
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual photoredox-nickel strategy for remote functionalization via iminyl radicals: radical ring-opening-arylation, -vinylation and -alkylation cascades†
Elizabeth M.
Dauncey‡
 a,
Shashikant U.
Dighe‡
a,
James J.
Douglas
b and
Daniele
Leonori
*a
a,
Shashikant U.
Dighe‡
a,
James J.
Douglas
b and
Daniele
Leonori
*a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: daniele.leonori@manchester.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, AstraZeneca, R&D, Macclesfield SK10 2NA, UK
First published on 27th June 2019
Abstract
A divergent strategy for the remote arylation, vinylation and alkylation of nitriles is described. These processes proceed through the photoredox generation of a cyclic iminyl radical and its following ring-opening reaction. The distal nitrile radical is then engaged in nickel-based catalytic cycles to form C–C bonds with aryl bromides, alkynes and alkyl bromides.
The selective functionalization of unactivated sp3 carbons streamlines access to molecules that can be difficult to prepare using classical disconnections.1 Radical strategies are powerful platforms to achieve this goal owing to the ability of odd-electron species to undergo fast transpositions by σ-bond cleavage.2 Processes based on 1,5-H-atom transfer (1,5-HAT) are frequently adopted as they allow selective functionalization at precise carbon sites in already assembled substrates.3 Methodologies centred on radical ring-opening reactions4 lead to skeletal rearrangements and as a result use different classes of starting materials. Most importantly they can functionalize sp3 carbons that are elusive by HAT-based protocols.
Following the pioneering work of Forrester5 and Zard,6 we have recently developed a photo-induced methodology where readily prepared oximes undergo SET (single-electron transfer) oxidation and fragmentation en route to cyclic iminyl radicals (Scheme 1A).7 With the correct juxtaposition of α-substituents and ring-size, efficient ring opening-functionalization cascades were developed accessing remotely fluorinated, chlorinated and azidated nitriles.
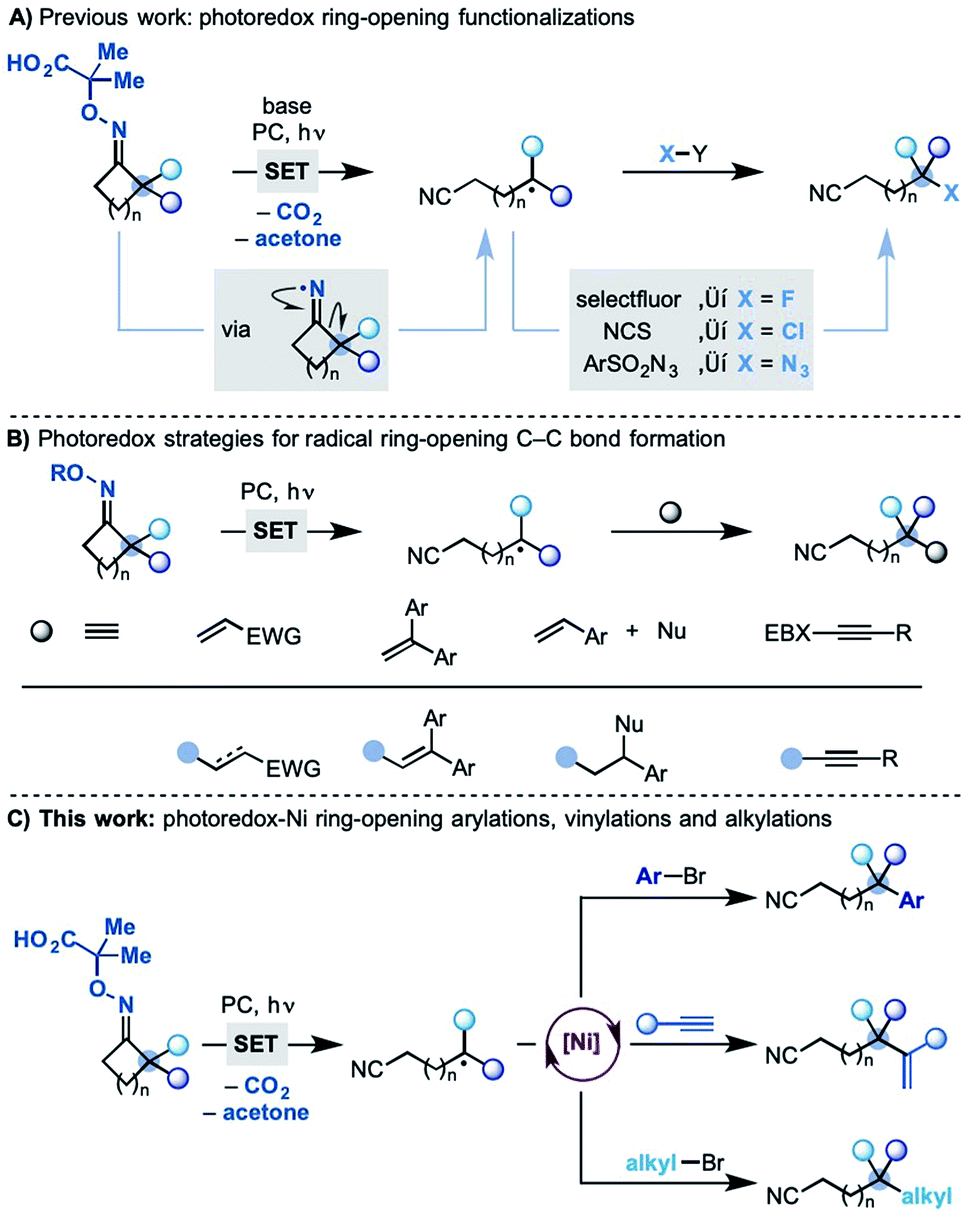 | ||
| Scheme 1 Strategies for the photo-induced ring-opening functionalizations of cyclic iminyl radicals. (A) Previous work: photoredox ring-opening functionalizations. (B) Photoredox strategies for radical ring-opening C–C bond formation. (C) This work: photoredox-Ni ring-opening arylations, vinylations and alkylations. | ||
Other groups have also been active in the area and several photo-induced strategies have been subsequently developed enabling radical ring-opening followed by reaction with, most notably, Michael acceptors,8 styrenes8c,9 and alkynylbenziodoxolone reagents10 (Scheme 1B).11 These methodologies have provided significant synthetic capacity as they enabled distal C–C bond formations, albeit with structurally specific coupling partners.
