
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic colloidal nanoparticle assembly triggered by aptamer–receptor interactions on live cell membranes†
Linlin
Yang
a,
Lingyan
Meng
c,
Jiaying
Song
a,
Yue
Xiao
a,
Ruowen
Wang
b,
Huaizhi
Kang
*ab and
Da
Han
 *b
*b
aPen-Tung Sah Institute of Micro-Nano Science and Technology, Xiamen University, Xiamen, Fujian 361005, China. E-mail: kang@xmu.edu.cn
bInstitute of Molecular Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, China. E-mail: dahan@sjtu.edu.cn
cCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China
First published on 28th June 2019
Abstract
Cells use dynamic systems such as enzyme cascades and signaling networks to control cellular functions. Synthetic dynamic systems that can be target-responsive have great potential to be applied for biomedical applications but the operation of such dynamic systems in complex cellular environments remains challenging. Here, we engineered an aptamer and DNA displacement reaction-based dynamic system that can transform its nanostructure in response to the epithelial cell adhesion molecule (EpCAM) on live cell membranes. The dynamic system consisted of a core nanoparticle and small satellite nanoparticles. With the modifications of different DNA hairpin strands and swing arm strands partially hybridized with an aptamer that specifically recognizes the EpCAM, the two separated particles can dynamically assemble into a core–satellite assembly by aptamer–receptor interactions on the cell membrane surface. The structural change of the system from separated particles to a core–satellite assembly generated plasmonic coupled hot spots for surface-enhanced Raman scattering (SERS) for sensitively capturing the dynamic structural change of the nanoassembly in the cellular environment. These concepts provide strategies for engineering dynamic nanotechnology systems for biological and biomedical applications in complex biological environments.
Introduction
Life is sustained by complex systems operating with dynamic reactions. Artificial dynamic systems that can be responsive to environmental stimuli and precisely adjust their physicochemical properties will greatly facilitate the development of biological and biomedical applications, such as imaging, diagnosis and disease treatment.1–4 For instance, a dynamic nanoassembly was reported to be able to alter the surface display of targeting ligands and thus control its cellular uptake properties.5 Another example is a nanoparticle system that was designed to dynamically shrink from 100 nm to 10 nm to achieve deep penetration into tumor tissue.6 Similarly, an Au nanoparticle assembly that was pH-sensitive was applied as a smart drug delivery platform with tumor microenvironmental responsive ability for anti-tumor therapy.7In recent years, dynamic DNA nanotechnology has been an appealing tool to engineer precisely controlled dynamic systems, which are based on the remarkable merits of high specificity, predictability and programmability of DNA Watson–Crick base pairing.8–10 In particular, a series of DNA nanomachines such as DNA robots,11–13 molecular motors,14–16 autonomous walkers17–20 and logical computers21–23 have been designed and implemented experimentally. The next step should be applying them for more complex biological applications. However, the design and implementation of dynamic DNA-based systems that automatically carry out functions in complex cellular environments present two critical challenges in molecular engineering. The first one is finding an effective driving force that can precisely control dynamic systems in complex biological environments. Compared to other reactions such as enzyme or DNAzyme-mediated hydrolysis reactions which need enzymes or cofactors (metal ions, etc.) that are either difficult to deliver or requiring strict conditions to functionalize in cells to provide energies for dynamic systems,15,24–26 DNA displacement reactions present some unique advantages. For instance, they are completely enzyme-free reactions, which means that these DNA strand displacement-based dynamic systems can be initiated and driven solely by the DNA molecules in cells. In addition, these DNA displacement reactions are well characterized by various physical or chemical methods and can be even simulated by well-designed in silico programs. All of these advantages of DNA displacement reactions provide good opportunities for controlling dynamic systems in complex biological environments. The second challenge is employing an effective component that can bridge dynamic DNA-based systems and interactive biomolecules such as proteins in cells without influencing the programmability and versatility. A common regulation paradigm used in previous reports is genetic editing, which requires sophisticated and cell-wide coordination. Aptamers, which are single-stranded oligonucleotides, have the promising properties of recognizing and regulating biological molecules in the cellular environment.27,28 In addition, since an aptamer is essentially a single-stranded oligonucleotide, it can be seamlessly encoded in dynamic DNA-based systems with an extended scope of functions, from simple DNA base-pairing reactions to more complicated biomolecular reactions such as DNA–protein interactions.29,30
Herein, we explored the use of DNA aptamer and DNA displacement reactions as molecular keys to operate the colloidal nanoparticle assembly in the cellular environment. The system consisted of a spatially separated core nanoparticle and small satellite nanoparticles. The assembly of the two types of particles can be controlled in response to the epithelial cell adhesion molecule (EpCAM) that is an overexpressed cell surface receptor of multiple adenocarcinomas cells.31,32 Compared to the intracellular genomic or transcriptional marker molecules, cell-surface receptors, mostly membrane proteins, are excellent candidates as molecular signatures for cellular applications such as smart control of the dynamic assembly process in situ due to their excellent specificity, accessibility and scalability. The conformational changes of the system from separated particles to a core–satellite assembly provided plasmonic coupled hot spots for SERS detection. Owing to its distinct advantages of low background, insensitivity to photobleaching or quenching, single-molecule sensitivity and fingerprint-like spectra,33–36 we are able to use SERS to monitor the performance of this nanoparticle assembly in the cellular environment. This dynamic nanoparticle system explored the use of aptamers and DNA displacement reactions to assemble the nanostructures in response to cell surface biomarkers. Therefore, it provides strategies for engineering dynamic systems for mediating cellular functions as well as intracellular imaging in complex biological environments.
