
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sterically encumbered photoredox catalyst enables the unified synthesis of the classical lignan family of natural products†
Edwin
Alfonzo
and
Aaron B.
Beeler
 *
*
Department of Chemistry, Boston University, Boston, Massachusetts 02215, USA. E-mail: beelera@bu.edu
First published on 5th July 2019
Abstract
Herein, we detail a unified synthetic approach to the classical lignan family of natural products that hinges on divergence from a common intermediate that was strategically identified from nature's biosynthetic blueprints. Efforts toward accessing the common intermediate through a convergent and modular approach resulted in the discovery of a sterically encumbered photoredox catalyst that can selectively generate carbonyl ylides from electron-rich epoxides. These can undergo concerted [3 + 2] dipolar cycloadditions to afford tetrahydrofurans, which were advanced (2–4 steps) to at least one representative natural product or natural product scaffold within all six subtypes in classical lignans. The application of those synthetic blueprints to the synthesis of heterolignans bearing unnatural functionality was demonstrated, which establishes the potential of this strategy to accelerate structure–activity-relationship studies of these natural product frameworks and their rich biological activity.
Introduction
Classical lignans (CLs) represent one of the oldest known and most sought after families of secondary metabolites found in planta.1 The interest in these molecules is well merited, as they possess a broad spectrum of promising biological actions, some of which have already significantly impacted society.2 Although their carbon frameworks are only composed of two phenylpropane units like 1 (Scheme 1), CLs exhibit wide structural diversity and oxidation patterns that extend to six different subtypes, namely, dibenzylbutane (CL1), dibenzylbutyrolactone (CL2), arylnaphthalene and its derivatives (CL3), dibenzocyclooctadiene (CL4), furan (CL5a–c), and furofuran (CL6). While these subtypes differ greatly in connectivity, they all share a common carbon–carbon β–β′ (8,8′) bond in their propane side chain, a birthmark that is the consequence of a common biosynthesis. | ||
| Scheme 1 Biosynthesis of the classical lignan family of natural products and proposed strategy to unify them in a laboratory setting. | ||
We became interested in developing a convergent and modular approach to access all CL subtypes that could be susceptible to structure–activity-relationship (SAR) studies. Although elegant biosynthetic and total synthetic routes have been established towards the synthesis of CLs, rarely have these strategies shown the flexibility that nature displays in its biosynthesis, resulting in routes that can only access a single or few subtypes and in some instances are not practical for intensive SAR investigation.3–6 Herein, we report a unified synthetic approach to CLs and demonstrate how the outlined synthetic blueprints are amenable to the synthesis of heterolignans, which can accelerate the investigation of promising biological functions that each subtype displays outside its natural framework.
Results and discussion
Our retrosynthetic analysis was greatly influenced by a bifurcation that occurs early in the biosynthesis of CLs. One electron oxidation and deprotonation of two units of 1 by an oxidative enzyme produce phenoxy radicals that are trapped by a dirigent protein forming a ternary complex 2.4a Within this complex a regio- and stereoselective carbon–carbon (8,8′) bond coupling event ensues, affording bis para-quinone-methide (pQM) 3 that is known to bifurcate into two CL subtypes. Two sequential 5-exo-trig cyclization steps of 3 furnish the furofuran (CL6), which has been shown in most cases to serve as the parent molecule for the remaining CLs.7 Alternatively, 3 can be trapped by an equivalence of water to deliver the furan (CL5a).8We hypothesized that CL5a could be accessed through a concerted [3 + 2] dipolar cycloaddition from the linear carbonyl ylide 4, derived from epoxides, and a dipolarophile 5 (Scheme 1). A strategic aspect of this reaction is that retention of the stereochemistry of the dipolarophile recapitulates the stereoselective carbon–carbon bond coupling event that occurs in the biosynthesis of CLs. We further posit that we could leverage CL5a into CL6 through the intermediacy of a bis-pQM via dehydration. Lastly, although CL6 and CL5a differ greatly in connectivity, they possess identical oxidation states. Thus, we thought that if nature could leverage CL6 into other CLs then we could find selective transformations to do the same with CL5a. With this outlined strategy, CLs can be segmented into their peripheral components (aryl groups and pendant oxygen), which would enable a modular approach to synthesize these molecules and facilitate SAR studies.
To this end, seminal work by Whiting established that 9,10-dicyanoanthracene (DCA, 11) (λmax = 422 nm, [DCA*/DCA˙−] = +1.99 V vs. SCE) (Scheme 2), a photoredox catalyst, can catalyze the formation of HOMO carbonyl ylides from epoxides, which can be trapped with LUMO dipolarophiles.9 An important feature of this reaction is that although carbonyl ylide formation proceeds through two distinct conformers (exo,exo and exo,endo), the resulting diastereomeric products retain the relative stereochemistry of the dipolarophile. In the context of CLs, we decided to enter CL5a with this reaction since conversion to CL6 would proceed through a stereoablative process, which would provide a single bis-pQM, like 3.
 | ||
| Scheme 2 Generation of electron-rich carbonyl ylides. | ||
In 2017, we reported our efforts on exploring the scope of carbonyl ylide formation and its [3 + 2] dipolar cycloaddition.10 We found some success using DCA, but when expanding the scope to more electron-rich epoxides bearing multiple aryl methoxy groups, characteristic of CL natural products, the reaction failed.11 For example, whereas the carbonyl ylide from epoxide 6 (Scheme 2a) could be trapped with modest efficiency with dimethyl fumarate (8), epoxide 7 would remain unreacted, only providing trace amounts of the desired product 10. We have since been able to identify 2,6-di-tert-butylanthracene-9,10-dicarbonitrile (DTAC, 12) (λmax = 431 nm, [DTAC*/DTAC˙−] = +1.81 V vs. SCE), a heretofore unknown catalyst in photoredox methodologies, which can selectively generate carbonyl ylides from electron-rich epoxides.12,13 When using DTAC as the catalyst for the cycloaddition of epoxide 6 or 7 with dipolarophile 8 the reaction proceeded in nearly quantitative yields (93–94%) with modest selectivity in both cases for the exo,exo conformer. Notably, after every reaction was performed the catalyst was recovered unscathed and recycled for future use.
