple test validates the cyclic nature of the reaction due to the possibility of producing the intended products starting even from the last released substance (i.e. NO), according to the proposed cycle (see Fig. 4). On the other hand, O2 represents a key intermediate of this transformation since this gas is proposed to regenerate the oxidizing agents in the last step of the cycle (step 7). Obviously, an inert atmosphere inhibits the reaction. As a matter of fact, in this case no conversion was observed because the absence of O2 prevented the propagation step to regenerate the NO2.
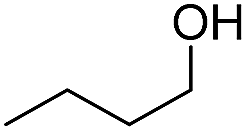
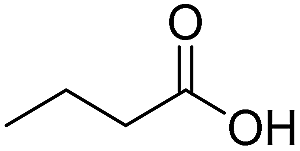
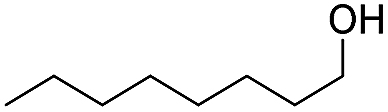
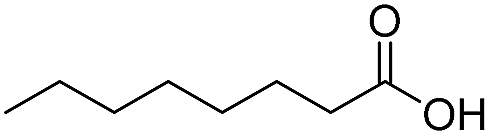
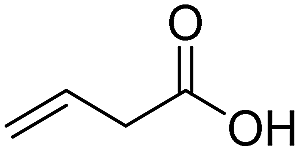
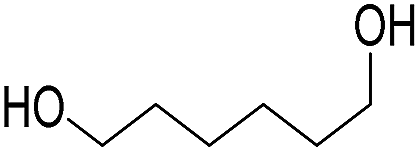
Finally, we also challenged the utility of GA in the catalytic oxidation of aliphatic alcohols (Table 3), which are less reactive than BA. Remarkably, after 2 h of reaction, the alcohols were fully transformed into the nitrites, as demonstrated by the detection of the sharp NMR peak at δ ≈ 6.0 ppm of the withdrawn aliquots. This suggests that the oxidation of the organic nitrite is likely the rate-determining step. Eventually, GA converted all alcohols listed in Table 3 with >99% selectivity to the corresponding acid in 16 h. Even the less reactive higher alcohols, such as 1-octanol, did not compromise GA activity. Furthermore, the reactive groups of the catalytic substrates were not affected during the oxidation: for example, 3-butenoic acid was produced with 98% selectivity starting from 3-butenol (i.e. less than 2% of the α,β-aldehyde was detected). Diols, like 1,6-hexanediol, were also oxidized to the corresponding adipic acid in 16 h.

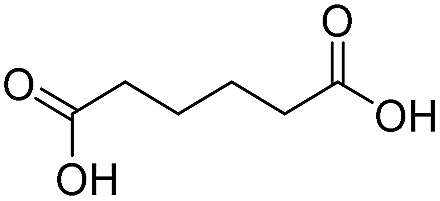
GA can successfully be recycled as the catalyst of BA oxidation. After reaction, the sample was recovered and washed profusely with acetone to remove any contaminant (see Fig. S9† for the UV-vis spectrum after the reaction), and submitted to a new catalytic cycle. This procedure can be repeated at least 10 times (Fig. S12†) without detecting an increase in the oxidation of the material. The metal impurities were also analyzed by ICP-MS during the cycling procedure. The initial crude catalyst (GA) displayed significantly higher content of metal contaminants than the catalyst after recycling (GA*, Table S5†) as a consequence of the additional washings and successive treatment with fresh HNO3. Nevertheless, the recycled catalyst showed identical activity to the “fresh” one even after ten recycling experiments, because there were no differences detected in conversion within the experimental error (Fig. S12†). These facts rule out again a potential effect of metal impurities on the catalytic performance of GA.
Conclusions
By a judicious choice of the experimental conditions, GA can catalyze with unprecedented efficiency the conversion of BA either to the aldehyde with catalytic amounts of HNO3 as the co-catalyst or to the carboxylic acid, under open air conditions and stoichiometric amount of HNO3. After thorough comparisons with other carbocatalysts (even with metal nanoparticles) as summarized in Table S3,† GA unequivocally delivers outstanding catalytic activity. On top of that, GA can be recovered after the reaction and reused in new catalytic runs with uncompromised activity for at least 10 cycles. Remarkably, GA demonstrated to be a versatile catalyst, performing flawlessly in the oxidation of a variety of alcohols. Accumulated experimental evidence and DFT calculations on the reaction mechanism unraveled key aspects of the reaction: (i) the redox decomposition of HNO3 on the GA surface; (ii) the generation of key NOx intermediates and organic nitrites; (iii) the evolution of the intermediates on the GA surface; (iv) the release of NO, which is oxidized by O2 to NO2 for re-entering the reaction cycle as the propagation step, and (v) equilibrium shifting by gas-release to direct the selectivity. Based on this understanding, the critical parameters were identified and optimized, turning the reaction fully selective to each of the two possible products of the oxidation: aldehyde or acid (when BA is employed), or carboxylic acids, in the case of aliphatic alcohols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the following projects: Italian MIUR (PRIN, SMARTNESS, 2015K7FZLH), and MAECI Italy-China bilateral project (GINSENG, PGR00953). M. B. gratefully acknowledges the Clarin CoFund Program (ACA17-29, 600196), funded by Gobierno del Principado de Asturias and Marie Curie Actions. M. O. acknowledges ERC Consolidator grant 683024 from the European Union's Horizon 2020 and the Operational Programme Research, Development and Education – European Regional Development Fund project (CZ.02.1.01/0.0/0.0/16_019/0000754) from the Ministry of Education, Youth and Sports of the Czech Republic. The authors also thank Mgr. Radka Pechancová and Dr David Milde for the digestion of solid samples and ICP-MS analyses.References
- C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef CAS PubMed
.
