DOI:
10.1039/C9SC02818H
(Edge Article)
Chem. Sci., 2019,
10, 7340-7344
Visible-light-mediated deuteration of silanes with deuterium oxide†
Received
10th June 2019
, Accepted 2nd July 2019
First published on 8th July 2019
Abstract
Isotopically labeled compounds are highly desirable as they can serve as both mechanistic probes in chemistry and diagnostic tools in medicinal research. Herein, we report an unprecedented visible-light-mediated metal-free deuteration of silanes using D2O as an inexpensive, readily available, and easy to handle deuterium source. A broad range of aryl- and alkyl-substituted silanes were deuterated with high deuterium incorporations and yields. Furthermore, a 100 gram-scale synthesis was demonstrated using continuous-flow micro-tubing reactors, where enhanced reaction efficiency was obtained. The photoredox-catalyzed polarity matched hydrogen atom transfer (HAT) between silanes and the thiol HAT catalyst was responsible for the efficient deuteration.
Introduction
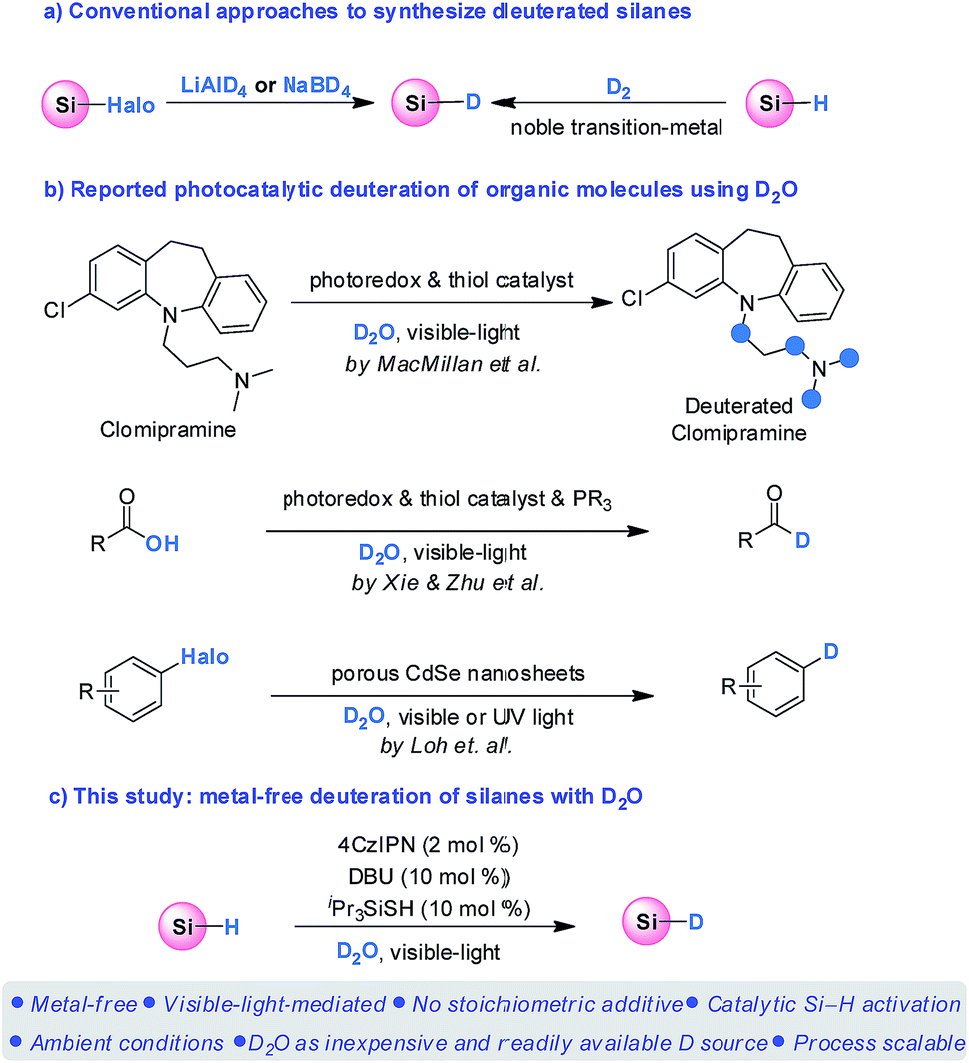
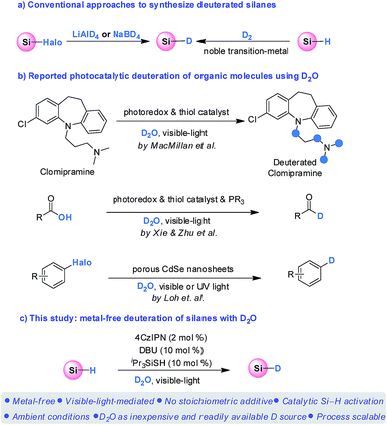
Deuterium-labeled compounds are of significant importance in organic synthesis,1 mechanistic investigations,2 mass spectrometric studies,3 and pharmaceutical discoveries.4 The increasing demand for deuterium-labeled compounds has recently stimulated the quest for convenient, selective, and especially catalytic approaches for their preparation.5 A number of valuable methods have therefore been developed.6 In this context, deuterated silanes not only serve as important probes for mechanistic studies in organic silicon chemistry, but are also versatile reagents for deuterium-labeling, which can add catalytically to C–X (X = C, N, O) multiple bonds through well-established hydrosilylations, as well as to reduce carbon–halogen and carbon–oxygen bonds.7,8 Despite these advantages, there are only limited protocols available for preparation of deuterated silanes. Conventionally, deuterated silanes are prepared by either reduction of halosilanes with LiAlD4 or NaBD4 (ref. 9) or isotopic exchange of the Si–H/D bond of silanes with D2 through noble transition-metal catalysis (Scheme 1a).10 However, the use of stoichiometric amounts of expensive metallo-deuteride reagents and the requirement of equipment-demanding D2 with noble metals undermine their wide application in synthetic chemistry. The development of a catalytic procedure using deuterium oxide (D2O) as the deuterium source for silanes deuteration therefore represents an appealing strategy in term of mildness, sustainability, and cost, which by far has not been realized.