Results and discussion
The overall concept and proposed strategy of constructing the aptamer-based dynamic colloidal nanoparticle assembly is depicted in Fig. 1. Au nanoparticles (AuNPs) with diameters of 50 and 13 nm (Fig. S1†) were used as the core and satellites to assemble the core–satellite assembly. The 13 nm satellite AuNPs were modified with a thiolated catalytic DNA hairpin (H) and a Raman reporter 4-ATP. The 50 nm core AuNPs were functionalized with another thiolated catalytic DNA hairpin (SS) and a swing arm single stranded DNA (SA) that was partially hybridized with an aptamer (Ap) which can specifically recognize the EpCAM. These two types of AuNPs functionalized with different components are named as Au13–H and Au50–SS–SA–Ap, respectively. When Au13–H and Au50–SS–SA–Ap were present together with the EpCAM-positive cells, the specific binding between the aptamer and EpCAM on the cell membrane surface can free the SA strand on the 50 nm AuNP surface. Meanwhile, SA possesses a toehold region that can hybridize with the adjacent SS strand to form an intermediate SA–SS complex. With the help of the H strand that was anchored on the 13 nm AuNP, these SA–SS intermediates can form a more stable SS–H duplex via toehold-mediated strand displacement and release the SA strand for the next cycle of catalyzation on the 50 nm AuNP surface. In this way, a single target molecule (EpCAM) on the live cell surface was able to trigger the autonomous assembly of the two hairpins and assemble multiple 13 nm AuNPs on the surface of 50 nm AuNPs, thus changing the state of the whole colloidal structures from spatially separated particles to a core–satellite assembly. Meanwhile, because of the interparticle plasmonic coupled hot spots generated in the core–satellite nanostructures, enhanced Raman scattering of 4-ATP molecules can be used to accurately capture the dynamic structural change of the nanostructures in response to the cell surface marker.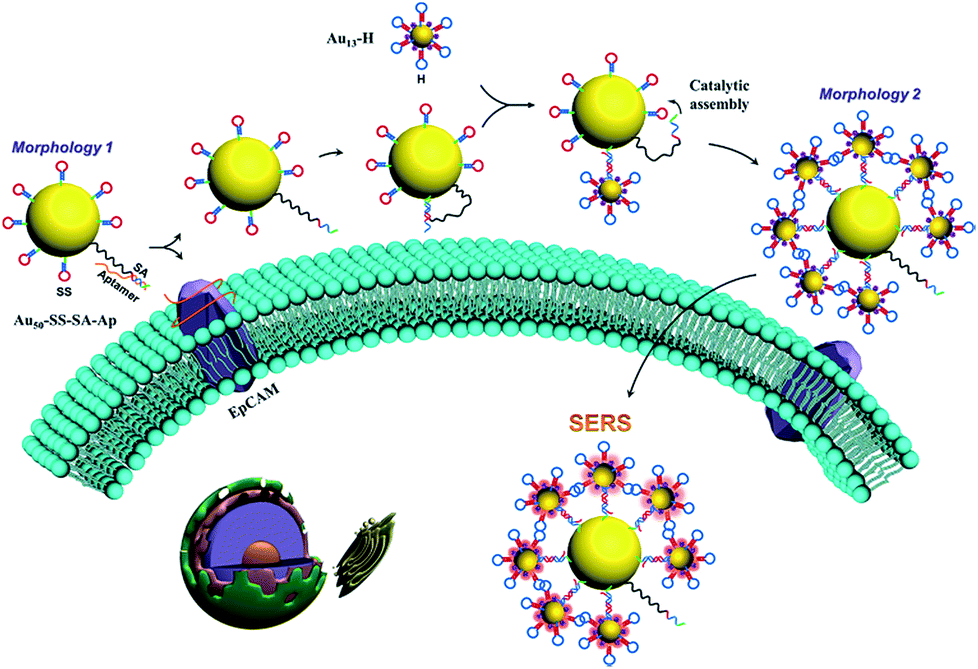 | ||
| Fig. 1 Schematic representation of the dynamic assembly of the core–satellite nanostructure from Morphology 1 to Morphology 2 triggered by EpCAM receptors on the cell membrane surface. | ||
We separately verified the functional components in this system step by step. First, to ensure that the partially hybridized SA–Ap duplex strand can be responsive to EpCAM protein,32 the FRET-based SA–Ap duplex with a fluorophore (SA-FAM) and quencher (BHQ1–Ap) pair was employed to monitor the competitive binding process between EpCAM–Ap and SA–Ap. The sequence length of SA0 (the functional domain of SA that can catalyze the subsequent reaction) plays an important role in ensuring the efficient separation of the SA and Ap duplex upon EpCAM as well as the low background “leaky” reaction without the presence of the EpCAM. In our design, 20 nucleotides (nts) were chosen as the length of the SA–Ap complementary duplex with a good signal to background ratio (Fig. S2†).