- D. R. Dreyer and C. W. Bielawski, Chem. Sci., 2011, 2, 1233–1240 RSC
- S. De, A. M. Balu, J. C. van der Waal and R. Luque, ChemCatChem, 2015, 7, 1608–1629 CrossRef CAS
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS
-
M. Monai, M. Melchionna and P. Fornasiero, in Advances in Catalysis, ed. C. Song, Academic Press, 2018, vol. 63, pp. 1–73 Search PubMed
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2015, 22, 3906–3924 CrossRef PubMed
- A. Lerf, H. He, M. Foster and J. Klinowski, J. Phys. Chem. B, 1998, 1998, 4477–4482 CrossRef
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813–6816 CAS
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron, 2011, 67, 4431–4434 CrossRef CAS
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Adv. Synth. Catal., 2011, 353, 528–532 CrossRef CAS
- A. V. Kumar and K. R. Rao, Tetrahedron Lett., 2011, 52, 5188–5191 CrossRef
- S. M. Islam, A. S. Roy, R. C. Dey and S. Paul, J. Mol. Catal. A: Chem., 2014, 394, 66–73 CrossRef CAS
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed
- S. Presolski and M. Pumera, Angew. Chem., Int. Ed., 2018, 57, 16713–16715 CrossRef CAS PubMed
- C. P. Vinod, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2010, 86, 161–171 CrossRef
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed
- C. Aellig, C. Girard and I. Hermans, Angew. Chem., Int. Ed., 2011, 50, 12355–12360 CrossRef CAS PubMed
- Y. Kuang, N. M. Islam, Y. Nabae, T. Hayakawa and M.-a. Kakimoto, Angew. Chem., Int. Ed., 2010, 122, 446–450 CrossRef
- Y. Cui, Y. H. Lee and J. W. Yang, Sci. Rep., 2017, 7, 3146 CrossRef PubMed
- S. R. Joshi, K. L. Kataria, S. B. Sawant and J. B. Joshi, Ind. Eng. Chem. Res., 2005, 44, 325–333 CrossRef CAS
- A. Bakandritsos, M. Pykal, P. Błoński, P. Jakubec, D. D. Chronopoulos, K. Poláková, V. Georgakilas, K. Čépe, O. Tomanec, V. Ranc, A. B. Bourlinos, R. Zbořil and M. Otyepka, ACS Nano, 2017, 11, 2982–2991 CrossRef CAS PubMed
- M. Pykal, P. Jurečk, F. Karlický and M. Otyepka, Phys. Chem. Chem. Phys., 2016, 18, 6351–6372 RSC
- D. Matochova, M. Medveď, A. Bakandritsos, T. Stekly, R. Zboril and M. Otyepka, J. Phys. Chem. Lett., 2018, 9, 3580–3585 CrossRef CAS PubMed
- D. Mosconi, M. Blanco, T. Gatti, L. Calvillo, M. Otyepka, A. Bakandritsos, E. Menna, S. Agnoli and G. Granozzi, Carbon, 2019, 143, 318–328 CrossRef CAS
- F. Carraro, L. Calvillo, M. Cattelan, M. Favaro, M. Righetto, S. Nappini, I. Píš, V. Celorrio, D. J. Fermín, A. Martucci, S. Agnoli and G. Granozzi, ACS Appl. Mater. Interfaces, 2015, 7, 25685–25692 CrossRef CAS PubMed
-
M. Frisch, et al, Gaussian 09.D01, Gaussian, Inc., Wallingord, CT, 2009 Search PubMed
- R. Ditchfield, W. J. Herhe and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- F. Carraro, L. Calvillo, M. Cattelan, M. Favaro, M. Righetto, S. Nappini, I. Píš, V. Celorrio, D. J. Fermín, A. Martucci, S. Agnoli and G. Granozzi, ACS Appl. Mater. Interfaces, 2015, 7, 25685–25692 CrossRef CAS PubMed
- M. Favaro, S. Agnoli, C. D. Valentin, C. Mattevi, M. Cattelan, L. Artiglia, E. Magnano, F. Bondino, S. Nappini and G. Granozzi, Carbon, 2014, 68, 319–329 CrossRef CAS
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed
- J. W. Larsen, M. Freund, K. Y. Kim, M. Sidovar and J. L. Stuart, Carbon, 2000, 38, 655–661 CrossRef CAS
- M. Blanco, B. Nieto-Ortega, A. d. Juan, M. Vera-Hidalgo, A. López-Moreno, S. Casado, L. R. González, H. Sawada, J. M. González-Calbet and E. M. Pérez, Nat. Commun., 2018, 9, 2671 CrossRef PubMed
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS PubMed
- K.-H. Wu, D.-W. Wang, X. Zong, B. Zhang, Y. Liu, I. R. Gentle and D.-S. Su, J. Mater. Chem. A, 2017, 5, 3239–3248 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Characterizations, oxidation of benzyl alcohol, computational details and monitoring of the catalysts. See DOI: 10.1039/c9sc02954k |
| This journal is © The Royal Society of Chemistry 2019 |