
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDispersing transition metal vacancies in layered double hydroxides by ionic reductive complexation extraction for efficient water oxidation†
Yang-Shan
Xie
a,
Zheng
Wang
a,
Min
Ju
a,
Xia
Long
*a and
Shihe
Yang
 *ab
*ab
aGuangdong Provincial Key Lab of Nano-Micro Material Research, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: xialong@pku.edu.cn; chsyang@pku.edu.cn
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 22nd July 2019
Abstract
Creating atomic defects in nanomaterials is an effective approach to promote the catalytic performance of a catalyst, but the defective catalysts are often prone to mechanical collapse if not properly synthesized. The uncontrollably formed defects also make it difficult to systematically investigate their effects on the catalytic performance. Herein, we report an efficient method of ionic reductive complexation extraction (IRCE) to fabricate atomic vacancies in a transition metal based nanomaterial without damaging its nanostructure, turning the otherwise catalytically inactive material to an advanced catalyst towards water oxidation in alkaline electrolyte. Here nickel based layered double hydroxide mixed with Cu(II) is used to demonstrate the concept. With a tunable content and uniform dispersion of Cu(II) on the brucite layer of the LDH, a suitable complexing agent could specifically combine with and remove the target Cu(II), thereby creating the desired vacancies. The resulting vacancy rich TM LDH is found to be an excellent OER electrocatalyst with a low overpotential and small Tafel slope, due to the purposely modulated geometric and electronic structures of the active sites, and the greatly decreased charge transfer resistance.
Introduction
Hydrogen production via water splitting is one of the most promising ways to tap renewable green energy and hence tackle energy crisis, global warming and environmental pollution problems. To accelerate the reaction kinetics and lower the energy barrier of water splitting, efficient electrocatalysts are greatly needed. The oxygen evolution reaction (OER) that involves a four-proton-coupled electron transfer process and one oxygen–oxygen double bond formation is regarded as the rate-determining half reaction of water splitting, highlighting the importance of OER catalysts, especially the non-noble metal ones.1 Among noble-metal free OER catalysts, transition metal based layered double hydroxides (TM LDHs),2–4 especially nickel-based LDHs such as NiFe LDH,5–7 NiV LDH,8,9 NiCr LDH,10 NiCoFe LDH,11,12etc., exhibit the most efficient performance toward water oxidation.3,13,14 However, little research has been done on NiCu LDH for the OER, due to the low catalytic activity of Cu ions and the weak synergistic effects between Ni and Cu.It has been recently reported that creating defects on the surface of catalysts could be an effective way to enhance the performance of catalysts, due to the increased number of catalytically active sites and modulated electronic properties of catalysts. As an example, the exfoliation process was used to reduce the thickness of a layered catalyst and hence edge defects were created.15 However, structural collapse also occurred upon defect formation due to mechanical deformation during the exfoliation process, leading to the formation of catalyst nanoparticles with deteriorated catalytic performance.16–18 More recently, plasma technology has been applied to fabricate defects by creating nano- or micro-pores. The greatly enlarged active sites around the defects greatly promoted the catalytic performance.19–23 Although in this case the structural collapse could be avoided by precisely controlling the plasma treatment condition, there is no control on the nature of the as-prepared defects, which contain both anion and cation vacancies with a distribution of defect sizes. Annealing metal oxides in a reductive atmosphere at high temperatures is an effective way to fabricate oxygen-deficient catalysts with copious oxygen-vacancies.24,25 Again, however, the nature and spatial distribution of the vacancies could not be pre-determined and controlled.
Herein, we report a mild and facile method to atomically disperse cation vacancies in NiCu LDH by etching the target Cu(II) ions via a preferential complexation process for the first time. Such an ionic reductive complexation extraction (IRCE) method judiciously combines the unique characteristics of tunable and uniformly distributed transition metal ions in the brucite layer of the TM LDH, and the variable difference in the equilibrium constant (K) of the transition metal ions with a suitably chosen complexing agent. The as-prepared NiCu LDH with atomically dispersed vacancies (SAV-NiCux LDH) exhibits advanced electrocatalytic performance toward water splitting, which has been verified to result from the modulated electronic structure of transition metal ions, the greatly decreased charge transfer resistance, and the adjusted surface properties that benefit the adsorption/desorption of intermediates during the OER process.
Results and discussion
NiCu LDH with low OER activity that is close to that of Ni(OH)2 was chosen as the model material. SCN− was used as the extractant because of the large lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K of Cu(I)-SCN (12.11) that is more than 6 times larger than that of Cu(II)-SCN (1.9) and Ni(II)-SCN (1.18). To achieve selective extraction of Cu from the NiCu LDH, Na2SO3 was added to the complexation solution to in situ reduce the Cu(II) to Cu(I) but keep the Ni(II) ions unchanged. The resulting Cu(I) was then further complexed with –SCN and removed from the LDH brucite layer via the IRCE process, as illustrated in Scheme 1.
