
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-neutral ortho-C–H amination of pinacol arylborates via palladium(II)/norbornene catalysis for aniline synthesis†
Shuqing
Chen‡
a,
Peng
Wang‡
a,
Hong-Gang
Cheng
 a,
Chihui
Yang
a and
Qianghui
Zhou
*ab
a,
Chihui
Yang
a and
Qianghui
Zhou
*ab
aSauvage Center for Molecular Sciences, Engineering Research Center of Organosilicon Compounds & Materials (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China
bThe Institute for Advanced Studies, Wuhan University, 430072, Wuhan, China. E-mail: qhzhou@whu.edu.cn
First published on 23rd July 2019
Abstract
A palladium(II)/norbornene cooperative catalysis enabled redox-neutral ortho-C–H amination of pinacol aryl- or heteroarylborates for the synthesis of structurally diverse anilines is reported. This method is scalable, robust (tolerance of air and moisture), phosphine ligand-free, and compatible with a wide range of functionalities. These practical features make this reaction amenable for industry. A plethora of synthetically very useful halogenated anilines, which often cannot be prepared via other transition-metal-catalyzed aminations, are readily produced using this method. Particularly, the orthogonal reactivity between pinacol arylborates and aryl iodides is demonstrated. Preliminary deuterium-labeling studies reveal a redox-neutral ipso-protonation mechanism of this process, which will surely inspire the future development of this field. Overall, the exceptionally broad scope (47 examples) and reliability of this procedure, together with the wide availability of pinacol arylborates, make this chemistry a valuable addition to the existing methods for aniline synthesis.
Functionalized anilines have attracted considerable attention as a result of their widely applications in the pharmaceutical industry and medicinal chemistry.1 For example, popular marketed medicines aripiprazole (antipsychotics),1b repaglinide (anti-type II diabetes),1c linezolid (antibacterial),1d and EphB4 kinase inhibitors1e all contain an aniline motif as the pharmacophore (Fig. 1). Therefore, general methods for the assembly of these motifs are highly desirable. Synthesis of anilines through C–N bond formation is the most widely adopted strategy, and substantial progress has been made over the past two decades.2–5 Among these approaches, palladium-catalyzed aminations play a significant role.3–5 For example, the famous Buchwald–Hartwig ipso-amination3 that allows the facile synthesis of anilines from aryl halides and nucleophilic amines (Scheme 1A). In 2013, the Dong group reported an elegant palladium(0)/norbornene (NBE) cooperative catalysis (the Catellani reaction)6 enabled ortho-C–H amination of aryl iodides (the Dong amination), utilizing electrophilic O-benzoyl hydroxylamines as the amination reagents and isopropanol as the proton source for ipso-termination.4 Later on, they extended this chemistry to ortho-C–H amination of aryl bromides (Scheme 1B).5 The Dong amination represents a nice complement to the Buchwald–Hartwig ipso-amination for the preparation of meta-substituted anilines, while it requires a stoichiometric reductant for the recovery of the palladium(0) catalyst. Recently, the group of Zhang and our group concurrently reported the ortho-C–H alkylation of the arylboronic acids and their derivatives (instead of aryl halides) via the palladium(II)-initiated Catellani-type reaction (Borono–Catellani reaction).7 Therein, a stoichiometric oxidant (Cu(OAc)2, air or oxygen) is required for regenerating the palladium(II) catalyst. Based on the chemistry, we envisaged a redox-neutral “Borono–Catellani amination” process for the facile preparation of anilines, with widely accessible arylboronic acids or their derivatives and O-benzoyl hydroxylamines as the reactants (Scheme 1C). Prior to the start of this project, redox-neutral Borono–Catellani reactions had not been reported. Until recently, the group of Dong reported elegant related research (Scheme 1D).8 As shown in Scheme 1C, it is surmised that the reaction is initiated through transmetalation between the palladium(II) catalyst and arylboronic acid derivative 1 to deliver the aryl palladium(II) intermediate I. The following steps involve insertion of NBE to I and subsequent ortho-C–H activation, generating the aryl/NBE palladacycle complex (ANP) II. The oxidative addition of amination reagent 2 to II gives the palladium(IV) complex III,9 which then undergoes reductive elimination and successive release of NBE to afford palladium(II) species IV. The final reaction termination by protonation10 of IV provides the desired aniline 3 and regenerate the palladium(II) catalyst. The intrinsic advantage of this “Borono–Catellani amination” lies in its “redox-neutral” property: this process involves a Pd(II)–Pd(IV)–Pd(II) catalytic cycle,11 thus no external oxidant and reductant are needed. This feature constitutes a nice extension of previous Catellani-type reactions.12 In addition, the widely accessible arylboronic species versus aryl halides will guarantee promising synthetic potential of this process for diversified aniline preparation. Although the above process is intriguing and mechanistically feasible, multiple challenges are foreseeable. First, protodepalladation can occur to arylpalladium species (I, II and IV) of the catalytic cycle, which may result in a selectivity issue. Second, transmetalation of arylborates and the following ortho-C–H activation are generally promoted by a base, while the final protodepalladation step requires an acid. Hence, the compatibility of these paradoxical steps could be another issue. Third, other competing side reactions related to arylboronic species (e.g. direct ipso-amination,13 protolytic deboronation,14 and homo-coupling15) will also constitute formidable challenges.