 |
| | Scheme 1 Silane deuteration and photocatalytic deuteration using D2O. | |
Significant developments have been achieved for photocatalysis over the past decade, which enables previously inaccessible transformations.11 In this context, photocatalytic deuteration of medicinally and synthetically valuable organic molecules using D2O has recently received considerable attentions (Scheme 1b). For instance, the MacMillan group have recently pioneered a photoredox-mediated HAT process for selective deuteration of α-amino C(sp3)–H bonds with D2O, enabling a powerful protocol to incorporate deuterium in pharmaceutical compounds.12 Very recently, the Xie and Zhu group realized an elegant deoxygenative deuteration of carboxylic acids with D2O for the synthesis of deuterated aldehydes, through the synergistic combination of photoredox catalysis, thiol catalysis and phosphoranyl radical chemistry.13 Both reactions relied on the HAT process between the carbon or acyl radical intermediates and the thiol catalyst which is in equilibrium with D2O. In addition, the Loh group have reported a deuteration of halogenated compounds using D2O catalyzed by porous CdSe nanosheets under visible or UV light irradiation through a photocatalytic D2O splitting process.14
We have recently reported a visible-light-mediated metal-free hydrosilylation of alkenes via Si–H activation.15 With the combination of an organo photocatalyst, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN), and triisopropylsilanethiol HAT catalyst, the hydrosilylation of electron-rich alkenes proceeded smoothly. In the proposed mechanism, the polarity matched HAT process between the thiyl radical and silane facilitates the generation of a silyl radical, which subsequently adds to alkenes to achieve the hydrosilylation products. Taking the significance of deuterated silanes into consideration, we speculated that if D2O is present instead of an alkene, the generation of deuterated thiol would occur reversibly,12,13 which might subsequently result in the formation of a Si–D bond through a deuterium atom transfer (DAT) between the silyl radical and the deuterated thiol.16 Herein, we report an unprecedented metal-free deuteration of silanes using D2O as an easily handled, inexpensive, and readily available deuterium source (Scheme 1c).