Once confirmed that our system can respond to the EpCAM and expose the active catalytic DNA domain (SA0) on SA effectively, we continued to optimize the subsequent DNA catalytic hairpin reactions to improve the nanoparticle assembly efficiency. As shown in Fig. 2A, SA0 contains toehold domain 1* that can hybridize with the exposed domain 1 on SS to initiate the strand displacement and domain 2* that can trigger the branch migration to the fully open hairpin structure of SS. In addition, loop domain 3 on the SS strand should also play important roles in the catalytic hairpin reactions. Therefore, we separately investigated the sequence length of domains 1, 2 and 3 on SS to achieve the highest catalytic efficiency as well as the lowest background signal of SS and H without SA. To ensure the structural stability of the SS strand, we chose 14 nts as the length of stem domain 2, and then studied 4 cases of different lengths of domain 1 and domain 3, respectively. As shown in Fig. 2B and C, we used the ratio of products (group with SA + SS + H) to “leaky” reaction byproducts (SS + H) to evaluate the hairpin catalytic reaction efficiency. It can be seen that case 1 (domain 1 = 6 nt, domain 3 = 15 nt) achieved the highest signal to noise ratio (Fig. 2D). Therefore, SS1 and H1 were chosen as the optimal sequences for the subsequent nanoparticle assembly.
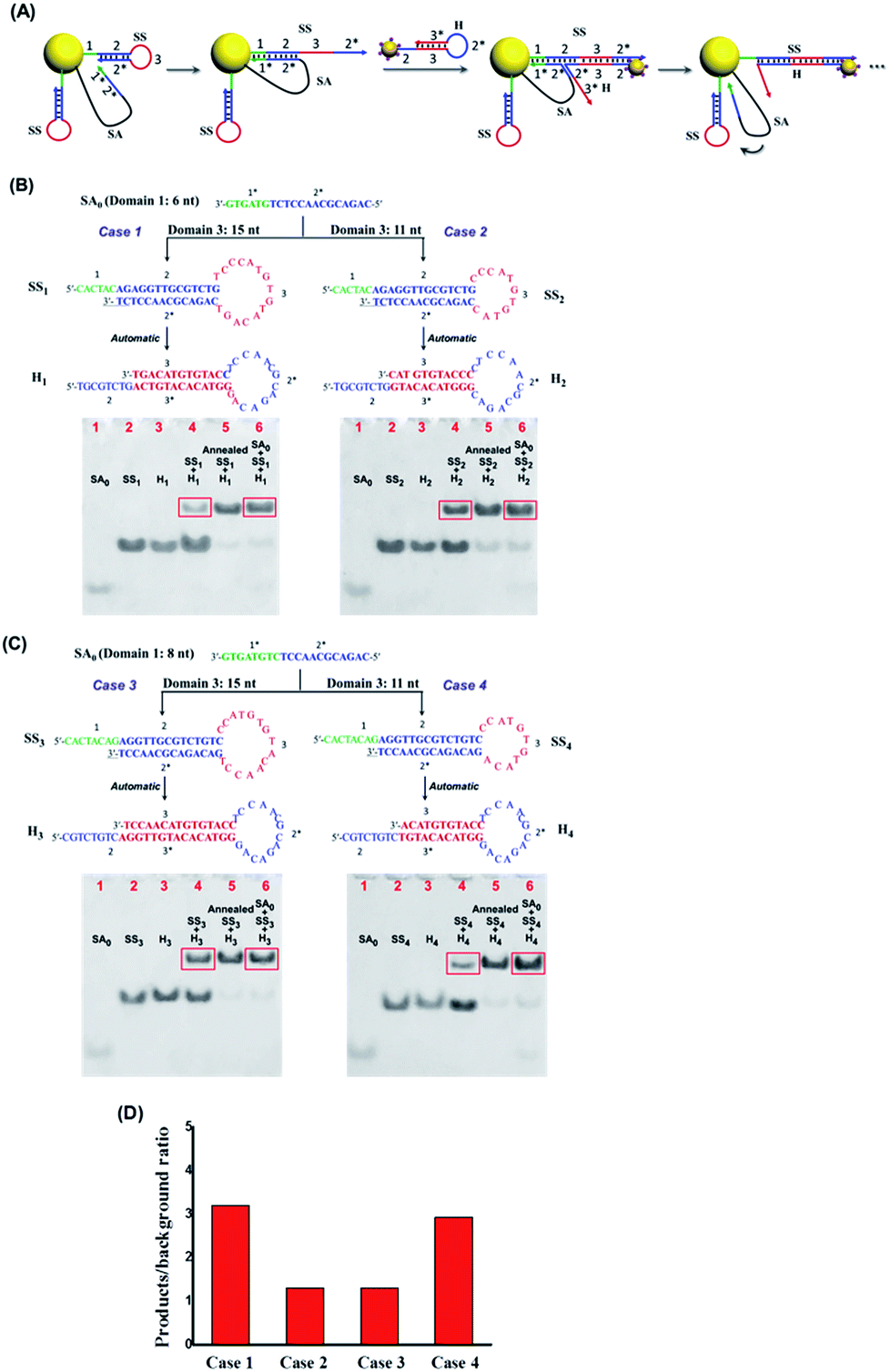 | ||
| Fig. 2 (A) Scheme of the catalytic hairpin assembly reaction triggered by toehold-mediated strand displacement. (B and C) Optimization and PAGE characterization of the sequences designed for the dynamic colloidal nanoparticle system. (D) Comparison of the ratio of products (group with SA + SS + H) to “leaky” reaction byproducts (SS + H). Here, we use NUPACK (http://www.nupack.org) to predict the secondary structures of the DNA strands. | ||
We subsequently performed a feasibility test to determine whether the DNA hairpin catalytic reaction can facilitate the assembly of Au nanoparticles to form the core–satellite structure in solution. The morphological changes of the system can be monitored by optical property measurements that are sensitive to the distance between neighboring particles, such as SERS and surface plasmon resonance (SPR). First, we used the SS1, H1 and an unblocked SA as the optimal sequence combination in terms of assembly efficiency to study the dynamic assembly of the core–satellite nanostructure by SPR (Fig. 3A). Meanwhile, a 20 bp complementary SA–Ap duplex was used as a control group to verify the assembly background signal of the system without the presence of the EpCAM. Notably, we used a molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of SS1 and SA to modify the 50 nm AuNP surface. Considering the catalytic efficiency of a similar hairpin system reported by Chen, one SA molecule should be able to catalyze 20–50 hairpins to form a stable duplex.37 However, the spatial steric effect generated by 13 nm AuNPs assembled on the surface of 50 nm AuNPs is another factor to consider, as too many 13 nm AuNPs would hinder the further assembly. Therefore, in this scenario, we chose a 10:1 ratio for SS1 and SA as the optimum condition to functionalize the 50 nm AuNPs. Following 12 h