K of Cu(I)-SCN (12.11) that is more than 6 times larger than that of Cu(II)-SCN (1.9) and Ni(II)-SCN (1.18). To achieve selective extraction of Cu from the NiCu LDH, Na2SO3 was added to the complexation solution to in situ reduce the Cu(II) to Cu(I) but keep the Ni(II) ions unchanged. The resulting Cu(I) was then further complexed with –SCN and removed from the LDH brucite layer via the IRCE process, as illustrated in Scheme 1.
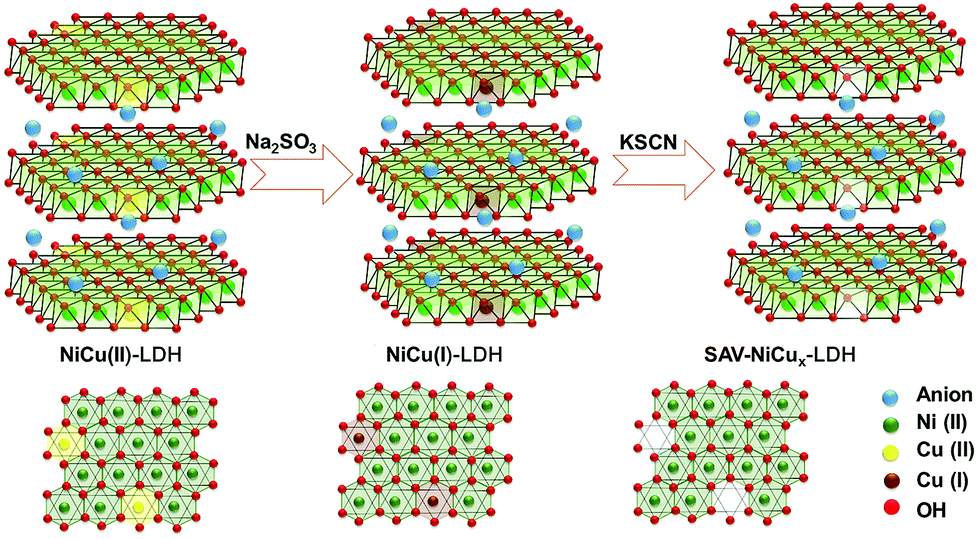 | ||
| Scheme 1 Schematic illustration of the IRCE process for the formation of NiCu LDH with atomic vacancies (SAV-NiCux LDH). | ||
From the scanning electron microscopy (SEM) images, we can see that SAV-NiCux LDH prepared by complexation treatment for 6 days showed nearly the same nanosheet structure (Fig. 1A and B) as that of NiCu LDH (Fig. S1†). Transition metal ions in SAV-NiCux LDH also exhibited a uniform dispersion (Fig. 1C), but a decreased Cu concentration when compared to that of the original LDH (Fig. S2 and S3†). Moreover, the XRD patterns (Fig. 1D) indicated that the SAV-NiCux LDH exhibited almost the same atomic structure as the NiCu LDH (Fig. 1D), consistent with the electron diffraction results (Fig. 1E and S4†). Further, from the high resolution transmission electron microscopy (HRTEM) image, it is clear that the SAV-NiCux LDH exhibited a continuous lattice fringe (Fig. 1F) with a fringe spacing of 0.27 nm, which could be well ascribed to the (110) plane of the LDH. Moreover, atomic vacancies denoted by red circles and arrows in Fig. 1F and S5† could also be found, suggesting successful fabrication of single atom vacancies without damaging the framework of the LDH by using the mild IRCE process.
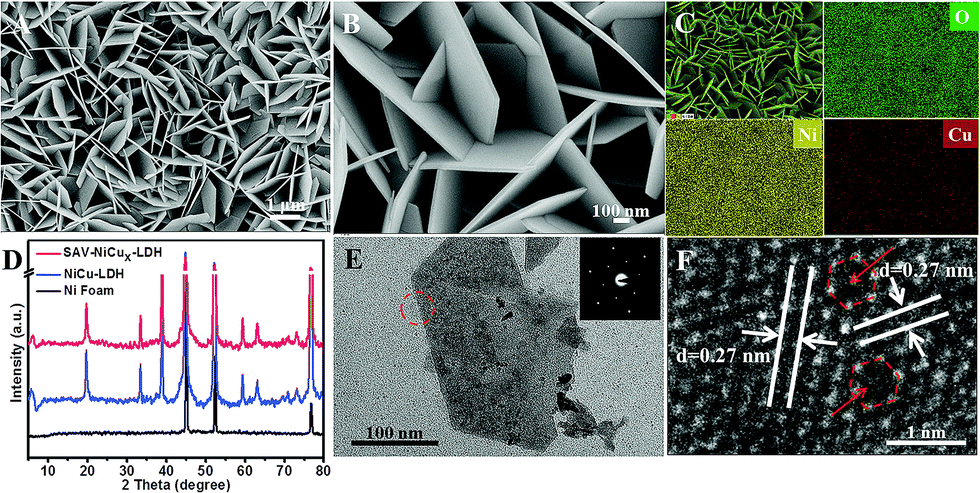 | ||
| Fig. 1 Morphology and structure characterization of the NiCu LDH with atomic cation vacancies (SAV-NiCux LDH). (A and B) SEM images, (C) elemental mapping images of O, Ni and Cu of the SAV-NiCux LDH, (D) XRD patterns, (E) TEM image and ED pattern (inset), and (F) HRTEM image of the SAV-NiCux LDH. | ||
From the X-ray photoelectron spectroscopy (XPS) results (Table S1†), the concentration of Cu in the original NiCu LDH was determined to be 1.84 at%, which was slightly reduced to 1.31 at% after the Na2SO3 treatment, probably due to the reduction of metal ions that somewhat destabilized the brucite layers and hence led to the dissolution of the Cu ions. However, when the complexation agents of KSCN and Na2SO3 were both introduced into the NiCu LDH suspension, the Cu concentration was greatly decreased to 0.01 at%. Moreover, from the inductively coupled plasma (ICP) results, the leakage of Cu2+ in the complexation solution was determined to be 1.21 mg L−1, proving the extraction effect of the complexing agent and thus the