Evidence for the superior efficacy of DTAC to DCA was in two forms. First, isolation of adduct 13 under catalytic conditions (Scheme 2b) established the capacity of DCA to participate in the reaction as a dipolarophile, alluding to less undesired reactivity in this reaction setting. Secondly, in the case of epoxide 7, where only a trace amount of product was detected, we observed a distinct colour formation when it was mixed with DCA which is representative of CT complex formation.14 Subsequent UV-Vis spectroscopy studies of DCA in the presence of epoxide 6 or 7 verified the presence of a CT band (λCT) in the case of epoxide 7 but not 6 (Scheme 2c, left). When performing an identical UV-Vis experiment with DTAC (Scheme 2c, right), CT band formation was not observed with epoxide 7. We speculate that in the event where a CT complex is formed, inner-sphere SET and sequential back electron transfer preclude cage escape and carbonyl ylide formation, a known unproductive pathway of catalyst 11.15a Thus, DTAC functions by sterically inhibiting CT complex formation and primarily engaging epoxides through an outer-sphere SET, leading to efficient cage escape and, consequently, carbonyl ylide formation.15b
Synthesis of furofuran (CL6)
We began our synthetic efforts towards validating our hypothesis for the dehydration of CL5a by targeting methyl piperitol (19) (Table 1).16 Leveraging previous biomimetic strategies that have relied on bis-pQM formation to access CL6, it was reasoned that if we could generate the bis-pQM from 18 then the cyclization that would follow would prefer to proceed through the conformer where the quinone methide is oriented away from the first formed tetrahydrofuran (18dvs.18c) to afford 19.17|
|
|||
|---|---|---|---|
| Entry | Conditionsa | d.r. (19, C7,C7′) | % Conversionb (IY, RSM) |
| a 23 °C. b Isolated yield (IY) and recovered starting material (RSM). c 2 M HCl in Et2O (3 equiv.), CHCl3. d RSM from entry 4 was used. | |||
| 1 | FeCl3 (20 mol%), DCM, 12 h | 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :29:26 :29:26 |
100 |
| 2 | HCl (10 mol%), DCM/HFIP, 12 h | 59:21:19 |
100 |
| 3 | TMSCl (5 equiv.), DCM, 72 h | 67:28:1 |
72 |
| 4c | 2 M HCl (3 equiv.), 48 h (cycle 1) | 93:7:0 |
40 (40,58) |
| 5c,d | 2 M HCl (3 equiv.), 48 h (cycle 2) | 97:3:0 |
33 (30,60) |

Epoxide 17 was synthetized on a decagram scale (95%, d.r. 6:1), using a one-pot sulfonium-ylide (15) mediated epoxidation of aldehyde 16 with alcohol 14 (Table 1).18 Sequentially, 17 was subjected to the [3 + 2] dipolar cycloaddition with 8, and the resulting product was reduced with LiAlH4, providing diol 18 in 90% yield as a mixture of four inconsequential diastereomers (d.r. 3:3:2:1), which served as the substrate for the investigation of the proposed bis-pQM formation.
Early success was found when performing the reaction with catalytic amounts of FeCl3 (Table 1, entry 1), but unfortunately the poor selectivity, providing the desired product and two of its epimers at C7 and C7′ (which themselves are natural products), led us to consider Brønsted acids over Lewis acids. Under catalytic HCl (10 mol%) in a binary solution of DCM and hexafluoroisopropyl alcohol (HFIP), we observed full conversion of diol 18 with a minor improvement in the selectivity (Table 1, entry 2). A more thorough screening of Brønsted acids led to the discovery that the presence of water significantly slowed the reaction but efforts to remove water with drying agents were futile, as they inhibited reactivity.19 We found that generating HCl in situ with TMSCl provided higher conversions but only with