incubation of Au13–H1 with Au50–SS1–SA–Ap and Au50–SS1–SA, respectively, the products were examined by UV-vis absorption spectra and dynamic light scattering (DLS) (Fig. 3B and C). Compared to Au13–H1 only (curve a in Fig. 3B), Au50–SS1–SA only (curve b in Fig. 3B) and mixtures of Au50–SS1–SA–Ap and Au13–H1 (curve c in Fig. 3B), the absorption peak of Au50–SS1–SA + Au13–H1 mixtures showed a significant red shift from about 530 nm to 552 nm (curve (d) in Fig. 3B), indicating strong surface plasmon resonance happening in the assembly. Combined with the obvious increase of hydrodynamic diameters from 66.44 ± 2.27 nm (curve b in Fig. 3C) with Au50–SS1–SA only to 102.40 ± 8.19 nm (curve d in Fig. 3C), it is believed that the core–satellite assemblies were formed. We further used transmission electron microscopy (TEM) to visualize the structures. When mixing Au50–SS1–SA–Ap with Au13–H1, it can be seen that 50 nm AuNPs were surrounded by only six 13 nm AuNPs in close proximity on average, which is believed to be induced by the “leaky” reaction of the hairpin assembly (Fig. 3D and F). However, the number of “satellites” surrounding the “core” increased to 14 on average when mixing Au50–SS1–SA with Au13–H1 (Fig. 3E and F). These observations verified that the dynamic assembly of Au nanoparticles can be formed and controlled by the SA strand.
:1 of SS1 and SA to modify the 50 nm AuNP surface. Considering the catalytic efficiency of a similar hairpin system reported by Chen, one SA molecule should be able to catalyze 20–50 hairpins to form a stable duplex.37 However, the spatial steric effect generated by 13 nm AuNPs assembled on the surface of 50 nm AuNPs is another factor to consider, as too many 13 nm AuNPs would hinder the further assembly. Therefore, in this scenario, we chose a 10:1 ratio for SS1 and SA as the optimum condition to functionalize the 50 nm AuNPs. Following 12 h incubation of Au13–H1 with Au50–SS1–SA–Ap and Au50–SS1–SA, respectively, the products were examined by UV-vis absorption spectra and dynamic light scattering (DLS) (Fig. 3B and C). Compared to Au13–H1 only (curve a in Fig. 3B), Au50–SS1–SA only (curve b in Fig. 3B) and mixtures of Au50–SS1–SA–Ap and Au13–H1 (curve c in Fig. 3B), the absorption peak of Au50–SS1–SA + Au13–H1 mixtures showed a significant red shift from about 530 nm to 552 nm (curve (d) in Fig. 3B), indicating strong surface plasmon resonance happening in the assembly. Combined with the obvious increase of hydrodynamic diameters from 66.44 ± 2.27 nm (curve b in Fig. 3C) with Au50–SS1–SA only to 102.40 ± 8.19 nm (curve d in Fig. 3C), it is believed that the core–satellite assemblies were formed. We further used transmission electron microscopy (TEM) to visualize the structures. When mixing Au50–SS1–SA–Ap with Au13–H1, it can be seen that 50 nm AuNPs were surrounded by only six 13 nm AuNPs in close proximity on average, which is believed to be induced by the “leaky” reaction of the hairpin assembly (Fig. 3D and F). However, the number of “satellites” surrounding the “core” increased to 14 on average when mixing Au50–SS1–SA with Au13–H1 (Fig. 3E and F). These observations verified that the dynamic assembly of Au nanoparticles can be formed and controlled by the SA strand.