Despite these advances, the possibility to use related radical transpositions as a tool for the general and modular construction of C–C bonds at remote sites is still an unmet goal.
We speculated that one way to partially address this fundamental challenge would be to incorporate the concept of metallaphotoredox catalysis12 in these radical transpositions. In principle, this strategy would enable the use of readily available coupling partners, like aryl/alkyl bromides and alkynes, and access products currently elusive by other methodologies. In this paper, we demonstrate the successful development of a divergent dual photoredox–nickel process that gives access to remotely arylated, vinylated and alkylated nitriles by means of radical ring-opening–cross coupling cascades (Scheme 1C).
The proposed mechanism for this divergent strategy was centred on our previously developed reductive quenching photoredox cycle for iminyl radical generation.13 As shown in Scheme 2, we would use a visible-light excited photocatalyst (*PC) to promote the SET oxidation of oxime-carboxylate A. Following extrusion of CO2 and acetone, the iminyl radical B should undergo facile ring-opening delivering the distal nitrile radical C. At this point, we hoped that a Ni(0) co-catalyst would simultaneously undergo oxidative addition on an aryl bromide coupling partner to give an aryl–Ni(II) species D.14 Radical transmetalation between C and D ought to be possible thus delivering an aryl,alkyl–Ni(III) complex E from which reductive elimination is facile. This step would generate the remotely arylated nitrile F and a Ni(I) species. A final SET between the reduced photocatalyst (PC˙−) and the Ni(I)-complex (Ered Ni(II)/Ni(0) = −1.2 V vs. SCE, DMF)15 would re-initiate both the Ni and the photoredox cycle. Related mechanistic frameworks should enable the use of alkynes and alkyl halides and therefore allow the remote installation of vinyl and alkyl groups respectively.
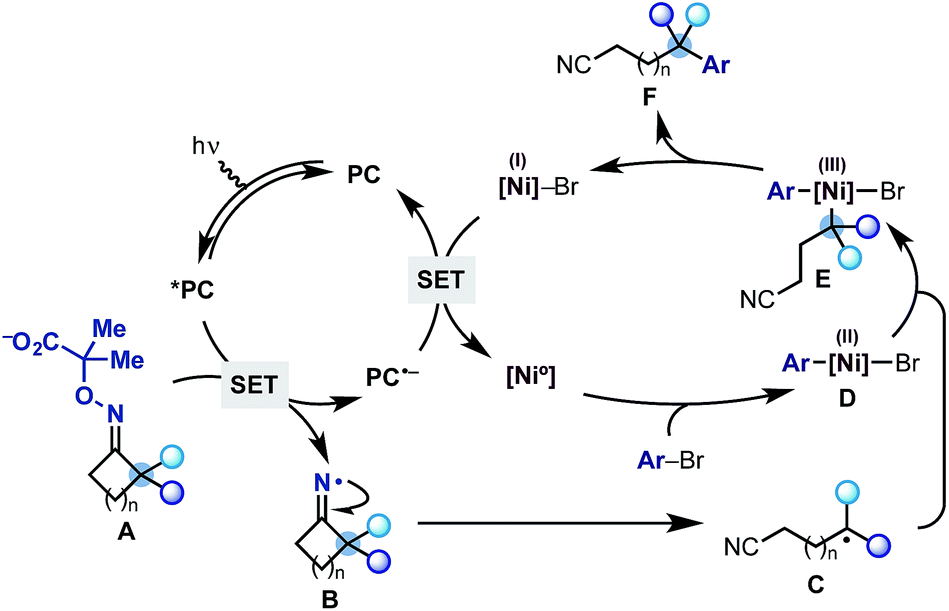 | ||
| Scheme 2 Proposed dual photoredox-Ni catalytic cycle for the radical ring-opening-arylation cascades. | ||
We started our investigation using the cyclobutanone-oxime 1 (one-step preparation on gram-scale)16 and p-Br-acetophenone 2 as the coupling partner (Scheme 3). Pleasingly, using the Ir-photocatalyst PC1, NiCl2·glyme, dtbbpy ligand and K2CO3 in DME under blue light irradiation, we obtained the desired product 3 in 15% yield (entry 1). Different photocatalysts were evaluated and while Fukuzumi's acridinium (PC2) was not suitable (entry 2), the organic dye 4CzIPN (PC3) and the Ir-complex PC4 provided 3 in 24 and 41% yield respectively (entries 3 and 4). Using PC4 we tested different inorganic and organic bases and identified tetramethyl guanidine (TMG) as optimum (entries 4–8). The final elements of reaction optimization involved the evaluation of a selection of Ni-catalysts (entries 9–11) as well as solvents from which EtOAc resulted ideal (entries 12–14). Control experiments confirmed the requirement for all reaction components (entries 15–18) and a quantum yield Φ = 0.19 is in agreement with the observed requirement for continuous light irradiation.