Results and discussion
Optimization of reaction conditions
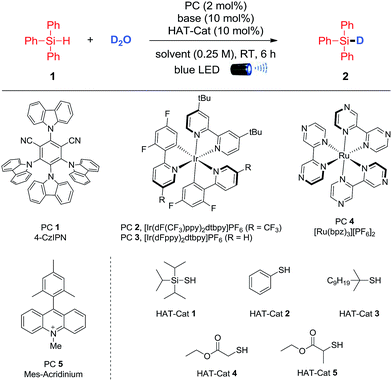
Our investigation of the proposed deuteration protocol was initiated by using triphenylsilane 1 as the model substrate. In the presence of photocatalyst 4CzIPN (PC 1, 2 mol%), DIPEA (10 mol%), and triisopropylsilanethiol (HAT-Cat 1, 10 mol%) in EtOAc, the deuteration of triphenylsilane 1 with D2O under blue LED irradiation (λmax = 470 nm) proceeded smoothly to give the product in 77% yield with 89% D-incorporation (Table 1, entry 1), demonstrating the viability of the hypothesis. A systematic survey on the reaction parameters including photocatalysts, solvents, bases and HAT catalysts was subsequently performed. Several common photocatalysts (PC 1–PC 5) were evaluated and 4CzIPN remained the most efficient with respect to both the yield and D-incorporation. Iridium(III) catalysts bearing similar redox potentials to 4CzIPN, such as Ir[dF(CF3)ppy]2(dtbpy)PF6 (PC 2) and Ir(dFppy)2(dtbpy)3PF6 (PC 3) gave comparable D-incorporation (entries 2 and 3). Other photocatalysts such as Ru(bpz)3[PF6]2 (PC 4) and 9-mesityl-10-methylacridinium perchlorate (PC 5) were not suitable for this transformation (entries 4 and 5). Among the solvents screened, EtOAc was the optimal, achieving the highest efficiency and D-incorporation (entries 6–8). The use of EtOAc as solvent was also green and economical. The use of base as additive had a big influence on the reaction outcome. Both K2CO3 and DBU resulted in excellent D-incorporations (entries 9 and 10), while other common bases such as Et3N and DMAP were less effective (entries 11 and 12). The deuteration could also occur in the absence of base, albeit in much lower D-incorporation (entry 13). Thiol HAT catalysts with different S–H bond dissociation energy (BDE) were examined. Triisopropylsilanethiol (HAT-Cat 1) was found to be the optimal HAT catalyst, whereas other catalysts such as PhSH (HAT-Cat 2), tert-dodecanethiol (HAT-Cat 3), ethyl 2-mercaptoacetate (HAT-Cat 4), and ethyl 2-mercaptopropanoate (HAT-Cat 5) gave moderate to good D-incorporations (entries 14–17). Finally, no deuterated silane 2 was generated in the absence of either the photocatalyst, the HAT catalyst, or light, demonstrating the necessity of all these components (entry 18).
Table 1 Survey of the model deuteration conditionsa
|
|
| Entry |
PC |
Base |
HAT-Cat |
Solvent |
Yieldb (%) |
D-inc.c (%) |
|
Typical conditions: silane (0.5 mmol), D2O (0.45 mL, 25 mmol), PC (0.01 mmol), base (0.05 mmol), and HAT-Cat (0.05 mmol) in solvent (2 mL), irradiated by 18 W blue LED.