effectiveness of the IRCE method. From the high resolution XPS spectra in the Ni 2p region (Fig. S6†), it is clear that the binding energy (BE) peaks of NiCu(I) LDH and SAV-NiCux LDH were positively shifted when compared with the pristine NiCu LDH, indicating the increased oxidation states of nickel ions in these two samples. Moreover, from the high resolution XPS spectra of the Cu 2p spin-orbital, it can be observed that the original NiCu LDH (Fig. S7,† blue curve) showed a split Cu 2p3/2 peak, indicating the coexistence of Cu(II) (∼934.8 eV)26,27 and Cu(I) (∼933 eV).28–30 For the NiCu(I) LDH (Fig. S7,† orange curve), it is not surprising to see that the peak of Cu 2p3/2 appeared at ∼933 eV that could be ascribed to Cu(I), while SAV-NiCux LDH showed a similar averaged peak position to that of original NiCu LDH, but with a much lower peak intensity (Fig. S7,† red curve) due to the much decreased Cu concentration after the IRCE process. It is worth meantioning that the oxygen vacancy peaks were negligible for both NiCu(I) LDH and SAV-NiCux LDH from XPS spectra (Fig. S8†), and no apparent signals at g = 2.004 (ref. 31 and 32) were detected from the electron paramagnetic resonance (EPR) spectra (Fig. S9†), indicating no detectable oxygen vacancy formed and the framework of LDH was well retained after the reduction and complexation treatments. More details could be derived from the deconvoluted spectra (Fig. 2A, B and S10†). The Ni(III) and Ni(II) peaks were fitted to both NiCu LDH and SAV-NiCux LDH at 855.7 eV and 855.1 eV, respectively. The calculated ratio of Ni(III)/Ni(II) of SAV-NiCux LDH is 4.32–2.03 (Table S2†), significantly larger than that of NiCu(I) LDH (3.87–2.23) and NiCu LDH (2.43–1.75) (Table S3†), further confirming the increased oxidation state of Ni ions with the introduction of cation vacancies. It is worth noting that Ni with the electronic structure of t62ge1g was at the peak position of the volcano shaped dependence of OER activity with the occupancy of the 3d electrons of transition metal ions.33–36 Moreover, Ni has been reported to be the direct active species for the OER.15,24,25,37,38 Therefore, it is reasonable to propose that the uniformly dispersed atomic vacancies effectively modulated the electronic structure and coordination number of the surface active atoms, and hence would idealize the adsorption/desorption characteristics of the catalysts towards the OER.
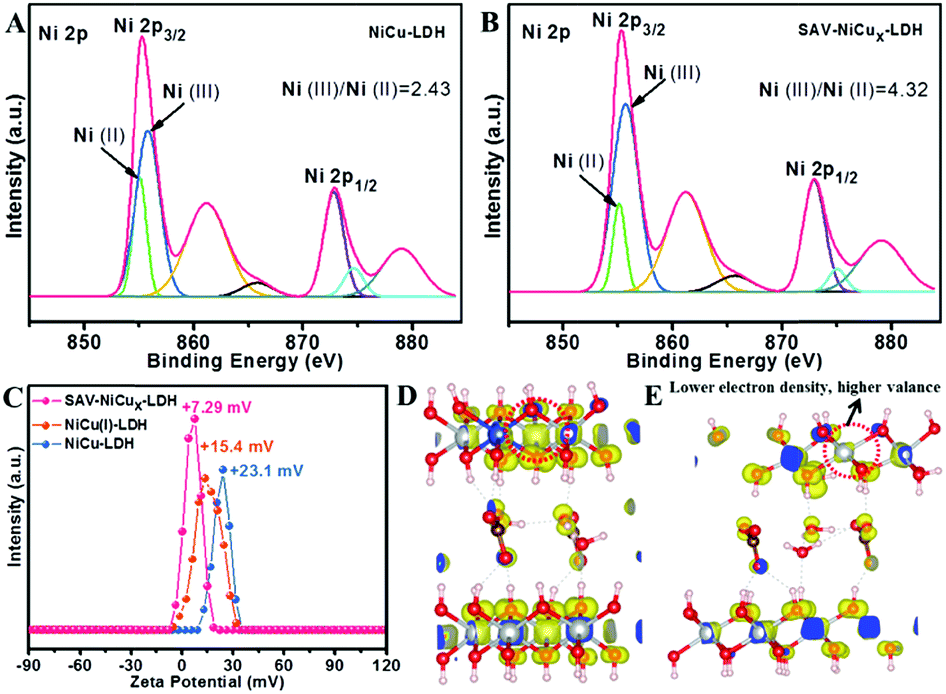 | ||
| Fig. 2 Electronic states and surface charge characterization. Deconvoluted XPS spectra of Ni 2p in (A) NiCu LDH and (B) SAV-NiCux LDH, (C) zeta potential tests, and partial charge density of (D) NiCu LDH and (E) SAV-NiCux LDH. | ||