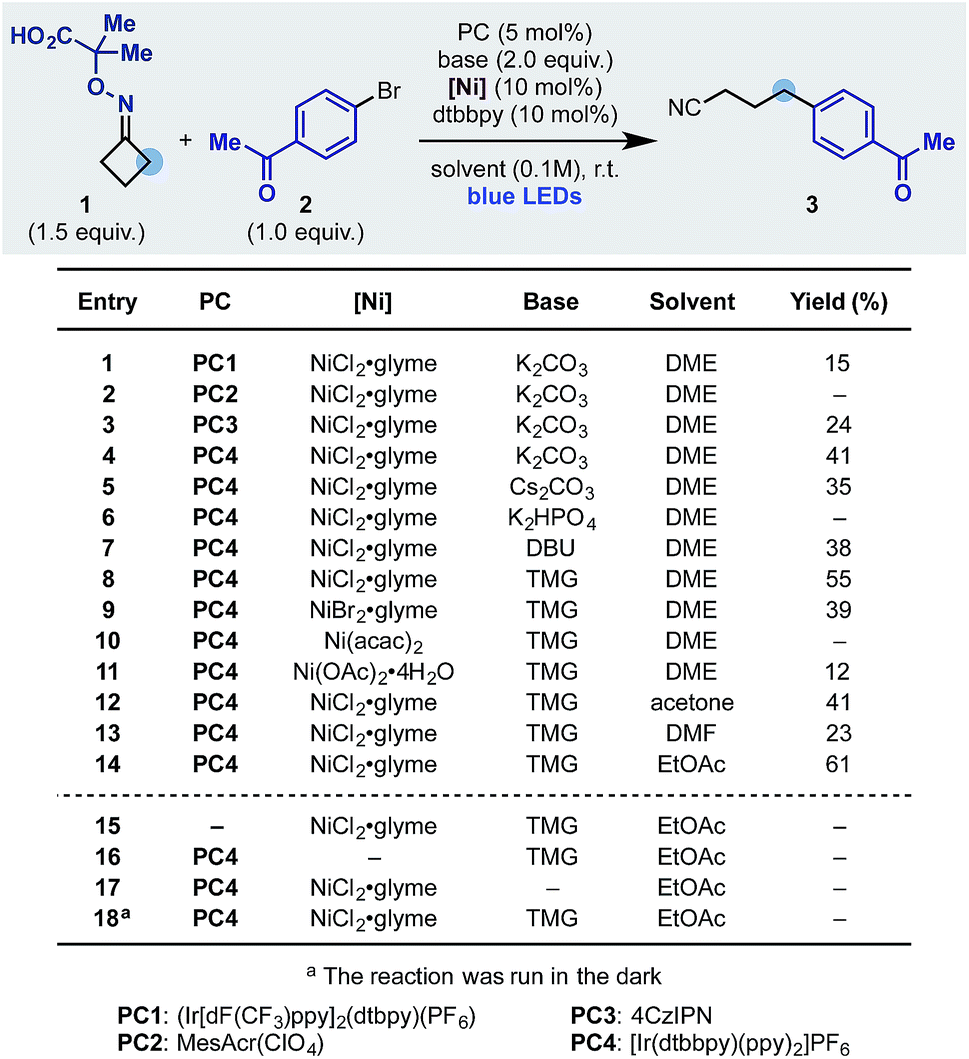 | ||
| Scheme 3 Development of dual photoredox–Ni ring-opening-arylation process. | ||
With these optimised reaction conditions, we evaluated the scope of the aromatic partner in conjunction with the cyclobutanone oxime 1 (Scheme 4). Pleasingly the process exhibited broad scope and tolerated a wide variety of common functionalities such as ketone (3 and 4), ester (5), nitrile (6), aldehyde (7), sulfone (8), trifluoromethyl (9) as well as groups that can be used as a handle for further functionalization like pinacol boronic ester (10) and aryl chloride (11) albeit in lower yields.17 Electron rich aryl bromides are a limitation of the protocol (12), possibly due to a more difficult oxidative addition process from the Ni(0) catalyst. Moreover, while meta-substituted aryl bromides were successfully engaged in this protocol (13 and 14), the presence of an ortho-substituent was detrimental to the reactivity and, for example, 15 was obtained in low yield. We then evaluated the use of hetero-aromatic coupling partners as these systems are frequently encountered in medicinal chemistry programs. Pleasingly the reaction enabled ring-opening-arylation with differentially functionalized pyridines at either C-2 (16 and 17) and C-3 (18), as well as quinoline (19), electron rich benzothiazole (20) and benzoimidazole (21) and Br–caffeine, which gave 22 in good yield.