a modest improvement in selectivity (Table 1, entry 3). Eventually, we found that the use of 2 M HCl in diethyl ether could reconcile the yield and selectivity providing 19 in 40% yield and excellent selectivity (d.r. > 10:1) after 48 h (Table 1, entry 4). More importantly, the remaining mass balance of the reaction (58% recovered diol) could be resubmitted to the same conditions and similar efficiency for formation of 19 was observed (Table 1, entry 5). Interestingly, resubmission of 19 to the same conditions resulted in the formation of C7 and C7′ epimers, which demonstrates that while the cyclization may be selective, prolonged exposure to the acidic media leads to epimerization.19 Ultimately, our synthetic approach provides (±)-methyl piperitol (19) in 4 steps and 32% overall yield.
Synthesis of dibenzylbutanes (CL1)
Continuing the theme of engaging CL5a in stereoablative processes, we identified Pd(OH)2, Pearlman's reagent,20 as a highly active catalyst that can indiscriminately reduce all stereoisomers obtained from the cycloaddition to CL1 scaffolds. A route was established that consists of [3 + 2] dipolar cycloaddition, LiAlH4 reduction, and Pd(OH)2 hydrogenation under atmospheric pressure that gives access to numerous CL1 scaffolds in a preparative scale (Scheme 3).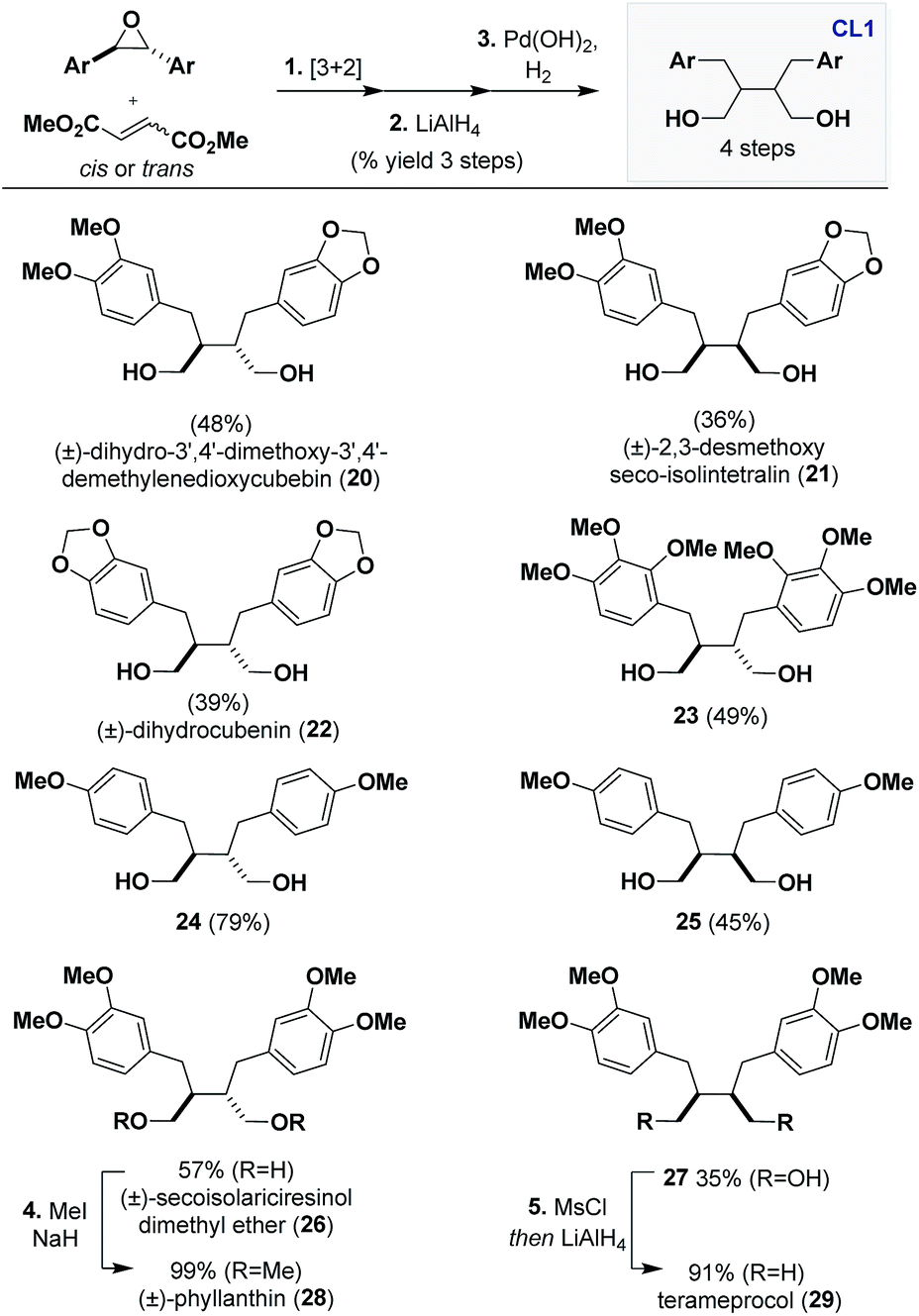 | ||
| Scheme 3 Synthesis of dibenzylbutanes (CL1) through hydrogenation of CL5a. Reagents and conditions: (1) DTAC (5 mol%), epoxide (1 equiv.), dipolarophile (3 equiv.), PhMe, Blue LEDs, 25–30 °C, N2, 1–10 days. (2) LiAlH4 (6 equiv.), THF, 0 to 23 °C, 1 h. (3) Pd(OH)2 (10–20 mol%), EtOH or 1:1 EtOH/EtOAc, H2 (balloon), 1–7 days. (4) MeI (10 equiv.), NaH (5 equiv.), THF, 0 to 23 °C. (5) MsCl (3 equiv.), TEA (3 equiv.), DCM, 0 to 23 °C then, LiAlH4 (8 equiv.), THF, 0 °C to Δ. | ||
An advantage of our strategy over previous methods to access CL1 is that it proceeds with retention of the dipolarophile stereochemistry, eliminating potential ambiguity of the β–β′ relative stereochemistry. For example, isolation and total synthesis of 20 through Stobbe condensation21 has been reported but our obtained 1H and 13C data did not match the reported values. Gratifyingly, (±)-dihydro-3′,4′-dimethoxy-3′,4′-demethylenedioxycubebin (20) has been reported elsewhere and our obtained spectra matched.22 We believe this is due to a misassignment in the earlier report, which occurred because the Stobbe condensation route proceeds through a non-selective hydrogenation, furnishing both the trans and cis scaffolds. Thus, the natural product isolated therein was (±)-2,3-desmethoxy seco-isolintetralin (21). We were able to confirm this through synthesis of 21 using dimethyl maleate as the dipolarophile, which indeed matched the reported spectral values, leading to its structural revision.
Other CL1 scaffolds that can be accessed through this sequence are (±)-dihydrocubenin (22)23 and 2,3,4-trimethoxy substituted 23. The latter example showcases the tolerability of the cycloaddition and hydrogenation to substitution in all positions of the aryl groups. Other notable examples include, 4-methoxy substituted trans24 and cis25 and 3,4-dimethoxy substituted (±)-secoisolariciresinol dimethyl ether (26)24 and 27, which were obtained in good yields over 3 steps (35–79%). The latter two examples can be further elaborated to other CL1 scaffolds. For example, 26 can be methylated to form (±)-phyllanthin (28),25 which has been found to have promising anti-hyperalgesia effects, and 27 can be deoxygenated to access the meso compound terameprocol (29), which is currently in a clinical trial for high grade glioma.26