Zeta potential was applied to investigate the charge characteristics of the LDH (Fig. 2C). Though the NiCu LDH, NiCu(I) LDH and SAV-NiCux LDH exhibited positive zeta potentials, the zeta potential of SAV-NiCux LDH (7.29 mV) is much smaller than that of the pristine NiCu LDH (23.1 mV) and NiCu(I) LDH (15.4 mV), respectively. This is consistent with the higher charge state of the Ni3+ in SAV-NiCux LDH, which binds more strongly with OH− and thus results in a lower zeta potential. Extensive density functional theory (DFT) calculations were carried out to verify this result. Comparing the partial charge densities of NiCu LDH (Fig. 2D) and SAV-NiCux LDH (Fig. 2E), one can see that the electron density on the selected nickel ion decreased for SAV-NiCux LDH vs. NiCu LDH, confirming that it is in a higher valence state that is in accordance with the XPS results.
The electrochemical performance of the as-fabricated catalysts was evaluated by using a typical three-electrode system in 1 M KOH if not otherwise indicated. Ag/AgCl, Pt wire and the catalysts coated with nickel foam were used as the reference, counter, and working electrodes, respectively. Besides, SAV-NiCux LDH, original NiCu LDH, NiCu(I) LDH (Fig. S11†) and Ni(OH)2 (Fig. S12†) with similar nanostructures were also subjected to the performance measurements. From the linear sweep voltammetry (LSV) curves, one can see that Ni foam (Fig. 3A) showed little OER activity with negligible current density (j) even at a high potential of 1.6 V (vs. RHE), while at the same potential, Ni(OH)2 and NiCu LDH showed a small j of 13 and 17 mA cm−2 (Fig. 3A), respectively, consistent with the reported values.39–41 Notably, the onset potential of the OER was greatly decreased to 252 mV on NiCu(I) LDH, and further lowered to 240 mV on SAV-NiCux LDH (Fig. 3A), indicating the critical roles of atomic vacancies in improving the OER activity. More interestingly, at a potential of 1.6 V (vs. RHE), the j of the OER catalyzed by SAV-NiCux LDH was 143 mA cm−2, 8.4 times higher than that of original NiCu LDH (17 mA cm−2). The overpotential at 10 mA cm−2, which is the approximate j for a 10% efficiency solar-to-fuel conversion device, was 290 mV, 289 mV, 343 mV, 347 mV, and 389 mV for SAV-NiCux LDH, NiCu(I) LDH, NiCu LDH, Ni(OH)2 and Ni foam, respectively (Fig. S13†) with the prepared SAV-NiCux LDH and NiCu(I) LDH showing the most smaller overpotentials that were even smaller than those of noble-metal catalysts of IrO2 (350 mV)42–45 and RuO2 (365 mV)46,47 (Fig. S14†). Moreover, at high current densities such as 100 mA cm−2 and 200 mA cm−2, the overpotentials were 355 mV and 385 mV for SAV-NiCux LDH, which were 17 mV and 26 mV smaller than that of NiCu(I) LDH, further demonstrating the effectiveness of the atomic vacancy engineering for improving the catalytic performance of nanocatalysts.
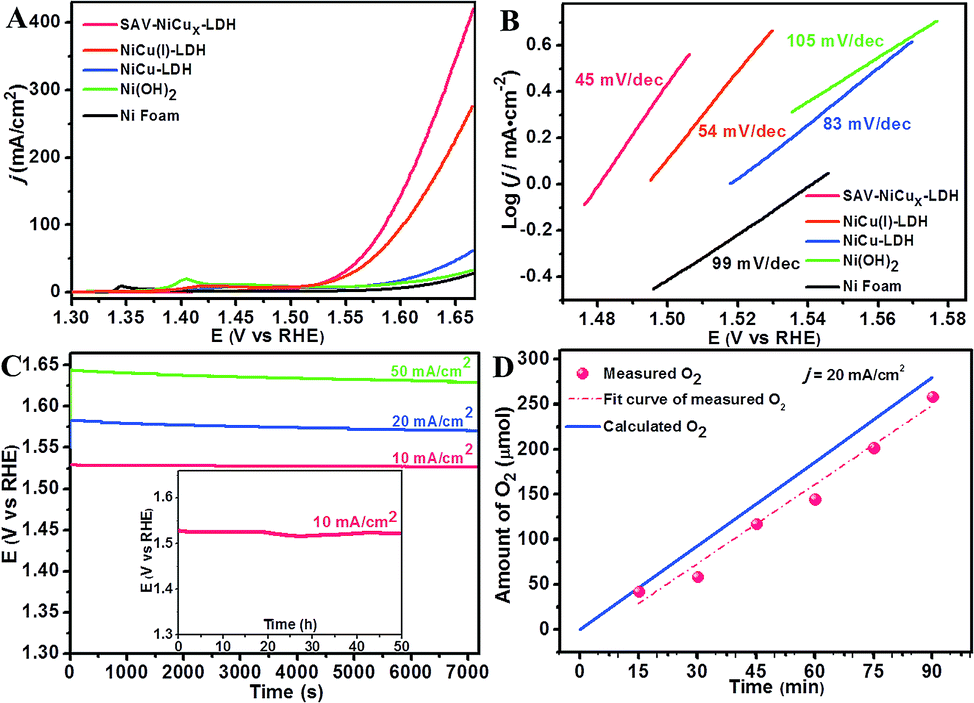 | ||
| Fig. 3 Electrochemical performance of the as-prepared catalysts towards the OER in alkaline solutions. (A) Linear sweep voltammetry (LSV) curves, (B) Tafel plots, (C) long-term stability test of SAV-NiCux LDH, and (D) theoretical and experimental tested O2 production on SAV-NiCux LDH. | ||
The Tafel slope describes the influence of potential/overpotential on the steady-state current density and rather reveals the mechanism and kinetics of the OER. From Fig. 3B, it is clear that SAV-NiCux LDH shows the smallest Tafel slope of 45 mV dec−1, when compared with that of NiCu(I) LDH, NiCu LDH, Ni(OH)2, and Ni foam (Fig. 3B) and also outperformed the state-of-the-art OER catalysts of IrO2 and RuO2 (Fig. S15†). In fact, the Tafel slope of SAV-NiCux LDH is also lower than that of the previously reported Cu-based OER catalysts48,49 and comparable to that of the most efficient transition metal based OER catalysts.6,11,50,51 Fig. S13† shows the comparison of both the overpotentials at j = 10 mA cm−2 and the Tafel slopes of the as-prepared catalysts. In all aspects, the SAV-NiCux LDH is a champion OER catalyst, exhibiting the most advanced OER performance with the smallest overpotential and Tafel slope. Taken together, the atomic vacancies are much more beneficial for the OER than the common defects widely used previously such as anionic vacancies.
Stability is an important factor to evaluate the potential of practical applications of a catalyst. Herein, the operational stability of the advanced OER catalyst of SAV-NiCux LDH was investigated by chronopotentiometry (CP). From Fig. 3C, it is obvious that the potentials for maintaining high j values of 20 mA cm−2 and 50 mA cm−2 were negligibly changed (<1%) over 7200 s and even for nearly 50 h at 10 mA cm−2. Moreover, both the micro-morphology (Fig. S16†) and atomic structure (Fig. S17†) of SAV-NiCux LDH after the stability tests were unchanged, confirming the long-term stability of SAV-NiCux LDH for OER catalysis.
From the EDX and XPS results, we know that there was still a little amount of Cu ions in SAV-NiCux LDH; therefore, SAV-NiCux LDH obtained after different complexation times was evaluated to investigate the effect of number of atomic vacancies on OER performance. From Fig. S18,† we can see that with the increase of complexation time, the overpotential of the OER decreased during the first 6 days, reaching the lowest onset potential of 240 mV (Fig. 3A and S18†), while it increased to 260 mV when the complexation time was prolonged to 8 days. The Tafel slope experiments (Fig. S19†) exhibited the same results with SAV-NiCux LDH (6 d) showing the smallest value (45 mV dec−1), hence the best OER performance. Therefore, it seems that the number of atomic vacancies was not proportional to the OER activity of SAV-NiCux LDH. To confirm this assumption, the electrochemical surface area (ECSA) was measured by using simple cyclic voltammetry (CV) methods52,53 (Fig. S20†), since the number of vacancies is related to the number of exposed catalytic active sites. By calculating the slope of the plotted curves of capacitive currents (Δj) as a function of scan rates, the electrochemical double layer capacitance (Cdl) was determined to be 13.25, 13.20, 12.29, 13.21, 7.80, 11.85, and 2.12 μF for SAV-NiCux LDH (3 d), SAV-NiCux LDH (6 d), SAV-NiCux LDH (8 d), NiCu(I) LDH, NiCu LDH, Ni(OH)2 and Ni foam, respectively (Fig. S21 and Table S4†), and all of the values were close to each other, suggesting a similar ECSA. This result confirmed the assumption that the active sites weren't the major reason for the improved OER performance of SAV-NiCux LDH, further suggesting that the mild IRCE process could precisely create the atomic vacancies on the surface of the LDH without damaging its nanostructure.
Conductivity is another critical factor that affects the performance of catalysts. From the electrochemical impedance spectroscopy results (EIS, Fig. S22†), it is clear that the diameter of Nyquist semicircles of SAV-NiCux LDH, NiCu(I) LDH, NiCu LDH, Ni(OH)2 and Ni foam decreased in sequence, with SAV-NiCux LDH showing the smallest one, suggesting the smallest charge transfer resistance. Again, the EIS data of SAV-NiCux LDH (3 d) and SAV-NiCux LDH (8 d) were also collected and compared with those of SAV-NiCux LDH (6 d) (Fig. S23†); again, SAV-NiCux LDH (6 d) exhibited the smallest diameter Nyquist semicircles, further confirming that the high conductivity of SAV-NiCux LDH came from the suitable concentration of atomic vacancies with undamaged nanostructures of the LDH. Therefore, the advanced OER performance of SAV-NiCux LDH was proposed to result from the increased concentration of Ni that enhanced the intrinsic catalytic activity of the catalyst, as well as the greatly reduced charge transfer resistance due to the retained nanosheet microstructure with atomic vacancies. Meanwhile, the faradaic efficiency (FE) of the OER on SAV-NiCux LDH was tested to be ∼95% (Fig. 3D), suggesting the high efficiency of energy conversion capability of SAV-NiCux LDH.
To better understand the OER process on SAV-NiCux LDH, the standard free energy of the OER was calculated (Fig. 4 and Table S5†). Evidently, the rate determining step was the formation of OOH* from O* for both NiCu LDH and SAV-NiCux LDH, with the overpotential of 0.73 and 0.53 V, respectively, suggesting an improved catalytic activity of SAV-NiCux LDH towards the OER, in line with the experimental results. The decreased overpotential of SAV-NiCux LDH is ascribed to the stabilization of OOH* and O* with respect to OH* (Fig. 4). Aiming at a deeper understanding of stabilization of OOH*, we derived the relationship between ΔG1 + ΔG2 (i.e. the adsorption free energy of OOH* refers to OH*) and the binding energy of OH*, Eb(OH), in eqn (1) (see ESI† for the derivation of eqn (1)). From eqn (1), we can learn that ΔG1 + ΔG2 would be smaller if Eads(OH) became less negative. Herein, the coordination number of OH* changed from three to two for SAV-NiCux LDH since copper was selectively removed from the lattice; therefore, it is reasonable for us to expect that the stabilization of OOH* referred to OH* for SAV-NiCux LDH can be due to the destabilization of OH*. Besides, the adjacent hydroxyl in SAV-NiCux LDH can also partly stabilize OOH* by forming an additional hydrogen bond due to its more flexible structure, as illustrated in Fig. S24.† A weaker binding energy of OH* can lead to stabilization of OOH* with respect to OH*; however, this would cause destabilization of O* (ΔG1) according to eqn (2). In addition, careful examination of structures of O* for NiCu LDH and SAV-NiCux LDH was carried out. From Fig. S24,† it can be observed that there is a larger structural change from OH* to O* for SAV-NiCux LDH that contributes to the stabilization of O*, benefiting from its more flexible structure. Therefore, we conclude that the decreased overpotential for SAV-NiCux LDH is due to the comparatively weaker binding energy of OH* than NiCu LDH, as well as the better stabilization of O* and OOH* with respect to OH* owing to its more flexible structure to accommodate the structural changes.
| ΔG2 + ΔG1 = (ξ − 1)Eads(OH) + C1(0 < ξ < 1, C1 > 0)C1 = E(OOH) − E(OH) + ZPE(OOH*) − ZPE(OH*) − TΔS(OOH*) + TΔS(OH*) − G(H2O) + G(H2) | (1) |
| ΔG1 = (λ − 1)Eads(OH) + C2 (1 < λ, C2 > 0)C2 = E(O) − E(OH) + ZPE(O*) − ZPE(OH*) − TΔS(O*) + TΔS(OH*) + 0.5G(H2) | (2) |
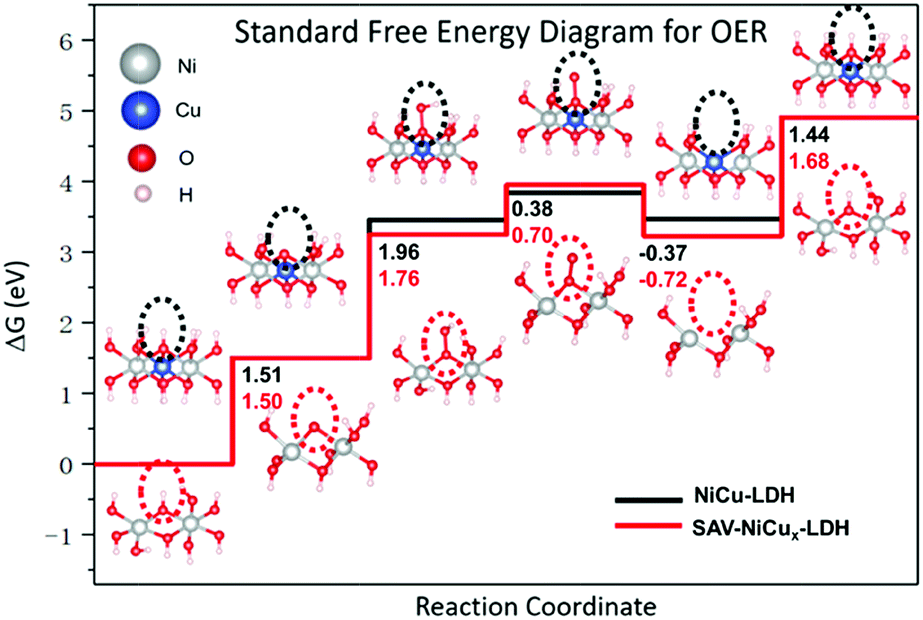 | ||
| Fig. 4 Standard free energy diagrams for the OER on NiCu LDH and SAV-NiCux LDH (gray balls represent Ni, blue balls represent Cu, red balls represent O and white balls represent H). | ||
Conclusions
In conclusion, we have developed a mild and facile IRCE method for creating atomic vacancies in NiCu LDH, transforming the otherwise catalytically inactive material into an advanced electrocatalyst towards water oxidation. Importantly, the controllability allows the atomic vacancies to be created without damaging the nanostructure of the TM LDH. This method takes advantage of the tunable chemical composition of the TM LDH brucite layer and the difference in equilibrium constants of transition metal ions with a judiciously chosen complexation agent as the ionic extractant. The overpotential at 100 mA cm−2 and Tafel slope of the synthesized SAV-NiCux LDH were evaluated to be as low as 355 mV and 45 mV dec−1, respectively, making it the best NiCu-based catalyst reported so far and comparable to the most efficient noble metal free OER catalysts. The greatly improved catalytic performance has been shown to result from the modulated electronic structure and adjusted surface adsorption/desorption characteristics, as well as the decreased charge transfer resistance of the catalysts. This work provides a promising direction for engineering atomic vacancies on demand in low dimensional nanomaterials to impart and to improve catalytic performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Shenzhen Peacock Plan (KQTD2016053015544057), the Science, Technology and Innovation Commission of Shenzhen Municipality – the Basic Research Program (JCYJ20180302153417057), the Nanshan Pilot Plan (LHTD20170001), the National Natural Science Foundation of China (21703003), China Postdoctoral Science Foundation Grant (No. 2018M631240), the Guangdong Science and Technology Program (2017B030314002) and the NSFC/Hong Kong RGC Research Scheme (N_HKUST610/14). The calculations were performed on the Tianhe-2 supercomputer system in Guangzhou.Notes and references
- Z. Wang, X. Long and S. H. Yang, ACS Omega, 2018, 3, 16529–16541 CrossRef CAS.
- Y. Y. Wang, D. F. Yan, S. El Hankari, Y. Q. Zou and S. Y. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef PubMed.
- X. Long, Z. L. Wang, S. Xiao, Y. M. An and S. H. Yang, Mater. Today, 2016, 19, 213–226 CrossRef CAS.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, J. Mater. Chem. A, 2019, 7, 5069–5089 RSC.
- M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- X. Y. Gao, X. Long, H. Yu, X. Y. Pan and Z. G. Yi, J. Electrochem. Soc., 2017, 164, H307–H310 CrossRef CAS.
- X. Long, J. K. Li, S. Xiao, K. Y. Yan, Z. L. Wang, H. N. Chen and S. H. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef CAS PubMed.
- K. Fan, H. Chen, Y. F. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. S. Li, Y. Luo and L. C. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- G. A. Caravaggio, C. Detellier and Z. Wronski, J. Mater. Chem., 2001, 11, 912–921 RSC.
- W. Ye, X. Y. Fang, X. B. Chen and D. P. Yan, Nanoscale, 2018, 10, 19484–19491 RSC.
- X. Long, S. Xiao, Z. L. Wang, X. L. Zheng and S. H. Yang, Chem. Commun., 2015, 51, 1120–1123 RSC.
- X. Y. Gao, X. Y. Pan, X. Long and Z. G. Yi, J. Electrochem. Soc., 2017, 164, H755–H759 CrossRef CAS.
- R. Patel, J. T. Park, M. Patel, J. K. Dash, E. B. Gowd, R. Karpoormath, A. Mishra, J. Kwak and J. H. Kim, J. Mater. Chem. A, 2018, 6, 12–29 RSC.
- M. Gong, D. Y. Wang, C. C. Chen, B. J. Hwang and H. J. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS.
- Y. W. Liu, H. Cheng, M. J. Lyu, S. J. Fan, Q. H. Liu, W. S. Zhang, Y. D. Zhi, C. M. Wang, C. Xiao, S. Q. Wei, B. J. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- J. F. Xie, H. Zhang, S. Li, R. X. Wang, X. Sun, M. Zhou, J. F. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- J. F. Xie, J. J. Zhang, S. Li, F. Grote, X. D. Zhang, H. Zhang, R. X. Wang, Y. Lei, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- Z. H. Li, H. H. Duan, M. F. Shao, J. B. Li, D. O'Hare, M. Wei and Z. L. Wang, Chem, 2018, 4, 2168–2179 CAS.
- L. Tao, C. Y. Lin, S. Dou, S. Feng, D. W. Chen, D. D. Liu, J. Huo, Z. H. Xia and S. Y. Wang, Nano Energy, 2017, 41, 417–425 CrossRef CAS.
- L. Tao, X. D. Duan, C. Wang, X. F. Duan and S. Y. Wang, Chem. Commun., 2015, 51, 7470–7473 RSC.
- Y. Y. Wang, Y. Q. Zhang, Z. J. Liu, C. Xie, S. Feng, D. D. Liu, M. F. Shao and S. Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
- L. Xu, Q. Q. Jiang, Z. H. Xiao, X. Y. Li, J. Huo, S. Y. Wang and L. M. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- Y. Y. Wang, C. Xie, Z. Y. Zhang, D. D. Liu, R. Chen and S. Y. Wang, Adv. Funct. Mater., 2018, 28, 1703363 CrossRef.
- J. Kim, X. Yin, K. C. Tsao, S. H. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed.
- F. C. Lei, Y. F. Sun, K. T. Liu, S. Gao, L. Liang, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- Y. Zheng, J. Qiao, J. Yuan, J. Shen, A.-j. Wang, P. Gong, X. Weng and L. Niu, Electrochim. Acta, 2018, 282, 735–742 CrossRef CAS.
- P. Zhang, L. Li, D. Nordlund, H. Chen, L. Fan, B. Zhang, X. Sheng, Q. Daniel and L. Sun, Nat. Commun., 2018, 9, 381 CrossRef PubMed.
- Z. He, H. Lin, P. He and Y. Yuan, J. Catal., 2011, 277, 54–63 CrossRef CAS.
- Z. Gao, C. Li, G. Fan, L. Yang and F. Li, Appl. Catal., B, 2018, 226, 523–533 CrossRef CAS.
- L. Chen, P. Guo, M. Qiao, S. Yan, H. Li, W. Shen, H. Xu and K. Fan, J. Catal., 2008, 257, 172–180 CrossRef CAS.
- V. Ischenko, S. Polarz, D. Grote, V. Stavarache, K. Fink and M. Driess, Adv. Funct. Mater., 2005, 15, 1945–1954 CrossRef CAS.
- S. Polarz, J. Strunk, V. Ischenko, M. W. E. van den Berg, O. Hinrichsen, M. Muhler and M. Driess, Angew. Chem., Int. Ed., 2006, 45, 2965–2969 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 647 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. G. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- D. A. Tompsett, S. C. Parker and M. S. Islam, J. Am. Chem. Soc., 2014, 136, 1418–1426 CrossRef CAS PubMed.
- X. K. Kong, C. L. Zhang, S. Y. Hwang, Q. W. Chen and Z. M. Peng, Small, 2017, 13, 1700334 CrossRef PubMed.
- M. R. Gao, W. C. Sheng, Z. B. Zhuang, Q. R. Fang, S. Gu, J. Jiang and Y. S. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- J. W. Nai, H. J. Yin, T. T. You, L. R. Zheng, J. Zhang, P. X. Wang, Z. Jin, Y. Tian, J. Z. Liu, Z. Y. Tang and L. Guo, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- A. C. Thenuwara, N. H. Attanayake, J. Yu, J. P. Perdew, E. J. Elzinga, Q. Yan and D. R. Strongin, J. Phys. Chem. B, 2018, 122, 847–854 CrossRef PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- C. Luan, G. Liu, Y. Liu, L. Yu, Y. Wang, Y. Xiao, H. Qiao, X. Dai and X. Zhang, ACS Nano, 2018, 12, 3875–3885 CrossRef CAS PubMed.
- M. Yu, S. Zhou, Z. Wang, J. Zhao and J. Qiu, Nano Energy, 2018, 44, 181–190 CrossRef CAS.
- X. Jia, S. Gao, T. Liu, D. Li, P. Tang and Y. Feng, Electrochim. Acta, 2017, 245, 59–68 CrossRef CAS.
- L. D. Y. Wang, X. B. Ge, Y. X. Li, J. Liu, L. Y. Huang, L. X. Feng and Y. Wang, J. Mater. Chem. A, 2017, 5, 4331–4334 RSC.
- S. M. Pawar, B. S. Pawar, B. Hou, J. Kim, A. T. A. Ahmed, H. S. Chavan, Y. Jo, S. Cho, A. I. Inamdar, J. L. Gunjakar, H. Kim, S. Cha and H. Im, J. Mater. Chem. A, 2017, 5, 12747–12751 RSC.
- H. J. Zhang, X. P. Li, A. Hahnel, V. Naumann, C. Lin, S. Azimi, S. L. Schweizer, A. W. Maijenburg and R. B. Wehrspohn, Adv. Funct. Mater., 2018, 28, 1706847 CrossRef.
- Z. L. Wang, S. Xiao, Y. M. An, X. Long, X. L. Zheng, X. H. Lu, Y. X. Tong and S. H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 13348–13359 CrossRef CAS PubMed.
- F. Song and X. L. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- X. Long, G. X. Li, Z. L. Wang, H. Y. Zhu, T. Zhang, S. Xiao, W. Y. Guo and S. H. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02723h |
| This journal is © The Royal Society of Chemistry 2019 |