Isolated yields of the Si–D/H mixtures.
Ratios determined by 1H NMR spectroscopy.
Recovery of the starting silane.
|
| 1 |
PC 1 |
DIPEA |
HAT-Cat 1 |
EtOAc |
77 |
89 |
| 2 |
PC 2 |
DIPEA |
HAT-Cat 1 |
EtOAc |
40 |
80 |
| 3 |
PC 3 |
DIPEA |
HAT-Cat 1 |
EtOAc |
48 |
70 |
| 4 |
PC 4 |
DIPEA |
HAT-Cat 1 |
EtOAc |
— |
—d |
| 5 |
PC 5 |
DIPEA |
HAT-Cat 1 |
EtOAc |
— |
—d |
| 6 |
PC 1 |
DIPEA |
HAT-Cat 1 |
CH2Cl2 |
50 |
13 |
| 7 |
PC 1 |
DIPEA |
HAT-Cat 1 |
THF |
20 |
73 |
| 8 |
PC 1 |
DIPEA |
HAT-Cat 1 |
CH3CN |
19 |
61 |
| 9 |
PC 1 |
K2CO3 |
HAT-Cat 1 |
EtOAc |
71 |
94 |
| 10 |
PC 1 |
DBU |
HAT-Cat 1 |
EtOAc |
78 |
95 |
| 11 |
PC 1 |
Et3N |
HAT-Cat 1 |
EtOAc |
48 |
86 |
| 12 |
PC 1 |
DMAP |
HAT-Cat 1 |
EtOAc |
81 |
34 |
| 13 |
PC 1 |
— |
HAT-Cat 1 |
EtOAc |
82 |
21 |
| 14 |
PC 1 |
DBU |
HAT-Cat 2 |
EtOAc |
70 |
52 |
| 15 |
PC 1 |
DBU |
HAT-Cat 3 |
EtOAc |
60 |
40 |
| 16 |
PC 1 |
DBU |
HAT-Cat 4 |
EtOAc |
42 |
84 |
| 17 |
PC 1 |
DBU |
HAT-Cat 5 |
EtOAc |
61 |
52 |
| 18 |
No PC or HAT-Cat or light |
— |
—d |
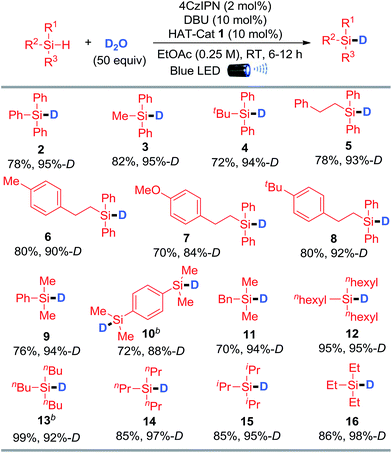
Scope of the deuteration reactions
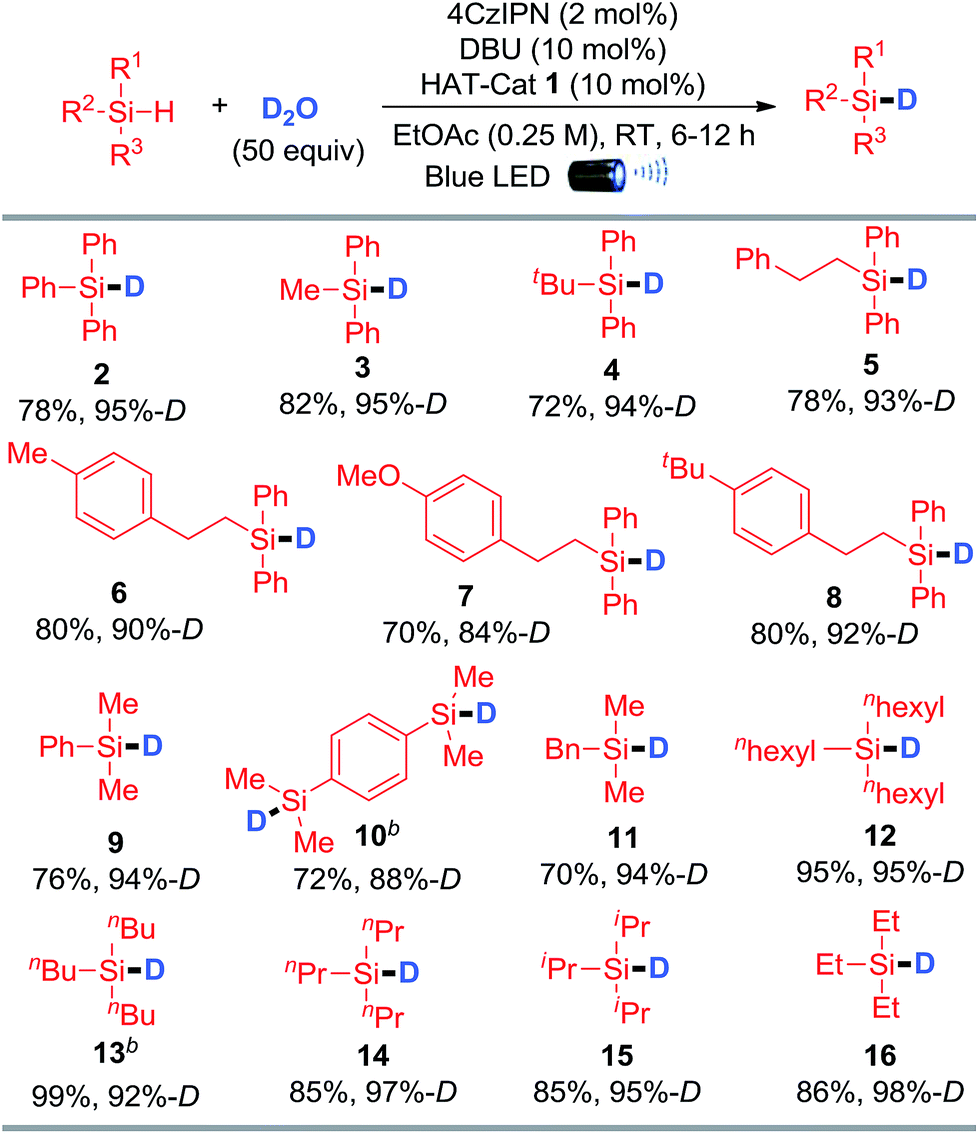
The generality of silane deuteration was evaluated employing the optimal reaction conditions (Table 1, entry 10). As depicted in Table 2, aryl-substituted silanes including tri-, di-, and mono-aryl silanes with different steric and electronic properties were well-tolerated to afford the corresponding deuterated products (2–10) in good to excellent D-incorporations. Disilanes such as para-phenylenebis(dimethylsilane) were also suitable substrates, smoothly delivering the product with two deuterium atoms incorporated (10). A series of trialkylsilanes with different sizes were effectively deuterated, affording the corresponding products with excellent D-incorporations (11–16). Notably, our protocol is efficient for the preparation of deuterated triethylsilane (16), a frequently utilized deuterated silane reagent in organic synthesis. It is worth mentioning that the labile benzylic C–H bonds were unperturbed during the deuteration event, highlighting the highly selective HAT for Si–H activation (5–8, 11). This visible-light-mediated metal-free protocol enables rapid and efficient deuteration of silanes using D2O under mild conditions, which is distinguished from the existing methods that require the use of either expensive and operational cumbersome LiAlD4 or equipment-demanding D2 and noble transition-metals. It therefore represents a green and economic method for the preparation of deuterated silanes.