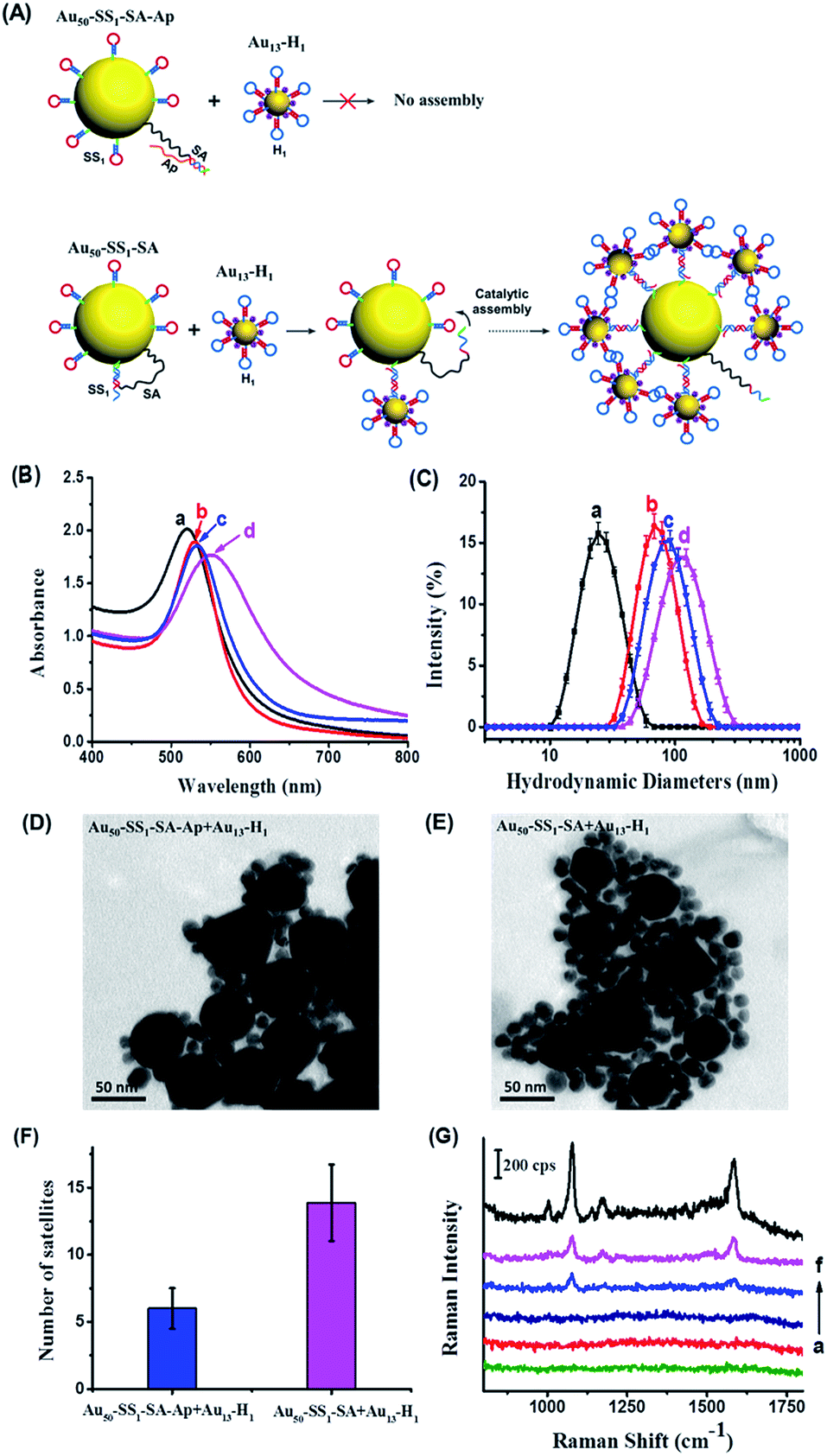 | ||
| Fig. 3 (A) Scheme of blocked and unblocked SA-triggered assembly of the core–satellite nanostructures. (B) UV-vis absorption spectra and (C) DLS intensity of (a) Au13–H1 only, (b) Au50–SS1–SA only, (c) mixtures of Au50–SS1–SA–Ap and Au13–H1, (d) mixtures of Au50–SS1–SA and Au13–H1. (D) TEM image of mixtures of Au50–SS1–SA–Ap and Au13–H1. (E) TEM image of mixtures of Au50–SS1–SA and Au13–H1. (F) Average numbers of 13 nm “satellite” nanoparticles of groups Au50–SS1–SA–Ap + Au13–H1 and Au50–SS1–SA + Au13–H1. The numbers were summarized by counting 30 “core” nanoparticles in each group in the TEM images. (G) SERS signals of (a) the mixture of SS1, H1, SA and Ap without AuNPs; (b) Au50–SS1–SA only; (c) Au50–SS1–SA–Ap only; (d) Au13–H1 only; (e) mixture of Au50–SS1–SA–Ap and Au13–H1; (f) mixture of Au50–SS1–SA and Au13–H1. | ||
Next, we used SERS to further monitor the dynamic assembly process of the system in solution. 4-ATP was adsorbed on the 13 nm AuNPs as a Raman reporter. As shown in Fig. 3G, Au50–SS1–SA and Au50–SS1–SA–Ap both displayed no Raman signal due to the absence of Raman reporters. Au13–H1 can generate a weak SERS signal because of the insufficient electromagnetic coupling on its surface. When mixing Au50–SS1–SA with Au13–H1 probes, strong characteristic peaks of 4-ATP at 1078 cm−1 and 1585 cm−1 were observed, indicating the formation of electromagnetic hot spots with proximity of Au nanoparticles. Meanwhile, as for the mixture of Au50–SS1–SA–Ap and Au13–H1, the SERS peak intensities were much weaker and just similar to those of Au13–H1 only. The finite difference time domain (FDTD) simulations were employed to theoretically calculate the enhanced electromagnetic field distribution of the core–satellite nanostructure. The electromagnetic field intensity in the gap between assembled 13 nm and 50 nm AuNPs was found to be much stronger than that on the 13 nm AuNP surface (Fig. S3†). Therefore, combining both the experimental and theoretical findings, the newly generated interparticle plasmonic coupled hot spots in the core–satellite nanostructures can provide strong SERS signals. It also paved ways for using SERS to reveal the dynamic assembly activated by aptamer–receptor interactions on live cell membranes.
Last, we determined whether we could activate the dynamic assembly of nanoparticles by aptamer–receptor interactions on live cell membranes. Specifically, we chose the EpCAM-positive cell line MCF-7, a cell line originating from human breast adenocarcinoma cells, as the target cell to test the assembly of the nanoparticle system. The EpCAM-negative cell line HEK-293T, derived from human embryonic kidney cells, was used as the control cell line. Prior to application of the nanoparticles to living cells, the cytotoxicity of Au13–H1 and Au50–SS1–SA–Ap was checked to investigate the influence of nanoparticles on the cell status after a relatively long incubation. The viabilities of the two cell lines were found to be almost unaffected by the concentrations and incubation time used for the cell experiment (Fig. S4†).
MCF-7 cells were incubated with Au50–SS1–SA–Ap and Au13–H1 for 12 h to make sure that the Au50–SS1–SA–Ap can sufficiently interact with EpCAM receptors on the cell membrane surface, assemble with Au13–H1 on the membranes and then internalize into the cells. Fig. 4 shows that the Raman signals of MCF-7 cells with different nanoparticle groups are acquired in the 1078 cm−1 channel. The typical Raman scattering peak of 4-ATP at 1078 cm−1 enhanced more significantly than that at 1585 cm−1, so the peak at 1078 cm−1 was used to map the cells. With addition of Au50–SS1–SA–Ap only, there was no signal due to the lack of Raman reporters. When MCF-7 cells were treated with Au13–H1 probes only, they exhibited a weak Raman signal which corresponded to the relatively insufficient enhancement of Raman scattering on the 13 nm AuNP surface. Notably, MCF-7 cells treated with both Au50–SS1–SA–Ap and Au13–H1 together generated a significantly enhanced Raman signal, indicating that core–satellite nanoassemblies were formed in the cellular environment. In addition, with the presence of Au50–SS1–SA and Au13–H1 probes, the Raman scattering of MCF-7 cells was also enhanced significantly, confirming that the unblocked SA is the key to initiate the strand displacement and change the dynamic system into a core–satellite structure in living cells (Fig. S5†).
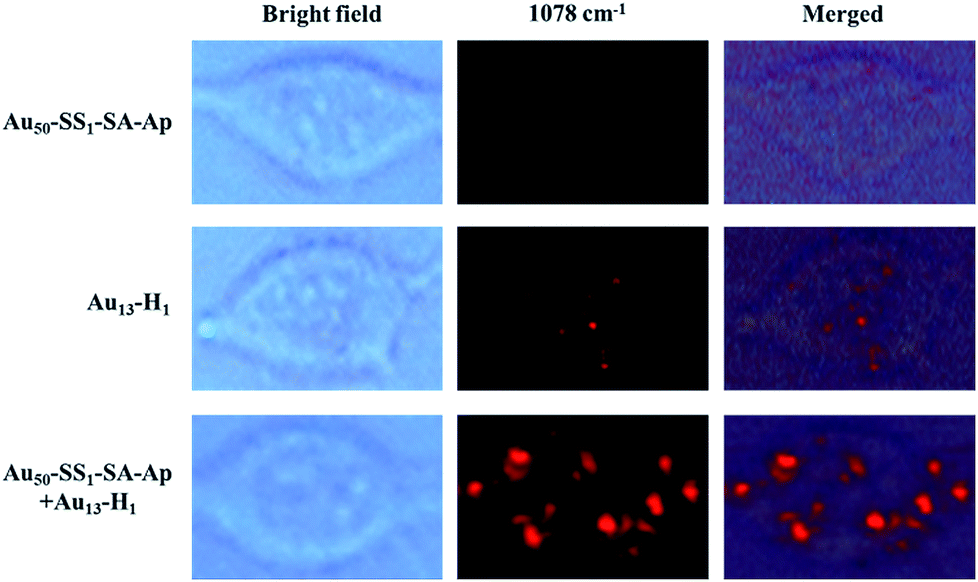 | ||
| Fig. 4 Bright field and Raman images of MCF-7 cells which were treated with Au50–SS1–SA–Ap (0.02 nM), Au13–H1 (0.5 nM) only and the mixture of Au50–SS1–SA–Ap (0.02 nM) and Au13–H1 (0.5 nM). | ||
Then we studied the nanoparticle assembly in EpCAM-negative cell line HEK-293T to determine whether the enhanced Raman signals were derived from the presence of the EpCAM receptor on the cell membrane surface. After 12 h incubation, a much weaker Raman signal compared to that of MCF-7 cells in the 1078 cm−1 channel was seen (Fig. 5A). We also summarized the mean Raman signal intensities of 10 different MCF-7 and HEK-293T cells. It can be seen that they are significantly different with a p value of 0.002. In addition, TEM images of 100 nm thick cell sections were compared and are shown in Fig. 5B and C. Core–satellite assembly structures were found in the MCF-7 cells, whereas mainly separated particles were seen in HEK-293T cells. These data verified that the cell-surface EpCAM can trigger the dynamic assembly of nanoparticles from separated particles to a core–satellite structure and also confirmed the credibility of our assembly strategy in complex biological environments.