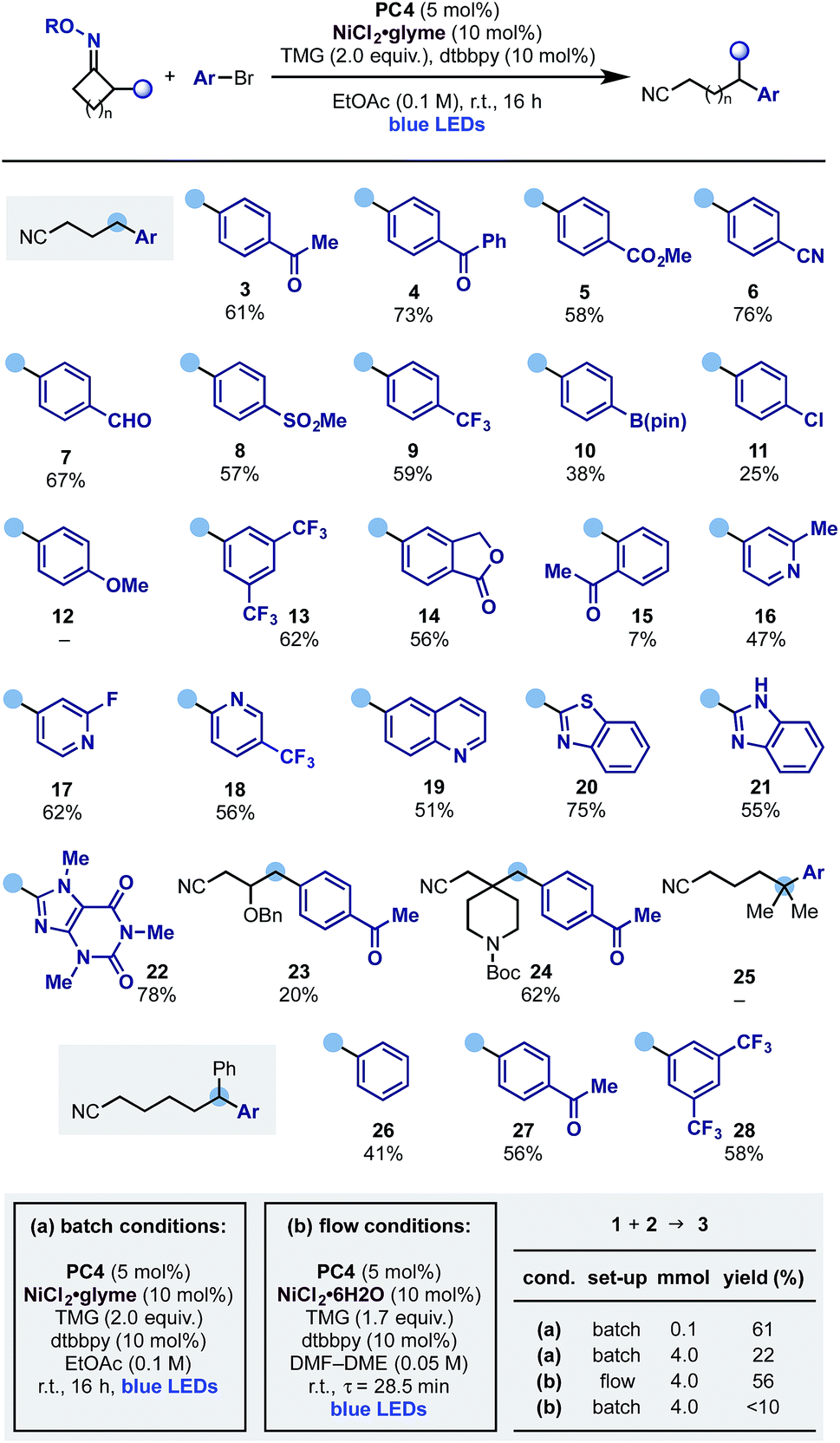 | ||
| Scheme 4 Scope of the dual photoredox–Ni ring-opening-arylation process. | ||
Other cyclobutanone oximes were tested and we used a substrate containing an OBn ether at C-3 (23) as well as a spirocyclic N-Boc-piperidine (24). This gave access to a C-4 benzylated piperidine, which is a common pharmacophoric unit in many NMDA antagonist drugs like ifenprodil.
As the ring-opening of cyclic iminyl radicals is thermodynamically feasible on larger rings with the correct juxtaposition of α-substituents,7 we evaluated the use of gem-dimethyl cyclopentanone and 2-Ph-cyclohexanone oxime starting materials. While the arylation cascade leading to 25 could not be implemented, we successfully extended it to the deconstruction of the six-membered ring and access the ε-di-arylated nitriles 26–28 in good to moderate yields. We propose the failure in obtaining 25 to be due to the known difficulty of tertiary C-radical to undergo transmetalation processes.