Table 2 Photo-mediated deuteration of silanes using D2Oa
|
Typical conditions: silane (0.5 mmol), D2O (0.45 mL, 25 mmol), DBU (7.5 μL, 0.05 mmol), triisopropylsilanethiol (10.7 μL, 0.05 mmol), and 4CzIPN (7.8 mg, 0.01 mmol) in ethyl acetate (2 mL), irradiated by 18 W blue LED. Isolated yields of the Si–D/H mixtures. Deuterium incorporation was determined by analysis of 1H NMR spectra.
DIPEA (8.2 μL, 0.05 mmol) was used instead of DBU.
|
|
Mechanistic rationale
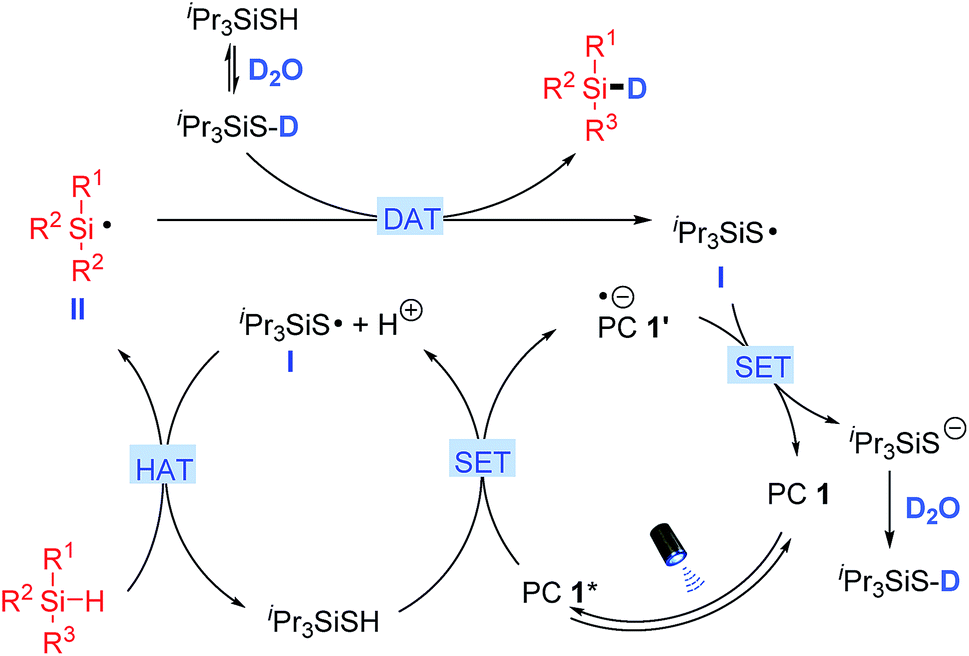
A proposed mechanism for silane deuteration is illustrated in Scheme 2 based on all experimental results. Initial photo-excitation of PC 1 (4CzIPN) generates its excited form PC 1* (E1/2 (*P/P−) = +1.35 V vs. SCE in MeCN),17 which is reductively quenched by triisopropylsilanethiol (Eox1/2 = +0.28 V vs. SCE in MeCN) via a single electron transfer (SET) process to give the reduced PC 1′ and the thiyl radical I after deprotonation.15 The presence of a catalytic amount of base presumably facilitates the deprotonation for the generation of thiyl radical I. The electrophilic thiyl radical then undergoes a polarity matched HAT to abstract a hydrogen atom from the hydridic Si–H bond of silane, generating the nucleophilic silyl radical II.18 In the presence of excess amounts of D2O, the H/D exchange of the thiol catalyst leads to dominant formation of the deuterated thiol (Fig. S7†).12,13 The silyl radical II then abstracts a deuterium atom from the deuterated thiol to deliver the deuterated silane product and the thiyl radical I.16 Single electron reduction of thiyl radical I (Ered1/2 = −0.82 V vs. SCE in MeCN)15 by PC 1′ (E1/2(P/P−) = −1.21 V vs. SCE in MeCN)17 regenerates the photocatalyst and the deuterated thiol after protonation with D2O. No reaction occurred when radical scavenger TEMPO was added, supporting the above radical-based process. Furthermore, the light on/off experiments illustrated a total interruption of the reaction process in the absence of light and recuperation of reactivity on further illumination (Fig. S8†). This indicates that light was essential for this transformation, and any chain-propagation process should be short-lived.
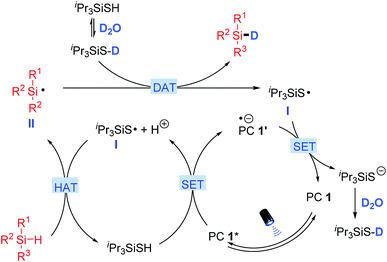 |
| | Scheme 2 Proposed mechanism for deuteration of organosilanes. | |
Synthetic utilities of the deuteration methodology
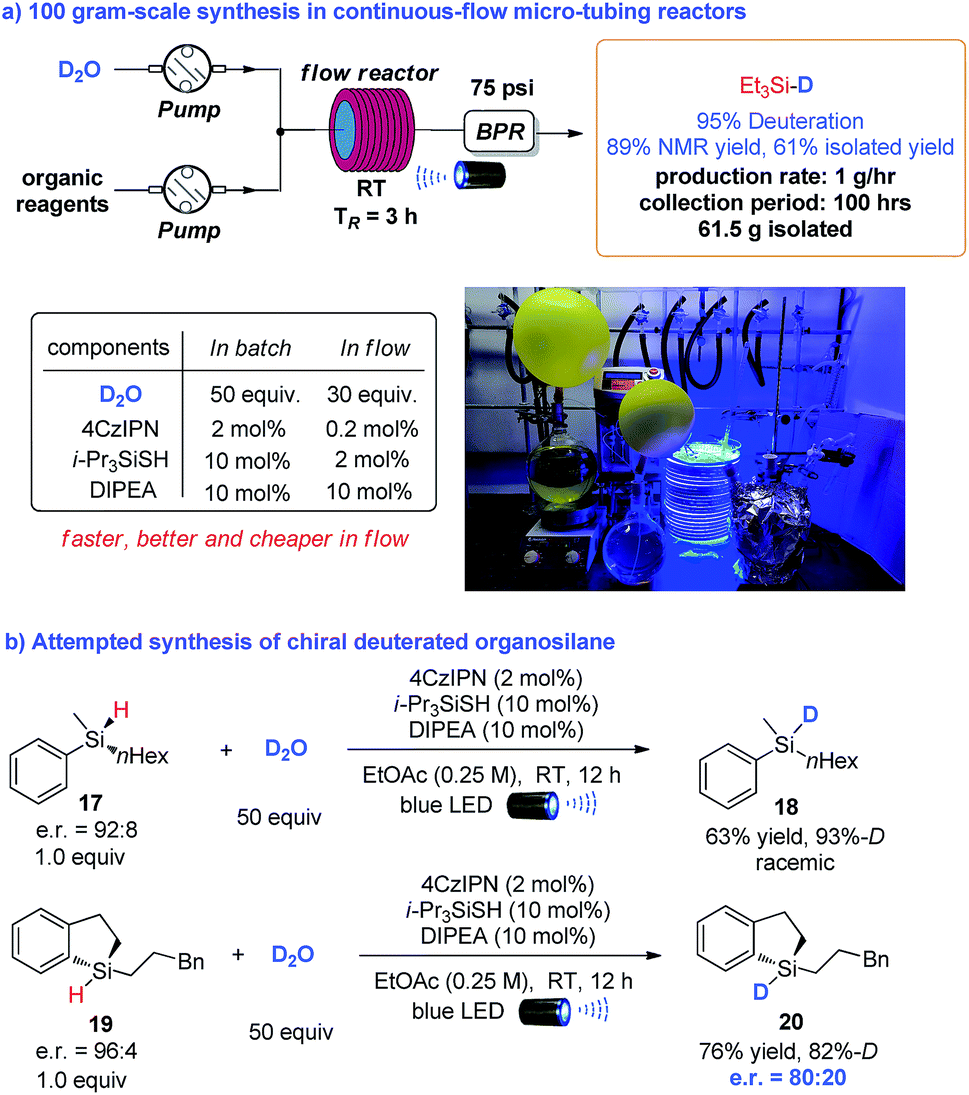
To further demonstrate the synthetic utility of this methodology, we examined the feasibility of large-scale synthesis using continuous-flow micro-tubing reactors. As illustrated in Scheme 3a, the transformation was amenable to scale-up with 100 grams of starting triethylsilane assisted by an operationally simple continuous-flow setup, which resulted in the deuteration of triethylsilane with excellent D-incorporation (95%) and good crude yield (89% based on analysis of the crude 1H NMR spectra). 61.5 grams of pure deuterated triethylsilane was isolated by a careful distillation of the crude product mixture, highlighting the potential for large scale synthesis. Notably, compared to the batch synthesis, the requirements of photocatalyst (4CzIPN), HAT catalyst (triisopropylsilanethiol) and D2O to achieve an efficient transformation were all dramatically decreased in continuous-flow micro-tubing reactors, from 2 mol%, 10 mol% and 50 equivalents to 0.2 mol%, 2 mol% and 30 equivalents, respectively. The residence time was shortened to 3 h instead of the 12 h required in the batch reaction. This provides an even more economic pathway for the synthesis of deuterated silanes,19 highlighting the superiority of micro-tubing flow reactors in photochemical synthesis.20 Moreover, considering the pyramidal configuration of silyl radicals,21 we envisioned that there might be an opportunity to access enantioenriched deuterated silanes starting from chiral organosilanes through memory of chirality.22 Even though the acyclic chiral silane 17 afforded racemic product 18, the chirality of cyclic chiral silane 19 was preserved in product 20 to a large extent via the photo deuteration protocol (Scheme 3b).23
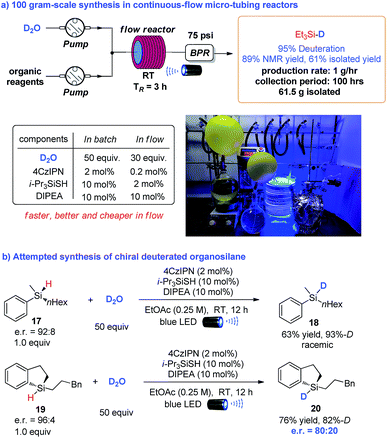 |
| | Scheme 3 Reaction scaling up in continuous-flow reactors and synthesis of chiral deuterated silanes. | |
Conclusions
In summary, we have developed a convenient visible-light-mediated deuteration of silanes using D2O by synergistic combination of an organophotocatalyst 4CzIPN and a thiol HAT catalyst in the presence of a catalytic amount of base additive. This protocol accommodates a wide range of silanes, achieving products with excellent D-incorporations, and is distinguished by its operational simplicity, metal-free character, mild reaction conditions, green solvent, and utilization of D2O as an easily handled and inexpensive deuterium source. Furthermore, a scaling-up process of 100 grams of starting silanes has been demonstrated using an operationally simple continuous-flow setup, enabling an even more efficient and economic synthesis. We anticipate that this convenient and economic synthetic protocol to prepare deuterated silanes will promote the wide usage of these important reagents in both academic and industrial settings.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support provided by the Ministry of Education (MOE) of Singapore (MOE2017-T2-2-081), GSK-EDB (R-143-000-687-592), National Natural Science Foundation of China (Grant No. 21702142, 21871205), and Japan Society for the Promotion of Science (a Grant-in-Aid for Scientific Research (S), 26220802).
Notes and references
-
(a) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396 CrossRef CAS PubMed
 ;
(b) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS PubMed .
;
(b) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS PubMed .
-
(a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed ;
(b) K. B. Wiberg, Chem. Rev., 1955, 55, 71 CrossRef .
- J. Atzrodt and V. Derdau, J. Labelled Compd. Radiopharm., 2010, 53, 674 CrossRef CAS .
- For selected reviews, see:
(a) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219 CrossRef CAS PubMed ;
(b) T. G. Gant, J. Med. Chem., 2014, 57, 3595 CrossRef CAS PubMed .
-
Synthesis and Application of Isotopically Labeled Compounds, ed. U. Pleiss and R. Voges, John Wiley, Chichester, New York, 2001, vol. 7 Search PubMed .
- For selected reviews, see:
(a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022 CrossRef CAS PubMed ;
(b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 CrossRef CAS PubMed . For representative recent examples, see:
(c) V. Soulard, G. Villa, D. P. Vollmar and P. Renaud, J. Am. Chem. Soc., 2018, 140, 155 CrossRef CAS PubMed ;
(d) X. Wang, M.-H. Zhu, D. P. Schuman, D. Zhong, W.-Y. Wang, L.-Y. Wu, W. Liu, B. M. Stoltz and W.-B. Liu, J. Am. Chem. Soc., 2018, 140, 10970 CrossRef CAS PubMed ;
(e) J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990 CrossRef CAS PubMed ;
(f) H. Yang, P. G. Dormer, N. R. Rivera and A. J. Hoover, Angew. Chem., Int. Ed., 2018, 57, 1883 CrossRef CAS PubMed ;
(g) R. P. Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195 CrossRef PubMed .
- For selected hydrosilylation reviews, see:
(a) X.-Y. Du and Z. Huang, ACS Catal., 2017, 7, 1227 CrossRef CAS ;
(b) J. Sun and L. Deng, ACS Catal., 2016, 6, 290 CrossRef CAS ;
(c) E. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729 CrossRef ;
(d)
B. Marciniec, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, 2009 CrossRef .
- For selected examples of reduction of carbon–halogen bonds, see:
(a) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188 CrossRef CAS PubMed ;
(b) M. Aizenberg and D. Milstein, Science, 1994, 256, 359 CrossRef PubMed For examples for reduction of carbon–oxygen bonds, see: Y. Nishibayashi, A. Shinoda, Y. Miyake, H. Matsuzawa and M. Sato, Angew. Chem., Int. Ed., 2006, 45, 4835 CrossRef CAS PubMed .
- For selected examples, see:
(a) A. Y. Khalimon, O. G. Shirobokov, E. Peterson, R. Simionescu, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Inorg. Chem., 2012, 51, 4300 CrossRef CAS PubMed ;
(b) S. C. A. Sousa and A. C. Fernandes, Adv. Synth. Catal., 2010, 352, 2218 CrossRef CAS ;
(c) P. D. Prince, M. J. Bearpark, G. S. McGrady and J. W. Steed, Dalton Trans., 2008, 271 RSC .
- For selected examples, see:
(a) Y. Kratish, D. Bravo-Zhivotovshii and Y. Apeloig, ACS Omega, 2017, 2, 372 CrossRef CAS ;
(b) K. A. Smart, E. Mothes-Martin, T. Annaka, M. Grellier and S. Sabo-Etienne, Adv. Synth. Catal., 2014, 356, 759 CrossRef CAS ;
(c) D. Schmidt, T. Zell, T. Schaub and U. Radius, Dalton Trans., 2014, 43, 10816 RSC ;
(d) G. C. Fortman, H. Jacobsen, L. Cavallo and S. P. Nolan, Chem. Commun., 2011, 47, 9723 RSC ;
(e) J. Campos, A. C. Esqueda, J. López-Serrano, L. Sánchez, F. P. Cossio, A. de Cozar, E. Álvarez, C. Maya and E. Carmona, J. Am. Chem. Soc., 2010, 132, 16765 CrossRef CAS PubMed .
- For selected reviews, see:
(a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed ;
(b) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed ;
(c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed ;
(d) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed ;
(e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed ;
(f) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed ;
(g) J. M. R. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC .
-
(a) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182 CrossRef CAS PubMed ;
(b) Foxr related photo-mediated HAT process, see: W. Xu, J. Ma, X.-A. Yuan, J. Dai, J. Xie and C.-J. Zhu, Angew. Chem. Int. Ed., 2018, 57, 10357 CrossRef CAS PubMed ;
(c) A. Hu, J.-J. Guo, H. Pan and Z.-W. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed ;
(d) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed ;
(e) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed .
- M. Zhang, X.-A. Yuan, C. Zhu and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 312 CrossRef CAS PubMed .
- C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G.-H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q.-H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef PubMed .
- R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 16621 CrossRef CAS PubMed .
- For reversible HAT involving the thiyl radical, see C. Schöneich and K.-D. Asmus, J. Chem. Soc., Faraday Trans., 1995, 91, 1923 RSC , and references cited therein.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS .
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC .
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef .
- The cost of preparation of Et3SiD using this flow protocol is estimated to ca. $10.5/1 g, which compared very favourably to commercially available Et3SiD (MOLBASE price: $156/500 mg, https://www.molbase.com/cas/1631-33-0.html) for details, see the ESI.†.
- C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229 CrossRef CAS .
- C. S. Gloor, F. Dénès and P. Renaud, Free Radical Res., 2016, 50, S102 CrossRef CAS PubMed .
- For the synthesis of chiral silanes, see: G. Zhan, H. Teng, Y. Luo, S. Lou, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2018, 57, 12342 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and NMR spectra of all new compounds. See DOI: 10.1039/c9sc02818h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Jiesheng
Li‡
a,
Han Wen
Cheo
a,
Rachel
Chua
a,
Gu
Zhan
d,
Zhaomin
Hou
abc,
Jiesheng
Li‡
a,
Han Wen
Cheo
a,
Rachel
Chua
a,
Gu
Zhan
d,
Zhaomin
Hou