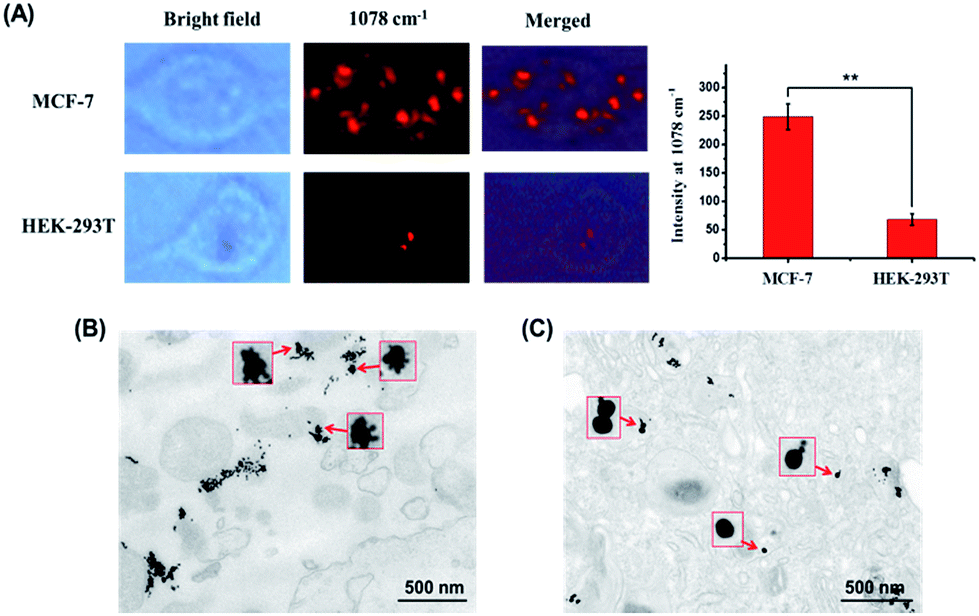 | ||
| Fig. 5 (A) Bright field and Raman images of MCF-7 cells and HEK-293T cells treated with both Au50–SS1–SA–Ap (0.02 nM) and Au13–H1 (0.5 nM) (** stands for p values < 0.01, n = 10). TEM images of 100 nm thick cell sections were derived from (B) MCF-7 and (C) HEK-293T cells incubated with both Au50–SS1–SA–Ap (0.02 nM) and Au13–H1 (0.5 nM). The insets show the enlarged pictures of the particles. | ||
Conclusions
Design of dynamic systems with such capabilities as conformation or function change in response to biological signals in biological environments is an important step toward intelligent diagnosis and disease treatment. In this study, we have engineered a dynamic system based on aptamer–receptor interactions and DNA displacement reactions to autonomously assemble core–satellite nanostructures in the cellular environment, owing to the combination of the high specificity of the aptamer and the stable performance of DNA catalytic reactions. The structural changes of the dynamic system have provided electromagnetic hot spots for SERS imaging to precisely characterize the systems in complex biological milieus.Compared with dynamic systems based totally on DNA base-pairing, the introduction of an aptamer provides a direct molecular bridge linking DNA with proteins on cell membranes, enabling precise sensing of the cellular environment and smart control of the dynamic assembly process in situ. Moreover, as a variety of aptamers are either available or can be obtained through SELEX to bind a broad range of targets with tunable binding ability, the proposed dynamic system also provided a versatile tool responsive to other biological targets in cellular context by simply changing the aptamers. Given the design modularity with DNA displacement reactions and target specificity with aptamers, the prototype aptamer–receptor interaction triggered dynamic system demonstrated here has the potential to enhance DNA nanotechnology with new insights and will broaden the utility of dynamic nanomaterials for applications in biology, biotechnology and biomedicine, such as development of future smart nanomaterials that combine autonomous delivery, transformation and activation functions in the blood stream to diagnose or treat targeted sites.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financially supported by the National Sciences Foundation of China (Grant No. 21675134). Da Han acknowledges start-up funding support from the Institute of Molecular Medicine of Shanghai Jiao Tong University and the 1000 Youth Talents Program, granted by the Chinese Central Government.Notes and references
- L. Lin, Y. Xie, S. Wang, W. Wu, S. Niu, X. Wen and Z. L. Wang, ACS Nano, 2013, 7, 8266–8274 CrossRef CAS PubMed.