Synthesis of dibenzylbutyrolactones (CL2)
Previously CL1 scaffolds have been oxidized to CL2 using stoichiometric Fétizon's reagent or using catalytic ruthenium complexes.27 We found that the catalytic protocol for aerobic oxidative lactonization of diols reported by Stahl28 was exceedingly effective in conversion of CL1 to CL2 products (Scheme 4a). The generality of this transformation is quite broad when varying the aryl oxidation patterns, as demonstrated by monomethoxy 30 and trimethoxy 33 as well as natural products (±)-dimethyl matairesinol (31) and (±)-hinokinin (32), which were obtained in excellent to good yields (84–98%).29 The operational simplicity of this reaction in comparison to previous methods is noteworthy and, to the best of our knowledge, represents its first use in the synthesis of bioactive natural products.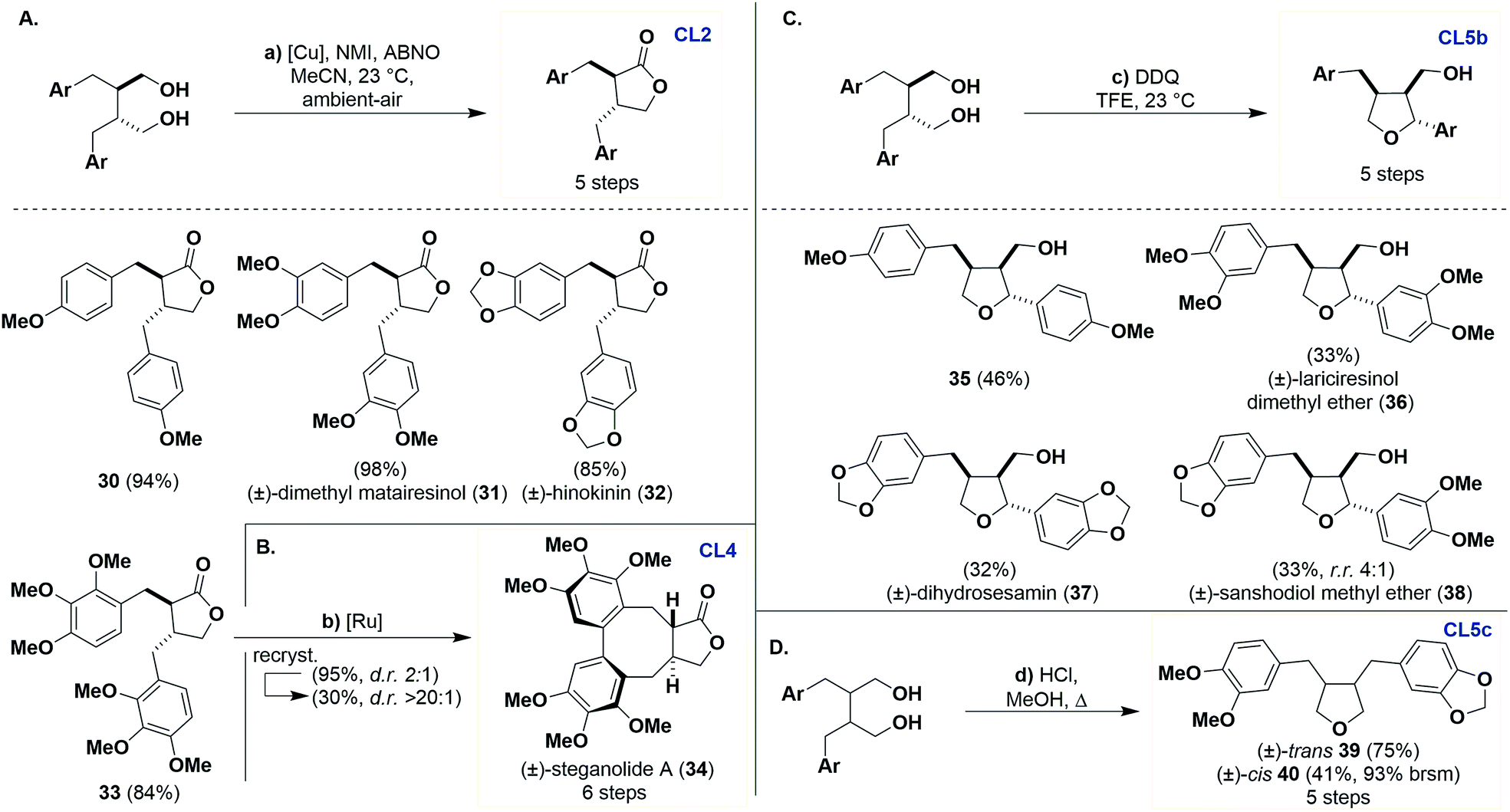 | ||
| Scheme 4 Synthesis of dibenzylbutyrolactones (CL2), dibenzocyclooctadienes (CL4) and furan (Cl5b and Cl5c) from dibenzylbutanes (CL1). Reagents and conditions: (a) Cu(MeCN)4OTf (5 mol%), 2,2′-bpy (5 mol%), ABNO (1 mol%), NMI (10 mol%), MeCN, ambient-air, 23 °C, 12 h. (b) RuO2·xH2O (2 equiv.), TFA (30 equiv.), TFAA (15 equiv.), BF3–OEt2 (8 equiv.), DCM, −10 to 23 °C, N2, 24 h. (c) DDQ (1.5 equiv.), TFE, 23 °C, 6–8 h. (d) HCl (5–10 drops), MeOH, Δ, 4–7 days. | ||
Synthesis of dibenzocyclooctadienes (CL4)
We can access CL4 products using oxidative coupling conditions reported by Robin,30 which is demonstrated by the synthesis of (±)-steganolide A (34) from lactone 33 in 95% yield (Scheme 4b). Although reported as atroposelective in our hands this reaction produces two atropisomers (d.r. 2:1). The selectivity of the product can be improved by recrystallization (30%, d.r. > 20:1).
Synthesis of furans (CL5a–c)
Entry into CL5b from CL1 has been previously described by Ward with the use of 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in acetic acid on a single substrate.31 This work was reproducible but the isolation of the desired product was tedious, requiring several purifications to access pure materials, resulting in low yields (<10%). A solvent screening revealed that 2,2,2-trifluoroethanol (TFE) gave full conversion of the starting material and a relatively cleaner reaction profile (Scheme 4c). With these conditions, we can access 4-methoxy substituted 35, (±)-lariciresinol dimethyl ether (36),32 and (±)-dihydrosesamin (37)33 in modest yields (32–46%). Interestingly, we can also use an unsymmetrical example and gain access to pure (±)-sanshodiol methyl ether (38)6b in 33% yield and with good selectivity (r.r. 4:1).34
Entry into CL5c can be accomplished from 20 and 21 through dehydration under refluxing methanolic HCl to afford 39 (75%) and 40 (41%, 93% brsm) in good yields (Scheme 4d).35 These examples again highlight the opportunities available for the stereodivergent synthesis of CLs controlled by the stereochemistry of the dipolarophile.
Lastly, CL5a represents the greatest challenge for the effective use of the [3 + 2] dipolar cycloaddition, as it can be found in nature possessing all possible variations in its relative stereochemistry.36 We set out to access the all-cisCL5a scaffold due to its known ability to promote neuronal differentiation and neurite growth.37 In particular, we targeted tetrahydrofuran 46 (Scheme 5), a reported natural product.38
 | ||
| Scheme 5 Total synthesis of a proposed CL5a natural product. Reagents and conditions: (1) DTAC (5 mol%), 17 (1 equiv.), 41 (2.5 equiv.), PhMe, Blue LEDs, 25–30 °C, N2, 8 days, then DDQ (1.6 equiv.), 24 h. (2) LiAlH4 (6 equiv.), THF, 0 to 23 °C. (3) Pd/C (10 mol%), 3:1 EtOH/EtOAC, H2 (balloon), 23 °C, 48 h. | ||
The synthesis commences with [3 + 2] dipolar cycloaddition between 17 and dimethyl acetylenedicarboxylate (DMAD) (41). At the completion of this reaction, in a one-pot fashion, DDQ is added to promote aromatization to furan 43 in 91% yield. Furan 43 was reduced to diol 44 with LiAlH4 in 84% yield on a gram scale. It was found that diol 44 could be directly hydrogenated to 46 with Pd/C under a hydrogen atmosphere, which proceeds through the intermediacy of furan 45, in 48% yield (35% yield over 4 steps). Unfortunately, the obtained 1H and 13C data of synthetic 46 did not match the natural product isolation values. This is due to misassignment of the isolated natural product as we observed C1 symmetry in the 1H NMR spectra of 46, which is characteristic of other known all-cisCL5a compounds.39 Although 46 was ultimately not a reported natural product, this route represents the shortest and most efficient to date to access all cis-CL5a scaffolds.36
Synthesis of arylnaphthalenes and their derivatives (CL3)
We wanted to take advantage of the oxidation state already present on CL5a (51) for the synthesis of aryltetralin natural products. Inspiration for the conversion of 51 into CL3 was reported by Haworth and co-workers (Scheme 6a), wherein they used an acid-mediated cyclization of bislactone 47, which is isoelectronic with 51, to synthesize dihydronaphthalene 48.40 Additionally, Hughes and Richie (Scheme 6b) demonstrated that galgravin (49), a CL5a natural product, undergoes rearrangement to form dihydronaphthalene 50 under acidic conditions.41 Seemingly, both 48 and 50 arise from pQM formation, Friedel–Crafts alkylation, and dehydration.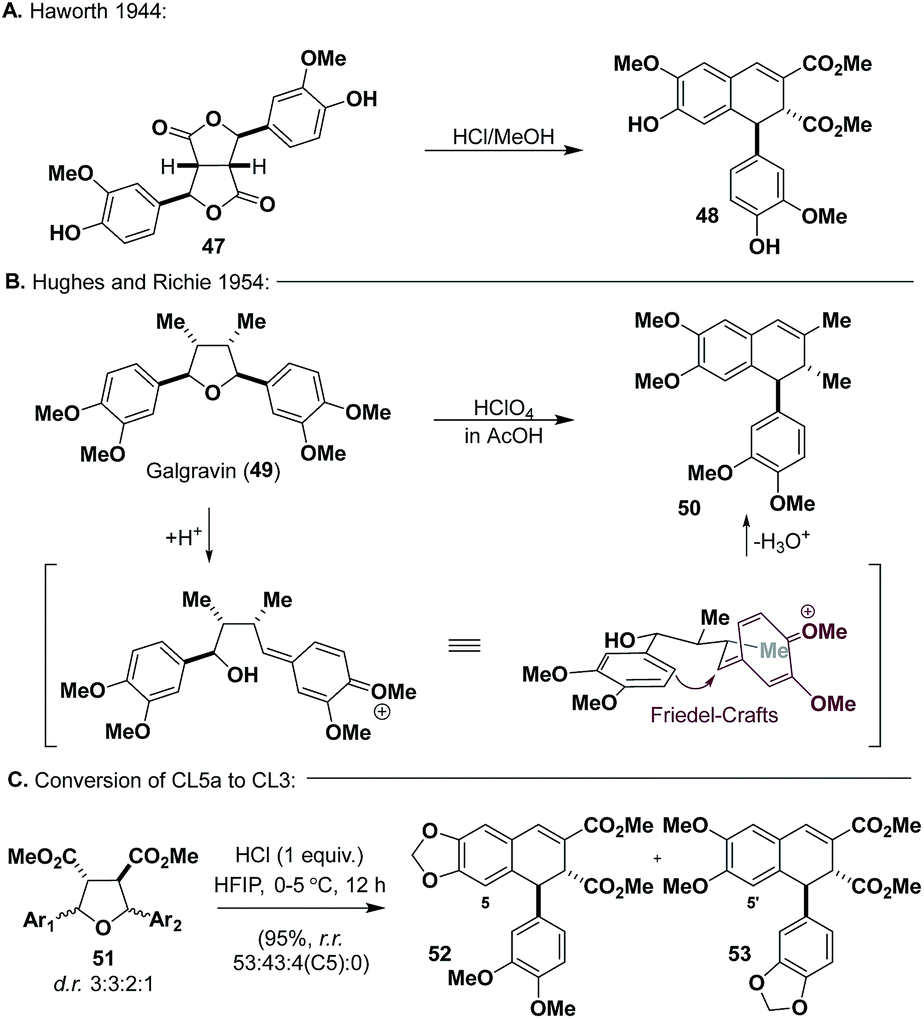 | ||
| Scheme 6 Instructive examples for conversion of CL5a to CL3. | ||
An extensive screening of Lewis and Brønsted acids19 revealed that HCl (1 equiv.) in HFIP could promote the desired reaction in high yields (Scheme 6c) (95%), affording 52 and 53 as the two major products (as single diastereomers) with trace amounts of a regioisomer of 52 (C5) from 51. Although inseparable through chromatography, it was eventually found that the mixture could be resolved through trituration with diethyl ether or after LiAlH4 reduction and flash chromatography. With access to both regioisomers secured, we proceeded to advance them to known aryl tetralin natural products, principally, (±)-galcatin (55) (from 52) and (±)-lintetralin (57) (from 53) (Scheme 7).
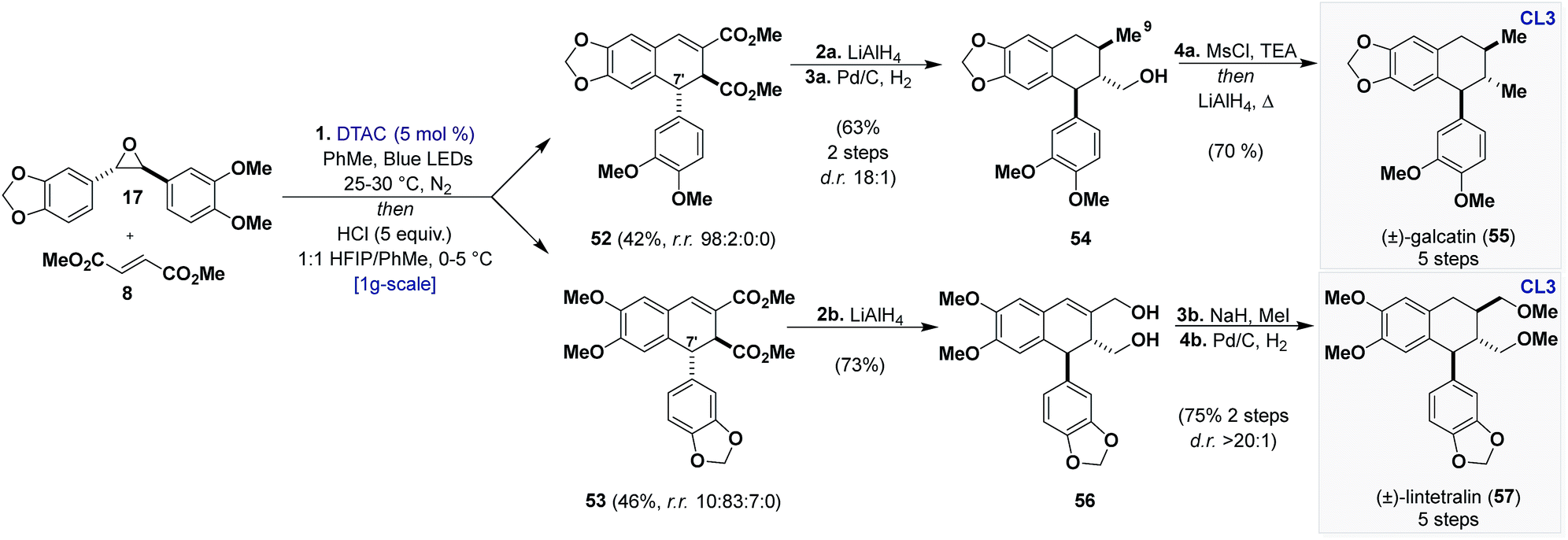 | ||
| Scheme 7 Total synthesis of aryltetralin (CL3) natural products (±)-galcatin (55) and (±)-lintetralin (57). Reagents and conditions: (1) DTAC (5 mol%), 17 (1 equiv.), 8 (3 equiv.), PhMe, Blue LEDs, 25–30 °C, N2, 5 days, then HCl (5 equiv.), 1:1 HFIP/PhMe, 0–5 °C, 36 h. (2a) LiAlH4 (6 equiv.), THF, 0 °C, 1 h. (3a) Pd/C (10 mol%), EtOH, H2 (balloon), 23 °C, 12 h. (4a) MsCl (1.5 equiv.), TEA (1.5 equiv.), THF, 0 to 23 °C, 12 h then LiAlH4 (6 equiv.), Δ, 1 h. (2b) LiAlH4 (6 equiv.), THF, 0 °C, 1 h. (3b) MeI (10 equiv.), NaH (5 equiv.), THF, 0 to 23 °C, 24 h. (4b) Pd/C (10 mol%), EtOAc, H2 (balloon), 23 °C, 12 h. | ||
We developed a one-pot procedure beginning with cycloaddition between 17 and 8, followed by addition of HFIP and HCl (5 equiv.) at 0 °C, which virtually gave a 1:1 mixture of 52 and 53. Flash chromatography followed by trituration afforded 52 in 42% yield and with excellent selectivity (r.r. 98:2:0:0) and 53 in 46% yield and with good selectivity (r.r. 10:83:7:0).
Sequential reduction of 52 with LiAlH4 and Pd/C under a hydrogen atmosphere reduces the esters to alcohols and the endocyclic alkene to the tetralin core, the latter occurs on the face opposite of the C7′ aryl group. This furnished the all-trans tetralin 54 in 63% yield over 2 steps with high selectivity (d.r. 18:1). Interestingly, the reduction of the alkene is also accompanied by deoxygenation of the allylic alcohol (C9). Although not planned, this deoxygenation was welcomed, as early attempts of a global deoxygenation proved cumbersome. To complete the synthesis deoxygenation of 54 was achieved through in situ activation with MsCl followed by reduction with LiAlH4, providing (±)-galcatin (55) in 5 steps in an overall 33% yield.42 Reduction of 53 with LiAlH4 gave diol 56 in 73% yield as a single regioisomer after flash chromatography. Successive methylation (MeI) and hydrogenation with Pd/C in EtOAc gave (±)-lintetralin (57) in 75% yield over 2 steps (45% overall yield over 5 steps).43
Application to the synthesis of heterolignans
To demonstrate a final application of this unified synthetic platform we sought to synthesize CL scaffolds bearing unnatural functionality. This is particularly difficult for most synthetic strategies because they rely on the reactivity of electron-rich aryl groups to access crucial intermediates. Pinerosinol (58) and isolariciresinol (60) are two CLs that have shown promising biological activities but both have also been shown to be metabolically unstable to human fecal microflora, affording enterolactone (59) and metabolite (61), respectively (Scheme 8a).44 A reasonable modification to improve their metabolic stability would be replacement of the oxygen appendages on the aryl groups with fluorine.45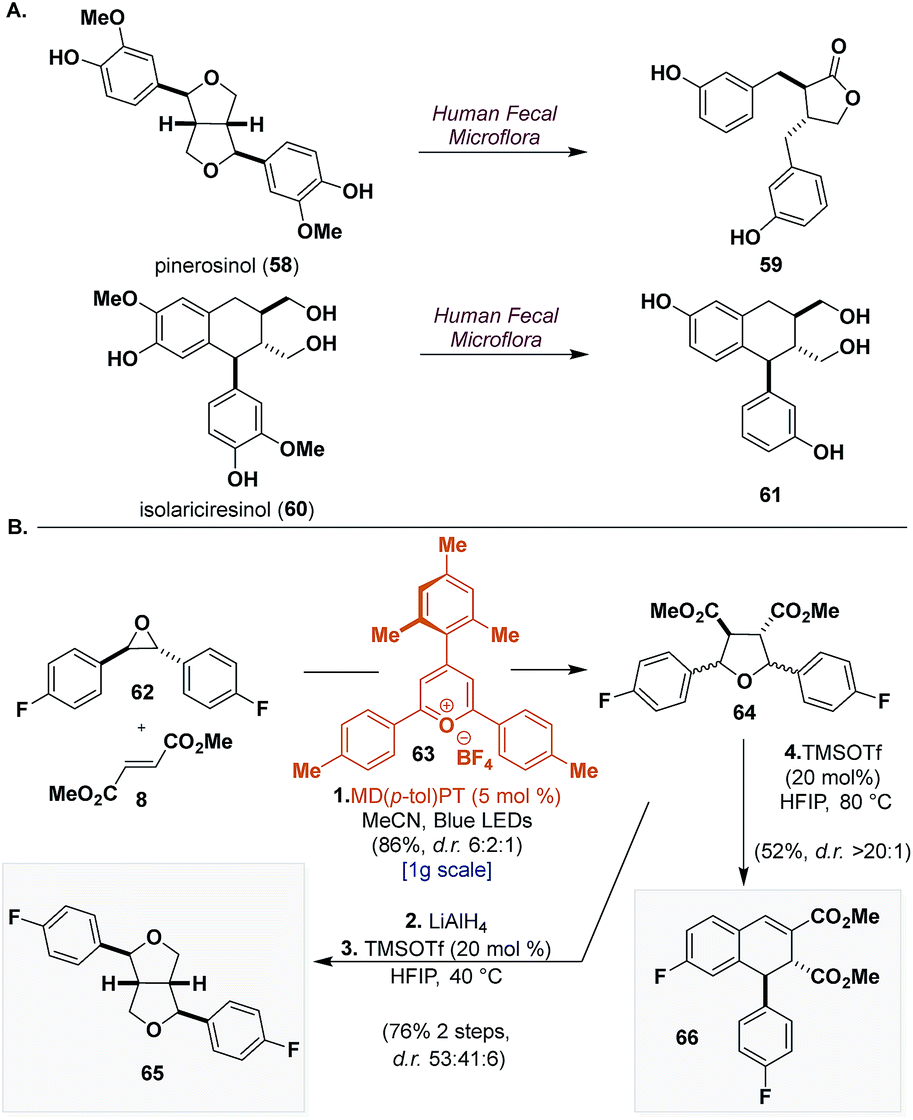 | ||
| Scheme 8 Application of the outlined synthetic blueprints to the synthesis of fluorinated heterolignans. Reagents and conditions: (1) MD(p-tol)PT (5 mol%), 62 (1 equiv.), 8 (1.2 equiv.), MeCN, Blue LEDs, 25–30 °C, N2, 12 h. (2) LiAlH4 (6 equiv.), THF, 0 to 23 °C, 1 h. (3) TMSOTf (20 mol%), HFIP, 40 °C, 24 h. (4) TMSOTf (20 mol%), HFIP, 80 °C, 24 h. | ||
To this end (Scheme 8b), using 4-mesityl-2,6-di-p-tolylpyrylium tetrafluoroborate (MD(p-tol)PT) (63) (λmax = 420 nm, [24+*/24˙] = +2.27 V vs. SCE), a catalyst that was identified in our earlier work that can generate electron-deficient carbonyl ylides,10 we can submit 62 to [3 + 2] dipolar cycloaddition with dimethyl fumarate (8) and isolate 64 in 86% yield as a mixture of inconsequential diastereomers (d.r. 6:2:1). LiAlH4 reduction and TMSOTf (20 mol%) catalyzed cyclization in HFIP of 64 furnish 65 in 76% yield over 2 steps. Although not selective (d.r. 53:41:6), the two major compounds can be easily separated providing two new notable analogues of the furofuran scaffold. Lastly, from 64 under similar acidic conditions but with elevated temperatures, we can induce rearrangement to form dihydronaphthalene 66 in 52% yield and with excellent selectivity (d.r. > 20:1). These molecules exemplify the modularity of our strategy to access CLs and demonstrate how our synthetic blueprints are suited to access natural product scaffolds possessing unnatural functionality.
Conclusion
We have identified short synthetic routes to all subtypes of the CL family of natural products from common building blocks. Key to the success of our strategy was the identification of a highly convergent [3 + 2] dipolar cycloaddition that is catalyzed by DTAC, a catalyst that was unknown for photoredox applications. Through the use of UV-Vis spectroscopy it was demonstrated that DTAC engages electron-rich epoxides through an outer-sphere SET by steric inhibition of a CT complex, which would otherwise funnel the reaction pathway through an unproductive inner-sphere SET. Finally, our synthetic strategies to access each subtype of CLs in a modular and convergent manner should make these molecules more available for SAR investigation. Such studies will be more effective due to their ability to introduce unnatural functionality, such as fluorine, which would be challenging using preexisting routes.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 5th July 2019, which contained errors in Scheme 4 and 5.Acknowledgements
Financial support from Boston University and the National Science Foundation (ABB CHE-1555300) is gratefully acknowledged. E. A. thanks Dr Felix S. Alfonso, Dr Randolph A. Escobar, and Dr Gabrielle Fleming for helpful discussions. We thank Dr Norman Lee (Boston University) and Dr Jeffrey Bacon (Boston University) for high-resolution mass spectrometry data and crystallographic data and analyses, respectively. NMR (CHE-0619339) and MS (CHE-0443618) facilities at Boston University are supported by the NSF.Notes and references
- For leading reviews, see: (a) R. D. Haworth, J. Chem. Soc., 1942, 448–456 RSC; (b) D. A. Whiting, Nat. Prod. Rep., 1985, 2, 191–211 RSC; (c) D. A. Whiting, Nat. Prod. Rep., 1987, 4, 499–525 RSC; (d) D. A. Whiting, Nat. Prod. Rep., 1990, 7, 349–364 RSC; (e) R. S. Ward, Nat. Prod. Rep., 1993, 10, 1–28 RSC; (f) R. S. Ward, Nat. Prod. Rep., 1995, 12, 183–205 RSC; (g) R. S. Ward, Nat. Prod. Rep., 1999, 16, 75–96 RSC; (h) M. Saleem, H. J. Kim, M. S. Ali and Y. S. Lee, Nat. Prod. Rep., 2005, 22, 696–716 RSC; (i) J.-Y. Pan, S.-L. Chen, M.-H. Yang, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251–1292 RSC; (j) R. B. Teponno, D. Kusari and M. Spiteller, Nat. Prod. Rep., 2016, 33, 1044 RSC.
- (a) H. F. Stähelin and A. von Wartburg, Cancer Res., 1991, 51, 5–15 Search PubMed; (b) M. Gordaliza, P. A. García, J. M. Miguel del Corral, M. A. Castro and M. A. Gómez-Zurita, Toxicon, 2004, 44, 441–459 CrossRef CAS PubMed; (c) J.-M. Lü, J. Nurko, S. M. Weakley, J. Jiang, P. Kougias, P. H. Lin, Q. Yao and C. Chen, Med. Sci. Monit., 2010, 16, RA93–R100 Search PubMed.
- For recent reviews of de novo synthesis of CLs, see: (a) X. Fang and X. Hu, Molecules, 2018, 23, 3385 CrossRef PubMed; (b) G. E. Magoulas and D. Papaioannou, Molecules, 2014, 19, 19769–19835 CrossRef PubMed.
- For selected biosynthetic approaches, see: (a) L. B. Davin, H.-B. Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362–367 CrossRef CAS PubMed; (b) W. Lau and E. Sattely, Science, 2015, 349, 1224–1228 CrossRef CAS PubMed; (c) B. Pickel, M.-A. Constantin, J. Pfannstiel, J. Conrad, U. Beifuss and A. Schalles, Angew. Chem., Int. Ed., 2010, 49, 202–204 CrossRef CAS PubMed.
- For selected strategies to access a single subtype, see: (a) C. P. Ting and T. J. Maimone, Angew. Chem., Int. Ed., 2014, 53, 3115–3119 CrossRef CAS PubMed; (b) J. A. Gaboury and M. P. Sibi, J. Org. Chem., 1993, 58, 2173–2180 CrossRef CAS; (c) D. J. Aldous, A. J. Dalençon and P. G. Steel, Org. Lett., 2002, 4, 1159–1162 CrossRef CAS PubMed; (d) D. Stadler and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 7557–7559 CrossRef CAS PubMed.
- For selected strategies that access multiple subtypes, see: (a) C. E. Rye and D. Barker, J. Org. Chem., 2011, 76, 6636–6648 CrossRef CAS PubMed; (b) S. C. Roy, K. K. Rana and C. Guin, J. Org. Chem., 2002, 67, 3242–3248 CrossRef CAS PubMed; (c) P. R. Jagtap, I. Císařová and U. Jahn, Org. Biomol. Chem., 2018, 16, 750–755 RSC; (d) A. van Oeveren, J. F. G. A. Jansen and B. L. Feringa, J. Org. Chem., 1994, 59, 5999–6007 CrossRef CAS; A. K. F. Albertson and J.-P. Lumb, Angew. Chem., Int. Ed., 2015, 54, 1–6 Search PubMed; (e) R. Ahmed, F. G. Schreiber and R. Stevenson, Tetrahedron, 1976, 32, 1339–1344 CrossRef CAS; (f) W. Li, J. Liu, J. Xu, P. Zang, Q. Liu and W. Li, Eur. J. Org. Chem., 2014, 3475–3482 CrossRef CAS; (g) Q. Wang, Y. Yang, Y. Li, W. Yu and Z. J. Hou, Tetrahedron, 2006, 62, 6107–6112 CrossRef CAS.
- (a) L. B. Davin and N. G. Lewis, Phytochem. Rev., 2003, 2, 257–288 CrossRef CAS; (b) S. Suzuki and T. Umezawa, J. Wood Sci., 2007, 53, 273–284 CrossRef CAS.
- L. B. Davin and N. G. Lewis, Curr. Opin. Biotechnol., 2005, 16, 398–406 CrossRef CAS PubMed.
- (a) P. Clawson, P. M. Lunn and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1990, 153–157 RSC; (b) A. Albini and D. R. Arnold, Can. J. Chem., 1978, 56, 2985–2993 CrossRef CAS.
- E. Alfonzo, F. S. Alfonso and A. B. Beeler, Org. Lett., 2017, 19, 2989–2992 CrossRef CAS PubMed.
- See ESI† for a summary of catalyst and reaction conditions explored.
- For characterization of DTAC (12) see ESI.†.
- Previous reports that have used DTAC in different contexts: (a) M. A. Reddy, A. Thomas, K. Srinivas, V. J. Rao, K. Bhanuprakash, B. Sridhar, A. Kumar, M. N. Kamalasanan and R. Srivastava, J. Mater. Chem., 2009, 19, 6172–6184 RSC; (b) J. G. Gillmore, J. D. Neiser, K. A. McManus, Y. Roh, G. W. Dombrowski, T. G. Brown, J. P. Dinnocenzo, S. Farid and D. R. Robello, Macromolecules, 2005, 38, 7684–7694 CrossRef CAS; (c) Y. Wang, O. Haze, J. P. Dinnocenzo and S. Farid, J. Org. Chem., 2007, 72, 6970–6981 CrossRef CAS PubMed.
- See ESI† for details. For CT-complex formation, see: (a) S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed; (b) C. G. S. Lima, T. d. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (c) R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- (a) F. D. Lewis, R. E. Dykstra, I. R. Gould and S. Farid, J. Phys. Chem., 1988, 92, 7042–7043 CrossRef CAS; (b) For examples of steric inhibition of the CT complex, see: R. Rathore, S. V. Lindeman and J. K. Kochi, J. Am. Chem. Soc., 1997, 119, 9393–9404 CrossRef CAS.
- A. Pelter, R. S. Ward, E. V. Rao and K. V. Sastry, Tetrahedron, 1976, 32, 2783–2788 CrossRef CAS.
- (a) M. Beroza and M. S. Schechter, J. Am. Chem. Soc., 1956, 78, 1242–1247 CrossRef CAS; (b) M. K. Syed, C. Murray and M. Casey, Eur. J. Org. Chem., 2014, 5549–5556 CrossRef CAS; (c) Y. Kawabe, R. Ishikawa, Y. Akao, A. Yoshida, M. Inai, T. Asakawa, Y. Hamashima and T. Kan, Org. Lett., 2014, 16, 1976–1979 CrossRef CAS PubMed.
- E. Alfonzo, J. W. L. Mendoza and A. B. Beeler, Beilstein J. Org. Chem., 2018, 14, 2308–2312 CrossRef CAS PubMed.
- See ESI† for details.
- W. M. Pearlman, Tetrahedron Lett., 1967, 8, 1663–1664 CrossRef.
- P. Satyanarayana and S. Venkateswarlu, Tetrahedron, 1991, 47, 8931–8940 CrossRef CAS.
- M. J. Kato, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1992, 31, 283–287 CrossRef CAS.
- R. S. Ward and A. Pelter, Tetrahedron, 1991, 47, 1275–1284 CrossRef.
- C.-C. Chen, W.-C. Hsin and Y.-L. Huang, J. Nat. Prod., 1998, 61, 227–229 CrossRef CAS PubMed.
- (a) R. Krithika, R. Mohankumar, R. J. Verma, P. S. Shrivastav, I. L. Mohamad, P. Gunasekaran and S. Narasimhan, Chem.-Biol. Interact., 2009, 181, 351–358 CrossRef CAS PubMed; (b) A. R. Chopade and F. J. Sayyad, Phytother. Res., 2015, 29, 1202–1210 CrossRef CAS PubMed.
- Terameprocol in Treating Patients with Recurrent High Grade Glioma, https://clinicaltrials.gov/ct2/show/NCT02575794, accessed May 24, 2019 Search PubMed.
- (a) Y.-M. Xia, X.-L. Liang, X.-L. Wang, X.-P. Cao and X.-F. Pan, Chin. J. Chem., 2003, 12, 1540–1542 Search PubMed; (b) M. Ito, A. Osaku, A. Shiibashi and T. Ikariya, Org. Lett., 2007, 9, 1821–1824 CrossRef CAS PubMed.
- X. Xie and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3767–3770 CrossRef CAS PubMed.
- (a) S. Duan, S. Huang, J. Gong, Y. Shen, L. Zeng, Y. Feng, W. Ren, Y. Leng and Y. Hu, ACS Med. Chem. Lett., 2015, 6, 386–391 CrossRef CAS PubMed; (b) M. L. A. e Silva, et al. , Bioorg. Med. Chem. Lett., 2017, 27, 176–179 CrossRef PubMed.
- D. Planchenault, R. Dhal and J.-P. Robin, Tetrahedron, 1993, 49, 5823–5830 CrossRef CAS.
- R. S. Ward and A. Pelter, Tetrahedron, 1991, 47, 1275–1284 CrossRef.
- S. M. H. Ayoub and D. G. I. Kingston, J. Nat. Prod., 1984, 47, 875–876 CrossRef CAS.
- Z. Lin-Gen, O. Seligmann, H. Lotter and H. Wagner, Phytochemistry, 1983, 22, 265 CrossRef CAS.
- C. A. Morales-Rivera, P. E. Floreancig and P. Liu, J. Am. Chem. Soc., 2017, 139, 17935–17944 CrossRef CAS PubMed.
- R. S. Ward and D. D. Hughes, Tetrahedron, 2001, 57, 2057–2064 CrossRef CAS.
- D. Soorukram, M. Pohmakotr, C. Kuhakarn and V. Reutrakul, Synthesis, 2018, 50, 4746–4764 CrossRef CAS.
- (a) K. Harada, M. Kubo, H. Horiuchi, A. Ishii, T. Esumi, H. Hioki and Y. Fukuyama, J. Org. Chem., 2015, 80, 7076–7088 CrossRef CAS PubMed; (b) K. Harada, K. Zaha, R. Bando, R. Irimaziri, M. Kubo, Y. Koriyama and Y. Fukuyama, Eur. J. Med. Chem., 2018, 148, 86–94 CrossRef CAS PubMed.
- A. Gonzáles-Coloma, P. Escoubas, J. Mizutani and L. Lajide, Phytochemistry, 1994, 35, 607–610 CrossRef.
- (a) A. M. Rimando, J. M. Pezzuto and N. R. Farnsworth, J. Nat. Prod., 1994, 57, 896–904 CrossRef CAS PubMed; (b) C. S. Ramos, H. V. Linnert, M. M. de Moraes, J. H. do Amaral, L. F. Yamaguchi and M. J. Kato, RSC Adv., 2017, 7, 46932–46937 RSC; (c) Y. Shen, P.-F. Yang, G. Yang, W.-L. Chen and Z. Chai, Org. Biomol. Chem., 2018, 16, 2688–2696 RSC.
- N. J. Cartwright and R. D. Haworth, J. Chem. Soc., 1944, 535–537 RSC.
- G. K. Hughes and E. Ritchie, Aust. J. Chem., 1954, 7, 104–112 CrossRef CAS.
- (a) J.-S. Liu, M.-F. Huang, Y.-L. Gao and J. A. Findlay, Can. J. Chem., 1981, 59, 1680–1684 CrossRef CAS; (b) J. C. T. Reddel, K. E. Lutz, A. B. Diagne and R. J. Thomson, Angew. Chem., Int. Ed., 2014, 53, 1395–1398 CrossRef CAS PubMed.
- (a) P. Satyanarayana, P. Subrahmanyam, K. N. Viswanatham and R. S. Ward, J. Nat. Prod., 1988, 51, 44–49 CrossRef CAS; (b) D. Enders, G. Del Signore and O. M. Berner, Chirality, 2003, 15, 510–513 CrossRef CAS PubMed.
- S. Heinonen, T. Nurmi, K. Liukkonen, K. Poutanen, K. Wähälä, T. Deyema, S. Nishibe and H. Adlercreutz, J. Agric. Food Chem., 2001, 49, 3178–3186 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1920090. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02682g |
| This journal is © The Royal Society of Chemistry 2019 |