Flow chemistry has emerged as an effective solution to the scale up of visible light-mediated processes.18 Dual photoredox–nickel catalysis can be challenging to conduct in flow, due to the typically heterogenous mixtures and long reaction times. We initiated the attempted scale-up of the ring-opening-arylation cascade by developing homogenous reaction conditions amenable to continuous flow (Scheme 4B). This allowed the preparation of 3 on a useful preparative scale (4 mmol over 4 h) with yield comparable to the small-scale (0.1 mmol) batch reaction (56% in flow vs. 61% batch). The developed flow process compared favourably to the original batch conditions employed at large-scale. Using these without further optimization gave reduced yield with increasing scale after 18 h (0.1 mmol scale 59%, 2 mmol 45%, 4 mmol 22%). The use of the developed homogenous conditions in batch gave a comparable 52% yield (0.1 mmol scale), however at increased scale resulted in <10% yield, indicating the benefit of the flow process.19
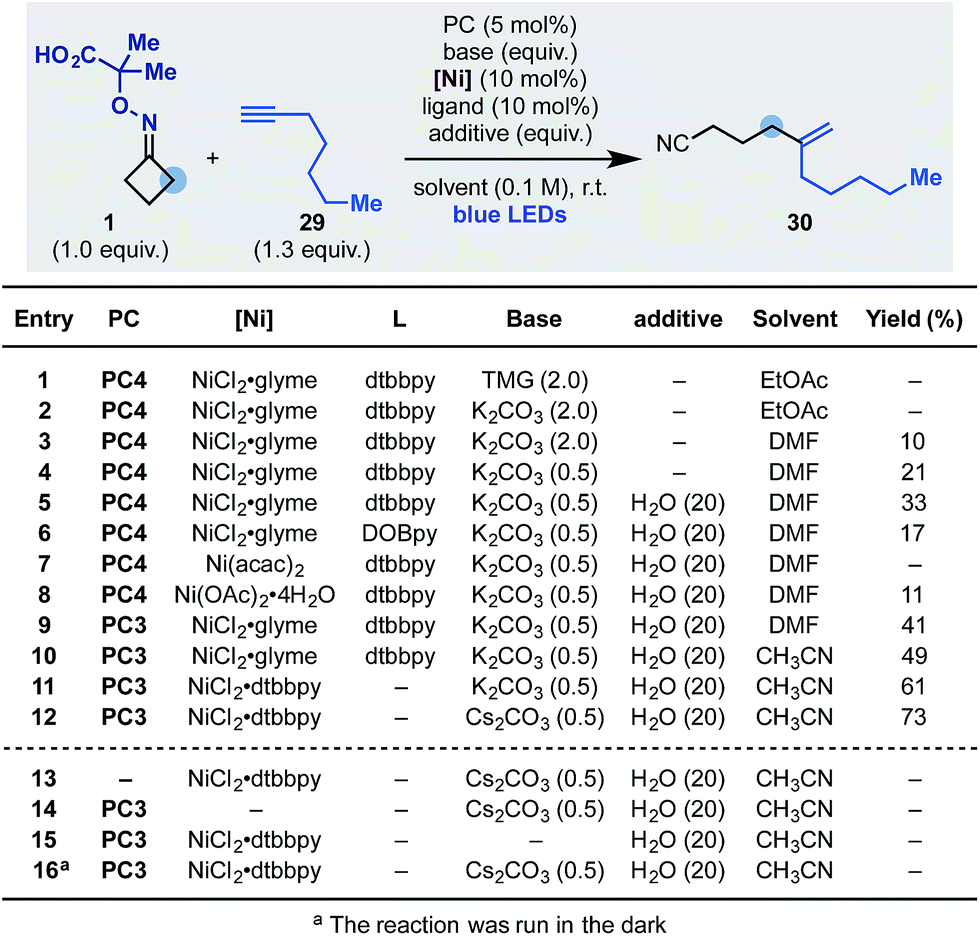
Having developed an oxidative cascade for ring-opening-arylation, we tested the feasibility of using terminal alkynes as coupling partners to construct gem-disubstituted olefins.20 The optimization of this process was performed using oxime 1 and 1-heptyne 29 (Scheme 5). In this case, conditions used for the arylation cascade failed to provide the desired product (entry 1) but, by switching the base to K2CO3 and the solvent to DMF, 30 was obtained in an encouraging 10% yield (entry 2 and 3). The efficiency of the process was improved to 33% by lowering the amount of base (entry 4) and adding 20 equiv. of H2O as an additive (entry 5). Other nickel–ligand combinations were tested but they provided 30 in generally lower yield, if any (entries 6–8). Evaluation of other photocatalysts and solvents identified PC3 (entry 9) and CH3CN (entry 10) as optimum. Finally, with the use of the preformed NiCl2·dttbpy catalyst and Cs2CO3 as the inorganic base, 30 was obtained in 73% yield (entries 11 and 12). Also in this case, control experiments confirmed the requirement for all reaction components as well as continuous blue-light irradiation (entries 13–16).
 | ||
| Scheme 5 Development of dual photoredox–Ni ring-opening-vinylation process. | ||
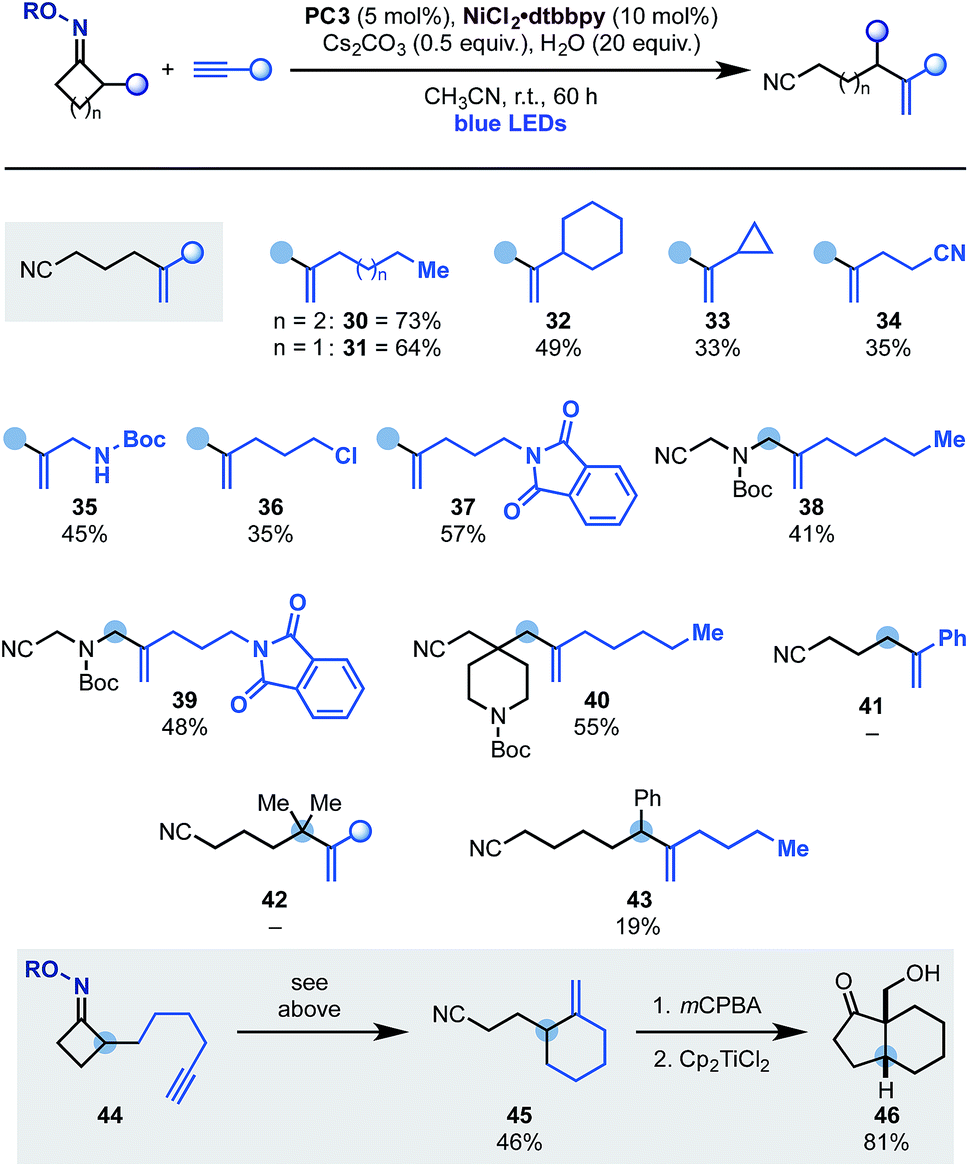