- M. Yasui, M. Hiroshima, J. Kozuka, Y. Sako and M. Ueda, Nat. Commun., 2018, 9, 3061 CrossRef PubMed.
- G. Yu, B. C. Yung, Z. Zhou, Z. Mao and X. Chen, ACS Nano, 2018, 12, 7–12 CrossRef CAS PubMed.
- J. Kim, D. Jang, H. Park, S. Jung, D. H. Kim and W. J. Kim, Adv. Mater., 2018, 30, 1707351 CrossRef PubMed.
- S. Ohta, D. Glancy and W. C. W. Chan, Science, 2016, 351, 841–845 CrossRef CAS PubMed.
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popović, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426–2431 CrossRef CAS PubMed.
- H. Park, J. Kim, S. Jung and W. J. Kim, Adv. Funct. Mater., 2018, 28, 1705416 CrossRef.
- N. C. Seeman, Trends Biotechnol., 1999, 17, 437–443 CrossRef CAS PubMed.
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- A. J. Thubagere, W. Li, R. F. Johnson, Z. Chen, S. Doroudi, Y. L. Lee, G. Izatt, S. Wittman, N. Srinivas, D. Woods, E. Winfree and L. Qian, Science, 2017, 357, eaan6558 CrossRef PubMed.
- R. A. Muscat, J. Bath and A. J. Turberfield, Nano Lett., 2011, 11, 982–987 CrossRef CAS PubMed.
- M. Hagiya, A. Konagaya, S. Kobayashi, H. Saito and S. Murata, Acc. Chem. Res., 2014, 47, 1681–1690 CrossRef CAS PubMed.
- T.-G. Cha, J. Pan, H. Chen, J. Salgado, X. Li, C. Mao and J. H. Choi, Nat. Nanotechnol., 2014, 9, 39–43 CrossRef CAS PubMed.
- H. Peng, X. F. Li, H. Zhang and X. C. Le, Nat. Commun., 2017, 8, 14378–14390 CrossRef PubMed.
- P. Q. Ma, C. P. Liang, H. H. Zhang, B. C. Yin and B. C. Ye, Chem. Sci., 2018, 9, 3299–3304 RSC.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. D. Mao, Angew. Chem., Int. Ed., 2005, 44, 4355–4358 CrossRef CAS PubMed.
- M. You, Y. Chen, X. Zhang, H. Liu, R. Wang, K. Wang, K. R. Williams and W. Tan, Angew. Chem., Int. Ed., 2012, 51, 2457–2460 CrossRef CAS PubMed.
- X. Qu, D. Zhu, G. Yao, S. Su, J. Chao, H. Liu, X. Zuo, L. Wang, J. Shi, L. Wang, W. Huang, H. Pei and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 1855–1858 CrossRef CAS PubMed.
- Y. Yang, M. A. Goetzfried, K. Hidaka, M. You, W. Tan, H. Sugiyama and M. Endo, Nano Lett., 2015, 15, 6672–6676 CrossRef CAS PubMed.
- Q. H. Liu, L. M. Wang, A. G. Frutos, A. E. Condon, R. M. Corn and L. M. Smith, Nature, 2000, 403, 175–179 CrossRef CAS PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS PubMed.
- L. Qian, E. Winfree and J. Bruck, Nature, 2011, 475, 368 CrossRef CAS PubMed.
- F. Chen, M. Bai, K. Cao, Y. Zhao, X. Cao, J. Wei, N. Wu, J. Li, L. Wang, C. Fan and Y. Zhao, ACS Nano, 2017, 11, 11908–11914 CrossRef CAS PubMed.
- J. Liu, M. Cui, H. Zhou and W. Yang, ACS Sens., 2017, 2, 1847–1853 CrossRef CAS PubMed.
- J.-J. Li, W.-N. Li, W.-F. Du, M.-M. Lv, Z.-K. Wu and J.-H. Jiang, Chem. Commun., 2018, 54, 10626–10629 RSC.
- X. Fang and W. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS PubMed.
- C. Liu, J. Zhao, F. Tian, L. Cai, W. Zhang, Q. Feng, J. Chang, F. Wan, Y. Yang, B. Dai, Y. Cong, B. Ding, J. Sun and W. Tan, Nat. Biomed. Eng., 2019, 3, 183–193 CrossRef CAS PubMed.
- D. Han, Z. Zhu, C. Wu, L. Peng, L. Zhou, B. Gulbakan, G. Zhu, K. R. Williams and W. Tan, J. Am. Chem. Soc., 2012, 134, 20797–20804 CrossRef CAS PubMed.
- H.-M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X.-B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- M. Munz, P. A. Baeuerle and O. Gires, Cancer Res., 2009, 69, 5627–5629 CrossRef CAS PubMed.
- Y. Song, Z. Zhu, Y. An, W. Zhang, H. Zhang, D. Liu, C. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS PubMed.
- J. Song, J. Zhou and H. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS PubMed.
- Y. Chen, L. Ding, W. Song, M. Yang and H. Ju, Chem. Sci., 2016, 7, 569–574 RSC.
- W. Zhou, Q. Li, H. Liu, J. Yang and D. Liu, ACS Nano, 2017, 11, 3532–3541 CrossRef CAS PubMed.
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02693b |
| This journal is © The Royal Society of Chemistry 2019 |