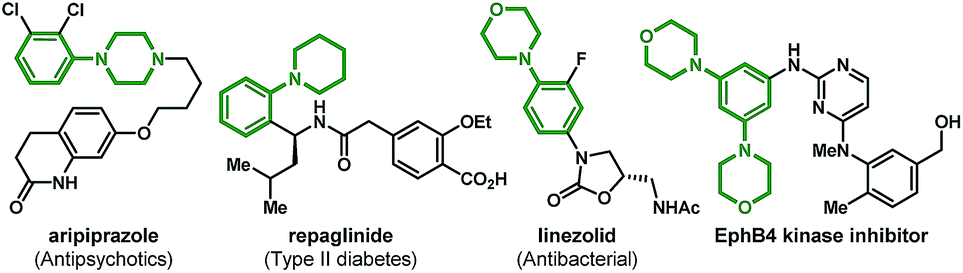 | ||
| Fig. 1 Biologically important aniline scaffolds. | ||
 | ||
| Scheme 1 Palladium-catalyzed amination strategies for aniline synthesis. | ||
To address the aforementioned challenges and realize this intriguing process, a model reaction was set up for the optimization of reaction conditions, with readily available 1-naphthylboronic acid pinacol ester (1a) and morpholino benzoate (2a) as the reactants. Selected results are summarized in Table 1. Initially, Pd(OAc)2 was chosen as the catalyst (10 mol%), 2-norbornene (NBE) as the mediator, K2CO3 as the base (2.5 equiv.), and DMF as the solvent. Gratifyingly, when the reaction was run at 70 °C in air for 16 h, the desired product 3a was obtained in 15% yield (entry 1). The following studies focused on screening of the reaction solvent, which was demonstrated to play a critical role in this process (entries 1–4). Only a trace amount of 3a was detected when nonpolar solvent toluene was used. In contrast, while switching to the polar solvent DMSO, a dramatic increase of yield of 3a was realized (56%, entry 3). In addition, the screening of other polar solvents did not provide further improvement in the reaction efficiency (see ESI for the details†). The use of DMSO as the solvent was beneficial for the desired reaction on the one hand, but on the other hand, a severe protodeboronation14 side reaction to yield naphthalene as well as quick decomposition of the amination reagent 2a also occurred. This was why an overall moderate yield (56%) was obtained. To our delight, after intensive optimizations, this problem was nicely solved by simply employing 1,4-dioxane as the cosolvent (DMSO/dioxane = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), and the yield of 3a was improved to 82% (entry 4). Additional optimizations regarding the base and palladium catalyst did not provide any significant improvement (entries 5–8). Thus, entry 4 was identified as the optimal conditions for this transformation. It is worth mentioning that these optimization studies were all performed in air,7b and a following control experiment revealed that air was beneficial since a tangible drop of yield was observed when running the reaction in argon (entry 9). This interesting phenomenon indicates that on the one hand the desired transformation is indeed a redox-neutral process (no external oxidant is needed). On the other hand, it shows that the presence of air could prohibit the possible palladium(0)-involved side reactions,15 thus favoring the expected reaction. Additionally, allowing us to perform the reaction in air is very attractive from both the practical and economic perspective. Moreover, this process does not require any phosphine or arsine ligand7b,c which is pivotal for previous palladium-catalyzed aminations.3–5,7a,8 Overall, these practical features make this reaction amenable for industry.
:5), and the yield of 3a was improved to 82% (entry 4). Additional optimizations regarding the base and palladium catalyst did not provide any significant improvement (entries 5–8). Thus, entry 4 was identified as the optimal conditions for this transformation. It is worth mentioning that these optimization studies were all performed in air,7b and a following control experiment revealed that air was beneficial since a tangible drop of yield was observed when running the reaction in argon (entry 9). This interesting phenomenon indicates that on the one hand the desired transformation is indeed a redox-neutral process (no external oxidant is needed). On the other hand, it shows that the presence of air could prohibit the possible palladium(0)-involved side reactions,15 thus favoring the expected reaction. Additionally, allowing us to perform the reaction in air is very attractive from both the practical and economic perspective. Moreover, this process does not require any phosphine or arsine ligand7b,c which is pivotal for previous palladium-catalyzed aminations.3–5,7a,8 Overall, these practical features make this reaction amenable for industry.
|
|
||||
|---|---|---|---|---|
| Entry | [Pd] | Base | Solvent | Yield (%)b |
|
a The reaction was performed on a 0.1 mmol scale.
b GC yield with biphenyl as an internal standard.
c DMSO/dioxane = 2:5.
d Isolated yield in parentheses on a 0.2 mmol scale.
e The reaction was performed under argon. TFA = trifluoroacetoxy, DMF = N,N-dimethylformamide, and DMSO = dimethylsulfoxide.
|
||||
| 1 | Pd(OAc)2 | K2CO3 | DMF | 15 |
| 2 | Pd(OAc)2 | K2CO3 | Toluene | 1 |
| 3 | Pd(OAc)2 | K2CO3 | DMSO | 56 |
| 4 | Pd(OAc)2 | K2CO3 | DMSO/dioxanec | 82 (84)d |
| 5 | Pd(OAc)2 | KHCO3 | DMSO/dioxanec | 46 |
| 6 | Pd(OAc)2 | KOH | DMSO/dioxanec | 70 |
| 7 | PdCl2 | K2CO3 | DMSO/dioxanec | 51 |
| 8 | Pd(TFA)2 | K2CO3 | DMSO/dioxanec | 74 |
| 9e | Pd(OAc)2 | K2CO3 | DMSO/dioxanec | 75 |

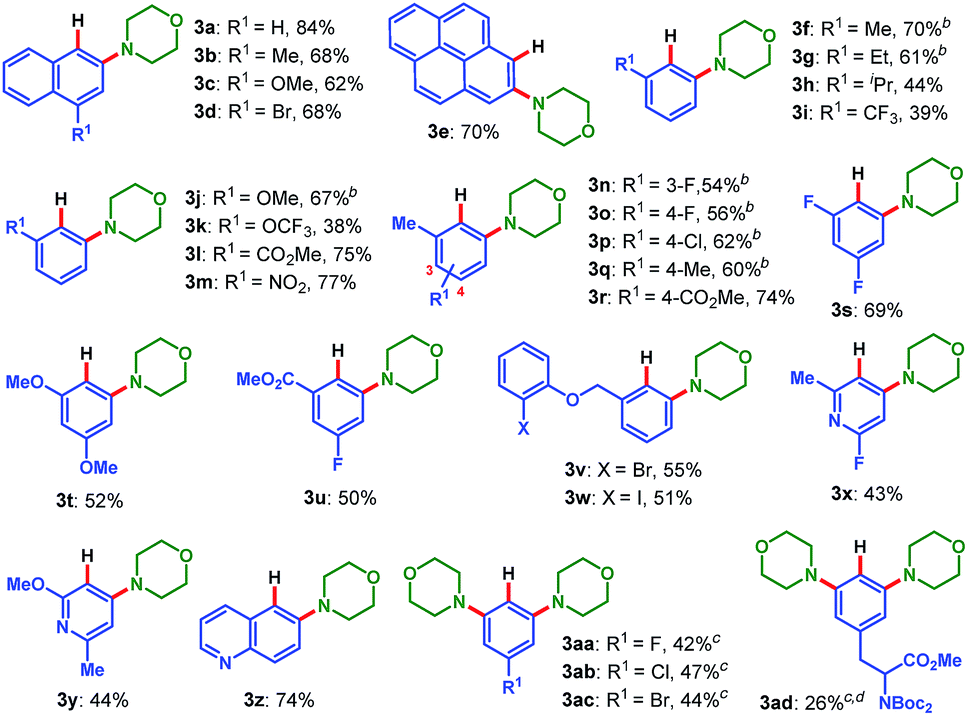
With the optimized conditions in hand, we first examined the scope of pinacol arylborates, employing 2a as the reaction partner. The results are summarized in Table 2. For ortho-substituted pinacol arylborates, both electron-donating (1f–h, 1j–k, 1q, and 1t) and electron-withdrawing substitutions (1i, 1l–p, 1r, and 1u) on the aromatic rings were compatible, providing meta-substituted anilines in moderate to good yields (38–77%). Polycyclic pinacol arylborates (1a–e) were also suitable substrates to deliver the corresponding anilines in good yields (62–84%). Notably, pinacol heteroarylborates (1x–1z) reacted well with 2a to afford the desired heteroanilines (3x–3z) in 43%, 44% and 74% yield, respectively. For pinacol arylborates without ortho substituents, the diaminated products (3aa–3ad) were obtained in moderate yields. In particular, the tyrosine-derived pinacol arylborate (1ad) could be applied to this reaction, delivering the corresponding unnatural amino acid-type product (3ad) in 26% yield. Furthermore, this transformation possesses excellent chemoselectivity, and a variety of functional groups were tolerated, including fluoro (3n–3o, 3s, 3u, 3x, and 3aa), chloro (3p and 3ab), bromo (3d, 3v, and 3ac), even iodo (3w), methoxyl (3c, 3j, 3t, and 3y), trifluoromethyl (3i), trifluoromethoxyl (3k), ester (3l, 3r, 3u, and 3ad), bis-Boc-protected amino (3ad), and nitro (3m). The compatibility of various functional groups is intriguing, since it would allow further manipulation of the obtained anilines. It is especially noteworthy that a variety of halogenated anilines (3d, 3p, 3v, 3w, and 3ab–3ac), which often cannot be prepared via other transition-metal-catalyzed aminations,3–5 enable further metal-catalyzed cross coupling reactions.16
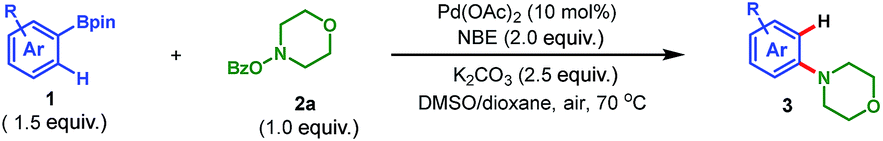
To illustrate the synthetic utility of this protocol, the scope of the amination reagent 2 was then investigated, with pinacol arylborate 1a as the reaction partner (Table 3). O-Benzoyl hydroxylamines derived from common cyclic amines, such as morpholine (2a), piperidine (2b–2k), piperazine (2l–2n), thiomorpholine (2o), pyrrolidine (2p) and azepane (2q) can all be used in the reaction to afford the corresponding products in moderate to good yields (29–84%). A gamut of functional groups in these amination reagents were tolerated during the process, such as primary alcohol (2g), secondary alcohol (2e), tertiary alcohol (2h), TBS ether (2f), ketal (2i), ester (2j and 2k), benzoylamino (2l) and carbamate (2m and 2n) groups. In addition, the uncyclized amination reagent N-benzyl N-benzoyl hydroxymethylamine (2r) also reacted uneventfully albeit with a moderate yield, which may serve as an alternative way to access the secondary aryl amines through a following debenzylation. It is worth mentioning that the incorporated heterocyclic motifs, e.g. morpholine, piperidine, piperazine, pyrrolidine and azepane, are ubiquitous and essentially important in small molecule drug discovery.17
|
|
|---|
| a All reactions were performed on a 0.2 mmol scale, and 2 was added in two portions. Isolated yields are reported. |

|
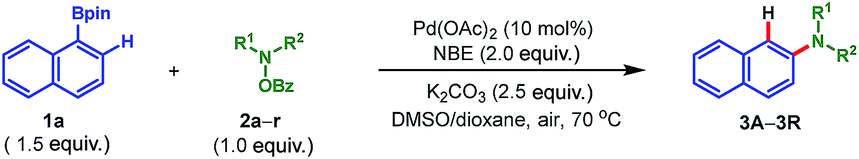
The practicality and robustness of this amination process are demonstrated by the 6.0 mmol scale experiment between 1a and 2a, as depicted in Scheme 2A, wherein application of the standard reaction conditions afforded product 3a in a good yield (72%, 0.92 g). The tolerance of the iodo group in the current Borono–Catellani amination process prompted us to investigate the possible orthogonal reactivity between pinacol arylborate and aryl iodides. The bifunctional reagent 1w was chosen as the model substrate to verify this hypothesis. Firstly, 1w reacted with 2a under the standard Borono–Catellani amination conditions, and the product 3w was indeed afforded in 51% yield while no classic Catellani-type product was observed. Then, the intact iodo group in 3w enabled a traditional Catellani reaction18 to provide the polysubstituted arene 4 in 71% yield. In stark contrast, subjecting 1w and 2a to traditional Catellani reaction conditions,4a the intramolecular Suzuki–Miyaura coupling product 5 was isolated as the major component, alongside the minor Catellani-type amination product 6 formed through intramolecular Suzuki–Miyaura termination.18 The Catellani-type ortho-C–H amination product 7 generated by protonation termination was not isolated (Scheme 2B). Hence, the orthogonal reactivity between these two types of Catellani reactions was demonstrated.