The scope of terminal alkynes that can be engaged in this reactivity pattern is illustrated in Scheme 6. Pleasingly, coupling partners containing linear (30 and 31) as well as cyclic (32) alkyl groups could be used including a substrate containing a cyclopropyl unit (33) albeit in lower yield.17 The methodology tolerated several important functionalities like nitrile (34), N-Boc-protected amine (35), alkyl chloride (36) and a phthalamide unit (37). Expansion of this chemistry to N-Boc-azetidone and a spirocyclic N-Boc-piperidine based oximes gave access to allylic (38 and 39) as well as a C-4 disubstituted piperidine (40), which are useful building blocks for further derivatization. Current limitations are the use of aryl-substituted alkynes (e.g.41) and also its expansion to higher ring-size iminyl precursors (42 and 43).
 | ||
| Scheme 6 Scope of the dual photoredox–Ni ring-opening-vinylation process. | ||
This radical ring-opening-vinylation strategy could also be performed in intra-molecular settings as demonstrated by the successful conversion of 44 into 45. Control experiments in the absence of the Ni-catalyst or H2O demonstrated that a radical ring-opening followed by 5-exo-dig and final H-abstraction was not operating,19 thus leaving the proposed dual photoredox–Ni-process as the likely pathway for the formation of 45. This reaction product is an interesting building block that, using literature methods, was converted into the bicyclic ketone 46 in good yield and as the single syn-diastereomer.
As this dual photoredox-Ni approach has enabled the construction of sp3–sp2 C–C bonds with both aromatic and vinyl substituents we questioned if alkyl halides could also be used and therefore achieve the very challenging assembly of sp3–sp3 C–C bonds.21 Indeed, remote alkylations via radical transposition are still very difficult to perform, with the exception of strategies involving the use of Michael acceptors as SOMOphiles.
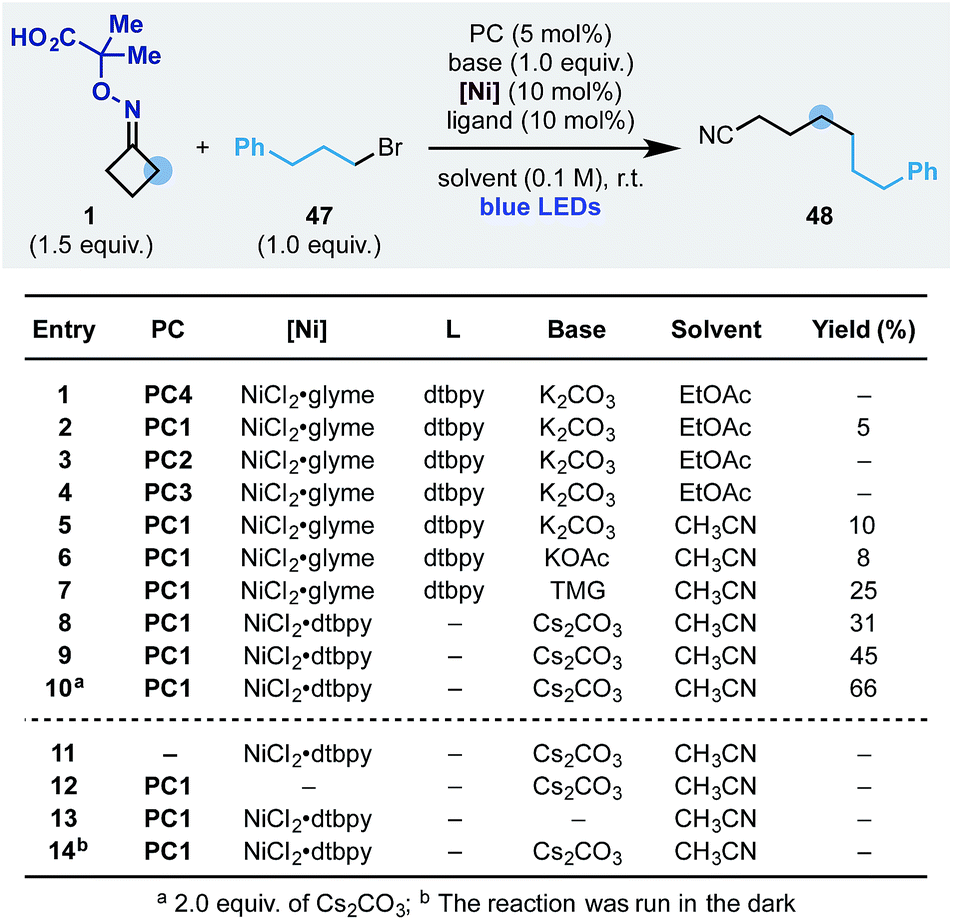
We started the optimization of this process using the oxime 1 and the simple alkyl bromide 47 (Scheme 7). Reaction conditions similar to the one developed for the ring-opening-arylation cascade did not provide the desired product (entry 1). However, evaluation of photoredox catalysts (entries 2–4), solvents (entry 5) and bases (entries 5–7) showed that the process could be achieved and 48 obtained in 25% yield using PC1 and Cs2CO3 in CH3CN (entry 9). Pleasingly, we have been able to increase the yield to 66% by adopting the preformed nickel catalyst NiCl2·dtbpy (entry 10) and by increasing the equivalents of base (entry 10). Also in this case, all reaction components as well as continuous blue-light irradiation were required (entries 11–14).
 | ||
| Scheme 7 Development of dual photoredox–Ni ring-opening-alkylation process. | ||
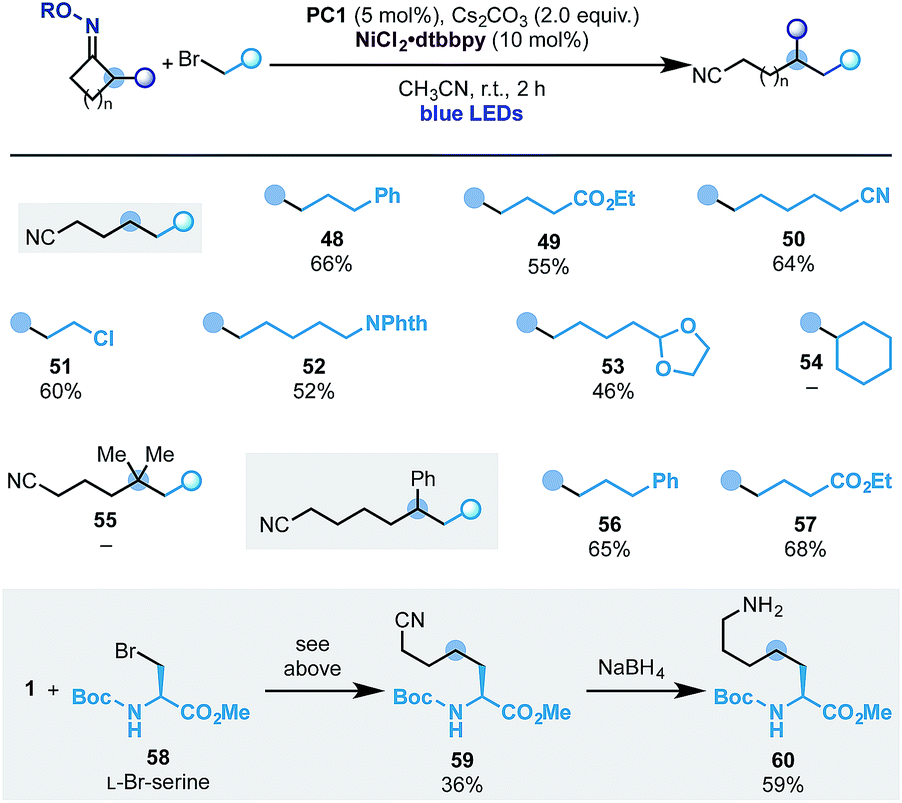
With this set of conditions in hand, we evaluated the scope of the process (Scheme 8). Pleasingly, using cyclobutanone oxime 1 we succeeded in engaging several primary alkyl bromides with different substitution patterns. This included ester (49), nitrile (50), alkyl chloride (51), N-phthalimide (52) and an acetal (53). At the moment this reactivity is limited to primary alkyl electrophiles as secondary ones (e.g.54) did not react.
 | ||
| Scheme 8 Scope of the dual photoredox–Ni ring-opening-alkylation process. | ||
In analogy to the ring-opening-arylation cascades we have not been able to engage the gem-dimethyl-cyclopentanone oxime (55) but we successfully used the 2-Ph-cyclohexanone starting material, which gave access to ε-alkylated nitriles 56 and 57 in good yield.
To showcase the potential of the methodology, we have used commercially available L-Br-serine 58 as the coupling partner. This functionalized building block provided access to the unnatural amino acid 59 that was converted into the 1-carbon elongated L-lysine analogue 60.22
Conclusions
In conclusion, we have reported the first example of a photoredox strategy where the ring-opening of iminyl radicals has been merged with Ni-catalysis. This divergent platform has enabled the development of distal arylation, vinylation and, for the first time, alkylation of nitrile-containing molecules. Current interest in our laboratory is towards rendering these processes asymmetric.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L. thanks EPSRC for a Fellowship (EP/P004997/1), and the European Research Council for a research grant (758427). E. M. D. thanks AstraZeneca for a PhD CASE Award. S. U. D. thanks the Marie Curie Actions for a Fellowship (791349). We thank S. Wells, T. Churchill and P. Gillespie (AstraZeneca Process Safety) for safety testing.Notes and references
- (a) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed; (b) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed.