 | ||
| Scheme 2 Scale-up experiment and synthetic applications. Reagents and conditions: (1) Pd(OAc)2, (2-furyl)3P, NBE, Cs2CO3, toluene, 80 °C, Ar, 20 h; (2) Pd(OAc)2, P(pOMe-C6H4)3, NBE, Cs2CO3, toluene, 100 °C, Ar, 24 h. | ||
Furthermore, the synthetic utility of the obtained ortho-C–H amination products is shown in Scheme 2C. The 3,5-diaminated aryl halides 3ab and 3ac from Table 2 were readily transformed to 3,5-di(morpholino)aniline 8, the useful common intermediate to prepare various potent EphB4 kinase inhibitors,19 in good yields through the Ullmann–Ma amination.20 It is noteworthy that 8 was previously prepared in three steps from expensive starting materials.21
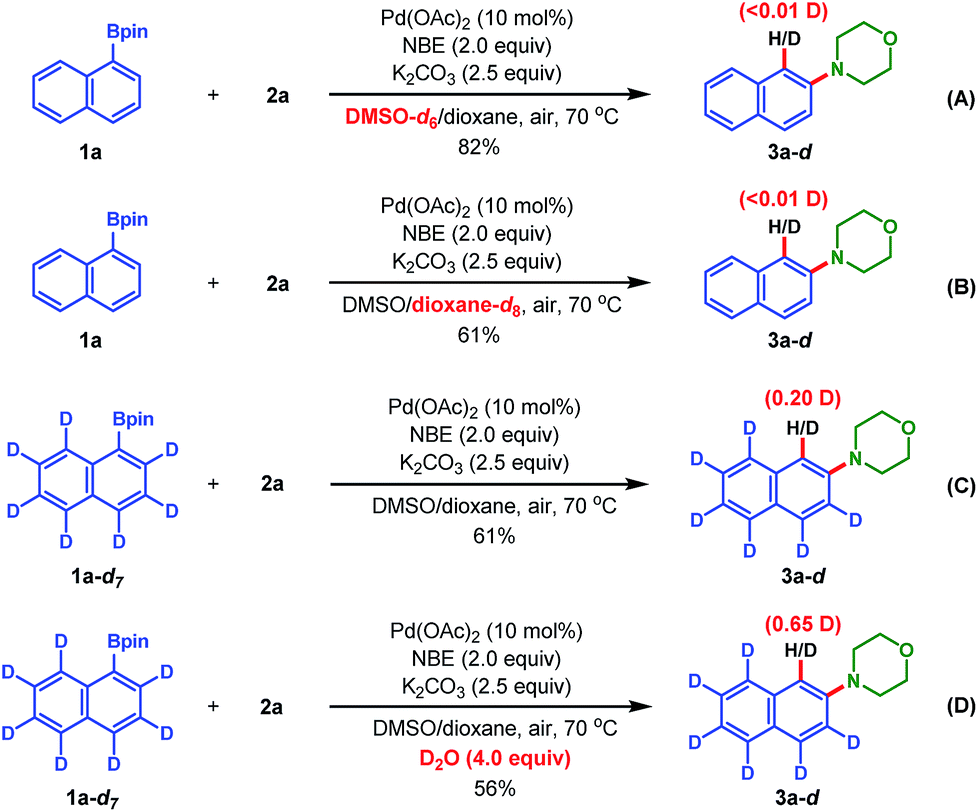
Finally, to probe the proton source of the termination step and gain mechanistic insights into this reaction, deuterium-labeling experiments were performed (Scheme 3). First, an experiment regarding the reaction between 1a and 2a in DMSO-d6 instead of normal DMSO under the standard reaction conditions led to the isolation of product 3a without any deuterium incorporation (eqn (A)); the same deuterium results were obtained when the 1,4-dioxane-d8 instead of 1,4-dioxane was used as the cosolvent (eqn (B)). These results indicated that the termination step didn't involve reductive elimination of [ArPdH] species with hydride from the solvents.22 Then, the fully deuterated 1a-d7 (ref. 23) was used as the substrate (see ESI for its preparation†), and 20% deuterium incorporation at the ipso position of product 3a was found (eqn (C)). Lastly, when 1a-d7 reacted with 2a under the standard reaction conditions with 4.0 equiv. of D2O as the additive, 3a was isolated in a lower yield (56%) but with a significantly increased deuterium incorporation (65%) at the ipso position (eqn (D)). These experimental results indicated that both the protons from the ortho-C–H of pinacol arylborate and the adventitious water in the reaction system are the sources for the Catellani termination step, but not the solvent DMSO. The reduction of the deuterium incorporation ratio in eqn (C) and (D) was probably due to the facile H–D exchange with water in the reaction system.8 Thus, these results support the redox-neutral ipso-protonation mechanism proposed in Scheme 1C.
 | ||
| Scheme 3 Preliminary mechanistic studies. | ||
Conclusions
In summary, we have developed a palladium(II)/norbornene cooperative catalysis enabled redox-neutral ortho-C–H amination of pinacol aryl- or heteroarylborates for the synthesis of structurally diverse anilines. This method is scalable, robust (tolerance of air and moisture), phosphine ligand-free, and compatible with a wide range of functionalities. These practical features make this reaction amenable for industry. It is especially noteworthy that a plethora of synthetically very useful halogenated anilines, which often cannot be prepared via other transition-metal-catalyzed aminations, are readily produced using this method. Particularly, the tolerance of the iodo function enables the orthogonal reactivity between pinacol arylborate and aryl iodides. Preliminary deuterium-labeling studies reveal a redox-neutral ipso-protonation mechanism of this process, which will surely inspire the future development of this research field. Overall, the exceptionally broad scope and reliability of this procedure, together with the wide availability of pinacol arylborates, make this chemistry a valuable addition to the existing methods for aniline synthesis.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grants 21602161, 21871213, and 21801193), the National “1000 Youth Talents Plan”, start-up funding from Wuhan University, and the China Postdoctoral Science Foundation (2016M602339 and 2018M642894) for financial support. We thank Prof. D. Ma (SIOC) for generously donating the oxalamide ligands, Prof. W.-B. Liu (WHU) and Prof. H.-D. Tang (WHU) for sharing their instruments. We thank Ms Q. Gao for checking the experiments, Ms N. Yang and Mr Z. Cui for measuring IR spectra.Notes and references
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS; (b) S. Basoglu, M. Yolal, A. Demirbas, H. Bektas, R. Abbasoglu and N. Demirbas, Turk. J. Chem., 2013, 36, 37–53 Search PubMed; (c) A. Inoue, S. Miki, M. Seto, T. Kikuchi, S. Morita, H. Ueda, Y. Misu and Y. Nakata, Eur. J. Pharmacol., 1997, 321, 105–111 CrossRef; (d) K. Prasad, K. Srinivas, A. Pallavi and K. Mukkanti, Drug Test. Anal., 2018, 10, 212–221 CrossRef; (e) B. Barlaam, R. Ducray, C. Lambert-van der Brempt, P. Ple, C. Bardelle, N. Brooks, T. Coleman, D. Cross, J. G. Kettle and J. Read, Bioorg. Med. Chem. Lett., 2011, 21, 2207–2211 CrossRef CAS.
- For aniline synthesis through copper catalyzed C–N bond formation, see: (a) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed; (b) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC; (c) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS . For aniline synthesis through nickel catalyzed C–N bond formation, see: ; (d) M. Marín, R. J. Rama and M. C. Nicasio, Chem. Rec., 2016, 16, 1819–1832 CrossRef PubMed . For aniline synthesis through cobalt catalyzed C–N bond formation, see: ; (e) M. Usman, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Synthesis, 2017, 49, 1419–1443 CrossRef CAS; (f) Y.-H. Chen, S. Grassl and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 1108–1111 CrossRef CAS . For aniline synthesis through transition metal-free C–N bond formation, see: ; (g) C. Zhu and J. R. Falck, Adv. Synth. Catal., 2014, 356, 2395–2410 CrossRef CAS PubMed.
- (a) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS; (b) A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS; (c) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed; (d) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- For seminal work, see: (a) Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350–18353 CrossRef CAS PubMed . For selected following studies, see: ; (b) Z.-Y. Chen, C.-Q. Ye, H. Zhu, X.-P. Zeng and J.-J. Yuan, Chem.–Eur. J., 2014, 20, 4237–4241 CrossRef CAS PubMed; (c) P.-X. Zhou, Y.-Y. Ye, J.-W. Ma, L. Zheng, Q. Tang, Y.-F. Qiu, B. Song, Z.-H. Qiu, P.-F. Xu and Y.-M. Liang, J. Org. Chem., 2014, 79, 6627–6633 CrossRef CAS PubMed; (d) F. Sun and Z. Gu, Org. Lett., 2015, 17, 2222–2225 CrossRef CAS; (e) H. Shi, D. J. Babinski and T. Ritter, J. Am. Chem. Soc., 2015, 137, 3775–3778 CrossRef CAS PubMed; (f) B. Luo, J.-M. Gao and M. Lautens, Org. Lett., 2016, 18, 4166–4169 CrossRef CAS PubMed; (g) P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed; (h) W. C. Fu, B. Zheng, Q. Zhao, W. T. K. Chan and F. Y. Kwong, Org. Lett., 2017, 19, 4335–4338 CrossRef CAS PubMed; (i) L. Fan, J. Liu, L. Bai, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2017, 56, 14257–14261 CrossRef CAS PubMed.
- Z. Dong, G. Lu, J. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2018, 140, 8551–8562 CrossRef CAS PubMed.
- For seminal work, see: (a) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS . For selected reviews, see: ; (b) M. Catellani, Top. Organomet. Chem., 2005, 14, 21–53 CrossRef CAS; (c) M. Catellani, E. Motti and N. Della Ca', Acc. Chem. Res., 2008, 41, 1512–1522 CrossRef CAS; (d) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef CAS PubMed; (e) N. Della Ca’, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed; (f) Z.-S. Liu, Q. Gao, H.-G. Cheng and Q. Zhou, Chem.–Eur. J., 2018, 24, 15461–15476 CrossRef CAS PubMed; (g) H.-G. Cheng, S. Chen, R. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2019, 58, 5832–5844 CrossRef CAS PubMed; (h) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed; For selected recent studies, see: (i) Y. Yamamoto, T. Murayama, J. Jiang, T. Yasui and M. Shibuya, Chem. Sci., 2018, 9, 1191–1199 RSC; (j) Z.-S. Liu, G. Qian, Q. Gao, P. Wang, H.-G. Cheng, Q. Wei, Q. Liu and Q. Zhou, ACS Catal., 2018, 8, 4783–4788 CrossRef CAS; (k) R. Li and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 1697–1701 CrossRef CAS PubMed; (l) Q. Zhao, W. C. Fu and F. Y. Kwong, Angew. Chem., Int. Ed., 2018, 57, 3381–3385 CrossRef CAS PubMed; (m) H.-G. Cheng, C. Wu, H. Chen, R. Chen, G. Qian, Z. Geng, Q. Wei, Y. Xia, J. Zhang, Y. Zhang and Q. Zhou, Angew. Chem., Int. Ed., 2018, 57, 3444–3448 CrossRef CAS PubMed; (n) L. Bai, J. Liu, W. Hu, K. Li, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2018, 57, 5151–5155 CrossRef CAS PubMed; (o) G. Qian, M. Bai, S. Gao, H. Chen, S. Zhou, H.-G. Cheng, W. Yan and Q. Zhou, Angew. Chem., Int. Ed., 2018, 57, 10980–10984 CrossRef CAS PubMed; (p) C. Wu, H.-G. Cheng, R. Chen, H. Chen, Z.-S. Liu, J. Zhang, Y. Zhang, Y. Zhu, Z. Geng and Q. Zhou, Org. Chem. Front., 2018, 5, 2533–2536 RSC; (q) F. Liu, Z. Dong, J. Wang and G. Dong, Angew. Chem., Int. Ed., 2019, 58, 2144–2148 CrossRef CAS; (r) Q. Gao, Z.-S. Liu, Y. Hua, L. Li, H.-G. Cheng, H. Cong and Q. Zhou, Chem. Commun., 2019, 55, 8816–8819 RSC.
- (a) G. Shi, C. Shao, X. Ma, Y. Gu and Y. Zhang, ACS Catal., 2018, 8, 3775–3779 CrossRef CAS; (b) S. Chen, Z.-S. Liu, T. Yang, Y. Hua, Z. Zhou, H.-G. Cheng and Q. Zhou, Angew. Chem., Int. Ed., 2018, 57, 7161–7165 CrossRef CAS PubMed; (c) P. Wang, S. Chen, Z. Zhou, H.-G. Cheng and Q. Zhou, Org. Lett., 2019, 21, 3323–3327 CrossRef CAS PubMed.
- Very recently, the Dong group reported an elegant ortho acylation of aryl boroxines via palladium/norbornene cooperative catalysis, see: R. Li, F. Liu and G. Dong, Chem, 2019, 5, 929–939 CAS . In their research, pinacol arylborates are unreactive substrates. They also reported the ortho amination of aryl boroxines (six examples), which required triphenyl phosphite as the ligand, and CsI and benzoquinone (BQ) as the additives.
- For palladium(IV)-involved aminations, see: (a) J. A. Jordan-Hore, C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS PubMed; (b) T. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806–10807 CrossRef CAS PubMed . However, another mechanism involving direct nucleophilic substitution of the amination reagent 2 by arylpalladium(II) species (II) can't be excluded, see related references: ; (c) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680–5681 CrossRef CAS; (d) T. J. Barker and E. R. Jarvo, J. Am. Chem. Soc., 2009, 131, 15598–15599 CrossRef CAS; (e) G. Cheng, T.-J. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 10950–10953 CrossRef CAS PubMed.
- For selected Catellani-type studies involving redox-neutral protonation as the termination step, see: (a) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990–12993 CrossRef CAS PubMed; (b) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2013, 134, 14563–14572 CrossRef PubMed; (c) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed; (d) Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed; (e) H. Zhang, H.-Y. Wang, Y. Luo, C. Chen, Y. Cao, P. Chen, Y.-L. Guo, Y. Lan and G. Liu, ACS Catal., 2018, 8, 2173–2180 CrossRef CAS . For a review about protodepalladation, see: ; (f) M. L. O'Duill and K. M. Engle, Synthesis, 2018, 50, 4699–4714 CrossRef PubMed.
- The oxidation state of the palladium complexes may be kept at two throughout the catalytic cycle. For selected examples, see: Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097–6100 CrossRef CAS PubMed ; and ref. 9e.
- For mechanistic studies on Catellani-type reactions, see: (a) M. Catellani and B. E. Mann, J. Organomet. Chem., 1990, 390, 251–255 CrossRef CAS; (b) G. Bocelli, M. Catellani and S. Ghelli, J. Organomet. Chem., 1993, 458, C12–C15 CrossRef CAS; (c) B. Martín-Matute, C. Mateo, D. J. Cárdenas and A. M. Echavarren, Chem.–Eur. J., 2001, 7, 2341–2348 CrossRef; (d) D. J. Cárdenas, B. MartínMatute and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 5033–5040 CrossRef PubMed; (e) G. Maestri, E. Motti, N. Della Ca’, M. Malacria, E. Derat and M. Catellani, J. Am. Chem. Soc., 2011, 133, 8574–8585 CrossRef CAS; (f) D. I. Chai, P. Thansandote and M. Lautens, Chem.–Eur. J., 2011, 17, 8175–8188 CrossRef CAS PubMed.
- (a) S. N. Mlynarski, A. S. Karns and J. P. Morken, J. Am. Chem. Soc., 2013, 134, 16449–16451 CrossRef PubMed; (b) C. Zhu, G. Li, D. H. Ess, J. R. Falck and L. Kürti, J. Am. Chem. Soc., 2013, 134, 18253–18256 CrossRef PubMed ; and ref. 2g.
- D. V. Partyka, Chem. Rev., 2011, 111, 1529–1595 CrossRef CAS.
- M. Kotora and T. Takahashi, in Handbook of Organopalladium Chemistry for Organic Synthesis, Vol. 1, ed. E.-i. Negishi, Wiley, New York, 2002, pp. 973–993 Search PubMed.
- J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- C. Ye, H. Zhu and Z. Chen, J. Org. Chem., 2014, 79, 8900–8905 CrossRef CAS PubMed.
- (a) C. Bardelle, B. Barlaam, N. Brooks, T. Coleman, D. Cross, R. Ducray, I. Green, C. Lambert-van der Brempt, A. Olivier and J. Read, Bioorg. Med. Chem. Lett., 2010, 20, 6242–6245 CrossRef CAS PubMed; (b) K. Ebert, J. Wiemer, J. Caballero, M. Köckerling, J. Steinbach, J. Pietzsch and C. Mamat, Bioorg. Med. Chem., 2015, 23, 6025–6035 CrossRef CAS PubMed . And ref. 1e.
- (a) M. Fan, W. Zhou, Y. Jiang and D. Ma, Org. Lett., 2015, 17, 5934–5937 CrossRef CAS PubMed; (b) J. Gao, S. Bhunia, K. Wang, L. Gan, S. Xia and D. Ma, Org. Lett., 2017, 19, 2809–2812 CrossRef CAS PubMed.
- For the general method for the synthesis of 8, see: ref. 19b.
- For a precedent using ethers (e.g. DME) as the hydride source, see: A. Martins, D. A. Candito and M. Lautens, Org. Lett., 2010, 12, 5186–5188 CrossRef CAS PubMed.
- F. Mo, J. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 2028–2032 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02759a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2019 |