- (a) P. Dowd and W. Zhang, Chem. Rev., 1993, 93, 2091 CrossRef CAS; (b) A. Gansäuer, T. Lauterbach and S. Narayan, Angew. Chem., Int. Ed., 2003, 42, 5556 CrossRef PubMed; (c) Z. Cekovic, J. Serb. Chem. Soc., 2005, 70, 287 CrossRef CAS; (d) M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034 CrossRef CAS PubMed; (e) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2017, 45, 62 Search PubMed.
- (a) C. G. Francisco, A. J. Herrera, A. Martin, I. Perez-Martin and E. Suarez, Tetrahedron, 2007, 48, 6384 CrossRef CAS; (b) C. G. Francisco, A. J. Herrera and E. Suarez, J. Org. Chem., 2003, 68, 1012 CrossRef CAS PubMed; (c) H. Zhang and K. Muniz, ACS Catal., 2017, 7, 4122 CrossRef CAS; (d) C. Martinez and K. Muniz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef CAS PubMed; (e) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed; (f) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- For a review, see: (a) S. P. Morcillo, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201905218. For selected examples, see: (b) S. Maity and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 9562 CrossRef CAS PubMed; (c) J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270 CrossRef CAS PubMed; (d) C. R. Pitts, M. S. Bloom, D. D. Bume, Q. A. Zhang and T. Lectka, Chem. Sci., 2015, 6, 5225 RSC; (e) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 4055 CrossRef CAS PubMed; (f) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 128, 15319 CrossRef PubMed; (g) H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef CAS PubMed; (h) J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 59, 12619 CrossRef PubMed.
- (a) A. R. Forrester, M. Gill, J. S. Sadd and R. H. Thomson, J. Chem. Soc., Perkin Trans. 1, 1979, 1, 612 RSC; (b) A. R. Forrester, M. Gill and R. H. Thomson, J. Chem. Soc., Chem. Commun., 1976, 677 RSC.
- (a) J. B. Boivin, E. Fouquet and S. Z. Zard, Tetrahedron, 1994, 50, 1745 CrossRef CAS; (b) J. Boivin, E. Fouquet, A.-M. Schiano and S. Z. Zard, Tetrahedron, 1994, 50, 1769 CrossRef CAS; (c) J. Boivin, E. Fouquet and S. Z. Zard, J. Am. Chem. Soc., 1991, 113, 1055 CrossRef CAS.
- E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 CrossRef CAS PubMed.
- (a) J.-F. Zhao, P. Gao, X.-H. Duan and L.-N. Guo, Adv. Synth. Catal., 2018, 360, 1775 CrossRef CAS; (b) J. Wu, J.-Y. Zhang, P. Gao, S.-L. Xu and L.-N. Guo, J. Org. Chem., 2018, 83, 1046 CrossRef CAS PubMed; (c) L. Li, H. Chen, M. Mei and L. Zhou, Chem. Commun., 2017, 53, 11544 RSC; (d) P.-Z. Wang, X.-Y. Yu, C.-Y. Li, B.-Q. He, J.-R. Chen and W.-J. Xiao, Chem. Commun., 2018, 54, 9925 RSC.
- (a) X.-Y. Yu, Q.-Q. Zhao, J. Chen, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505 CrossRef CAS PubMed; (b) S. Yao, K. Zhang, Q.-Q. Zhou, Y. Zhao, D.-Q. Shi and W.-J. Xiao, Chem. Commun., 2018, 54, 8096 RSC; (c) X. Shen, J.-J. Zhao and S. Yu, Org. Lett., 2018, 20, 5523 CrossRef CAS PubMed; (d) X.-Y. Yu, J.-R. Chen, P.-Z. Wang, M.-N. Yang, D. Liang and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738 CrossRef CAS PubMed.
- F. L. Vaillant, M. Garreau, S. Nicolai, G. Gryn'ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883 RSC.
- (a) H.-B. Yang and N. Selander, Chem.–Eur. J., 2017, 23, 1779 CrossRef CAS PubMed; (b) L. Yang, P. Gao, X.-H. Duan, Y.-R. Gu and L. N. Guo, Chem. Commun., 2018, 54, 10738 RSC; (c) J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161 RSC; (d) W. Ai, Y. Liu, Q. Wang, Z. Lu and Q. Liu, Org. Lett., 2018, 20, 409 CrossRef CAS PubMed; (e) D. Ding and C. Whang, ACS Catal., 2018, 8, 11324 CrossRef CAS.
- (a) J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (b) V. Corcé, L. M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414 CrossRef PubMed; (c) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (d) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361 CrossRef CAS PubMed.
- (a) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- M. Durandetti, M. Devaud and J. J. Perichon, New J. Chem., 1996, 20, 659 CAS.
- Starting material 1, and its precursor (aminooxy)-2-methylpropanoic acid hydrochloride, were subjected to safety testing and found not to be explosive. See ESI† for more information.
- In this case we have observed the formation of butyronitrile, possibly from radical ring-opening followed by H-atom abstraction from the solvent.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- See ESI† for more information.
- N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed.
- T. P. Boyle, J. B. Bremner, J. A. Coates, P. A. Keller and G. P. S, Tetrahedron, 2005, 61, 7271 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation. See DOI: 10.1039/c9sc02616a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |