
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Practical, metal-free remote heteroarylation of amides via unactivated C(sp3)–H bond functionalization†
Nana
Tang‡
a,
Xinxin
Wu‡
a and
Chen
Zhu
 *ab
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China. E-mail: chzhu@suda.edu.cn
bKey Laboratory of Synthesis Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Science, 345 Lingling Road, Shanghai 200032, China
First published on 11th June 2019
Abstract
Development of practical methods for the production of multi-functionalized amides is one of the most important topics in both synthetic chemistry and drug discovery. Disclosed herein is a new, efficient, site-selective heteroarylation of amides via C(sp3)–H bond functionalization. Amidyl radicals are directly generated from the amide N–H bonds under mild conditions, which trigger the subsequent 1,5-HAT process. A wide scope of aliphatic amides including carboxamides, sulfonamides, and phosphoramides are readily modified at remote C(sp3)–H bonds by installing diverse heteroaryl groups. Borne out of pragmatic consideration, this protocol can be used for the late-stage functionalization of amides.
Introduction
As a class of paramount chemical structures, amides consisting of carboxamides, sulfonamides, and phosphoramides are ubiquitous in natural and synthetic products (Scheme 1a). They constitute the basic framework of proteins in the form of peptides in life systems, and are engaged as key candidates for structure design in drug development. An analysis reveals that amide formation is one of the most frequently occurring reactions used in current medicinal chemistry.1 Generally, amides are readily obtained from the condensation of amines with carboxylic/sulfonic/phosphoric acids and derivatives (Scheme 1b, path a). Alternatively, direct installation of the target functional group at the aliphatic chain of amides via C(sp3)–H bond functionalization provides an ingenious and powerful access to the multi-functionalized amides (Scheme 1b, path b),2 in particular when the complex alkylamine precursors are inaccessible for the condensation approach.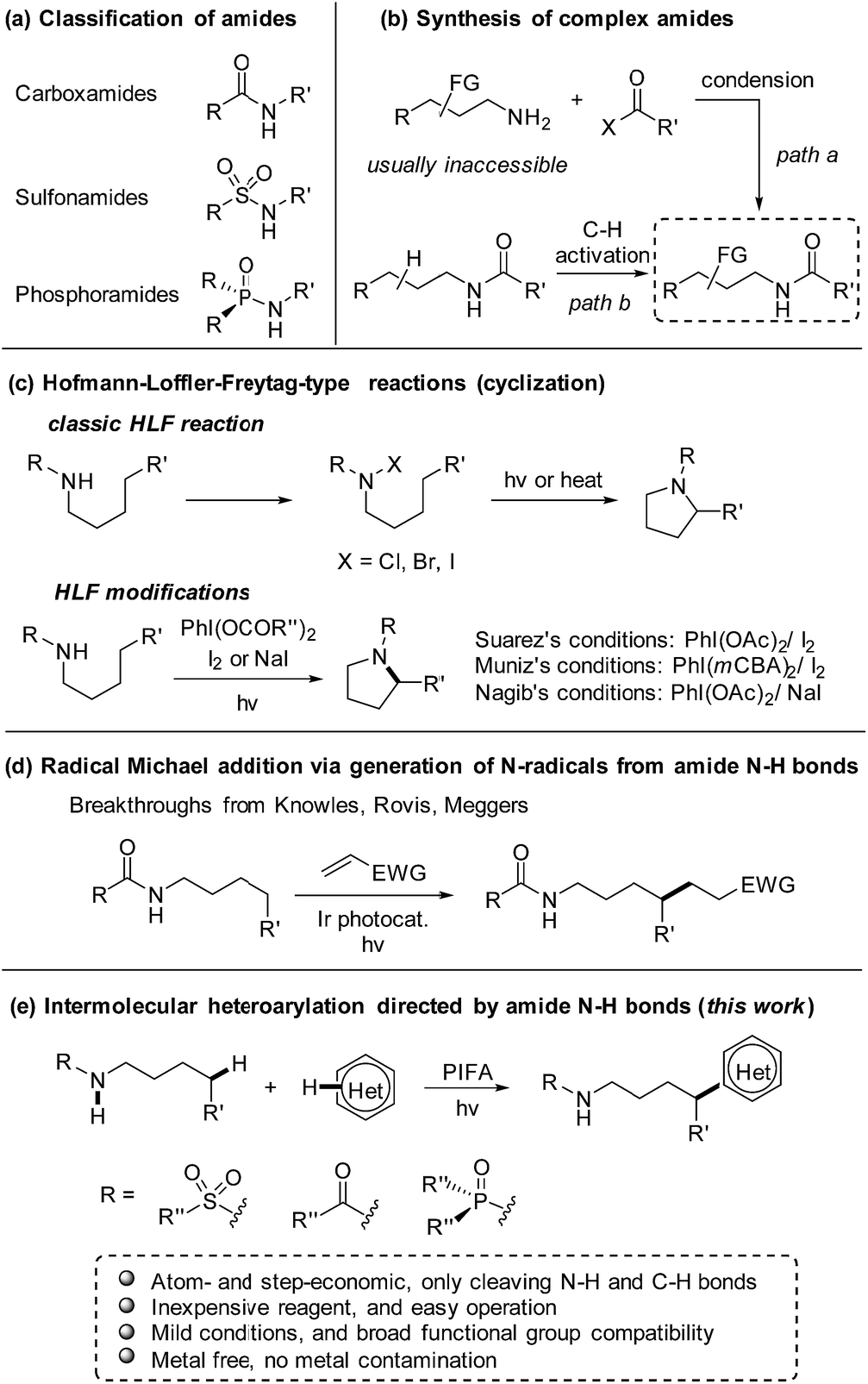 | ||
| Scheme 1 Amide N–H bond-directed functionalization of C(sp3)–H bonds. (a) Classification of amides. (b) Synthesis of complex amides. (c) Hofmann–Loffler–Freytag-type reactions. mCBA = 3-chlorobenzoate. (d) Intermolecular alkylation via generation of N-radicals from amide N–H bonds. (e) Intermolecular heteroarylation directed by amide N–H bonds. | ||
Inspired by the classic Hofmann–Löffler–Freytag (HLF) reaction,3 recently the regioselective functionalization of amides via C(sp3)–H functionalization enabled by hydrogen atom transfer (HAT) has gained intense interest.4 Due to the high bond-dissociation free energy (BDFE) of N–H bonds (107–110 kcal mol−1),5 it is a formidable challenge to directly generate amidyl radicals from N–H bonds. Therefore, amides are usually elaborated to other surrogates that deliver amidyl radicals under mild conditions.6,7 Elegant modifications of HLF reaction from Suárez8 and others9,10 convert the amide N–H bonds to amidyl radicals by homolysis of the in situ formed N–I species. But those reactions are limited to the cyclization to afford the pyrrolidine derivatives (Scheme 1c). The recent breakthroughs in the radical Michael addition achieved by Knowles,11 Rovis,12 and Meggers13 significantly increase the atom- and step-economy of the amide-directed C(sp3)–H functionalization (Scheme 1d). Despite the notable progress, the production of amidyl radicals direct from amide N–H bonds and applications in the intermolecular transformation of C(sp3)–H bonds still remains scarce.
Heteroarenes are extensively found in biologically active compounds, including almost half of the top 200 pharmaceuticals by prescriptions.14 It is of great synthetic value to introduce various heteroaryl groups to amides by C–H functionalization. To the best of our knowledge, only two examples from Zhu7g and Nagib,7l respectively, reported the remote C–H heteroarylation of amides. The preformed N–F derivative of amides were employed as starting materials. Both reports focused on the C–H arylation, while only a few examples on heteroarylation were involved. Inspired by our recent achievements in the alcohol-directed C(sp3)–H heteroarylation,15 we herein report a novel, metal-free regioselective heteroarylation of amides via C(sp3)–H bond functionalization (Scheme 1e). A broad range of heteroarenes as well as amides including carboxamides, sulfonamides, and phosphoramides are tolerated in the reaction. Amidyl radicals are readily generated from the amide N–H bonds under mild conditions, manifesting the step-economy of the protocol. The metal-free conditions avoid the metal contamination of products, boosting the potential use in medicinal chemistry. Furthermore, the method features the use of inexpensive reagent and easy operation, offering a practical approach for the late-stage functionalization of amides.
Results and discussion
At the outset, a systematic survey of reaction parameters was carried out to define the optimized conditions (for details, see ESI†). In the presence of visible-light irradiation and phenyliodine bis(trifluoroacetate) (PIFA), the reaction of tosylamide 1a with lepidine 2a proceeded smoothly to furnish the desired Minisci-type product 3a in 85% yield (Scheme 2). It should be noted that the use of excess amides significantly improved the yield, but the rest of amides could be recovered during the purification. The dual-role PIFA that acted as both the initiator of amidyl radical and oxidant was crucial to the reaction outcome. Variation of PIFA to either PIDA or the conditions valid for the HLF reactions involving hypervalent iodine reagents (Scheme 1c) did not efficiently afford the desired product, suggesting that the current reaction pathway should be distinct to the previous reports. The reaction did not take place without the photo-irradiation or while heating the reaction in dark.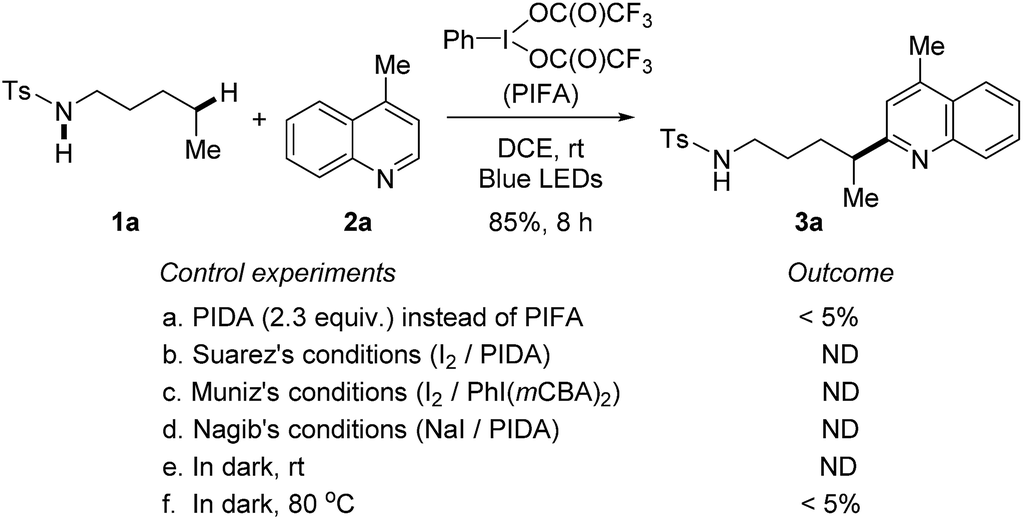 | ||
| Scheme 2 Control experiments. Standard reaction conditions: tosylamide 1a (0.6 mmol), lepidine 2a (0.2 mmol), and PIFA (0.46 mmol) in DCE (2 mL), irradiated by 2 × 50 W blue LEDs at rt. | ||
With the optimized reaction conditions in hand, we firstly examined the scope of heteroarenes (Scheme 3). A variety of six-membered nitrogen-containing heteroarenes were proved to be suitable reaction partners. Lots of functional groups were well tolerated under the mild conditions. Various quinolines were readily alkylated at either ortho- or para-position regardless of the electronic characteristics, leading to the corresponding products 3a–3h. The alkylation of isoquinolines only occurred at the 1-position (3i–3k). Aryl bromide remained intact during the reaction, reserving a platform for product processing by cross couplings (3c and 3k). While other heteroarenes such as phenanthridine (3l), acridine (3m), quinoxaline (3n), pyrazine (3o), and pyrimidine (3p) resulted in single alkylated products, the conversion of pyridine led to a mixture of regio-isomers (3q). The five-membered heteroarenes are generally challenging substrates for the Minisci reaction due to the disfavored high electron-density. In this reaction, the reaction of benzothiazoles and thiazoles afforded the desired products 3r–3t in synthetically useful yields.
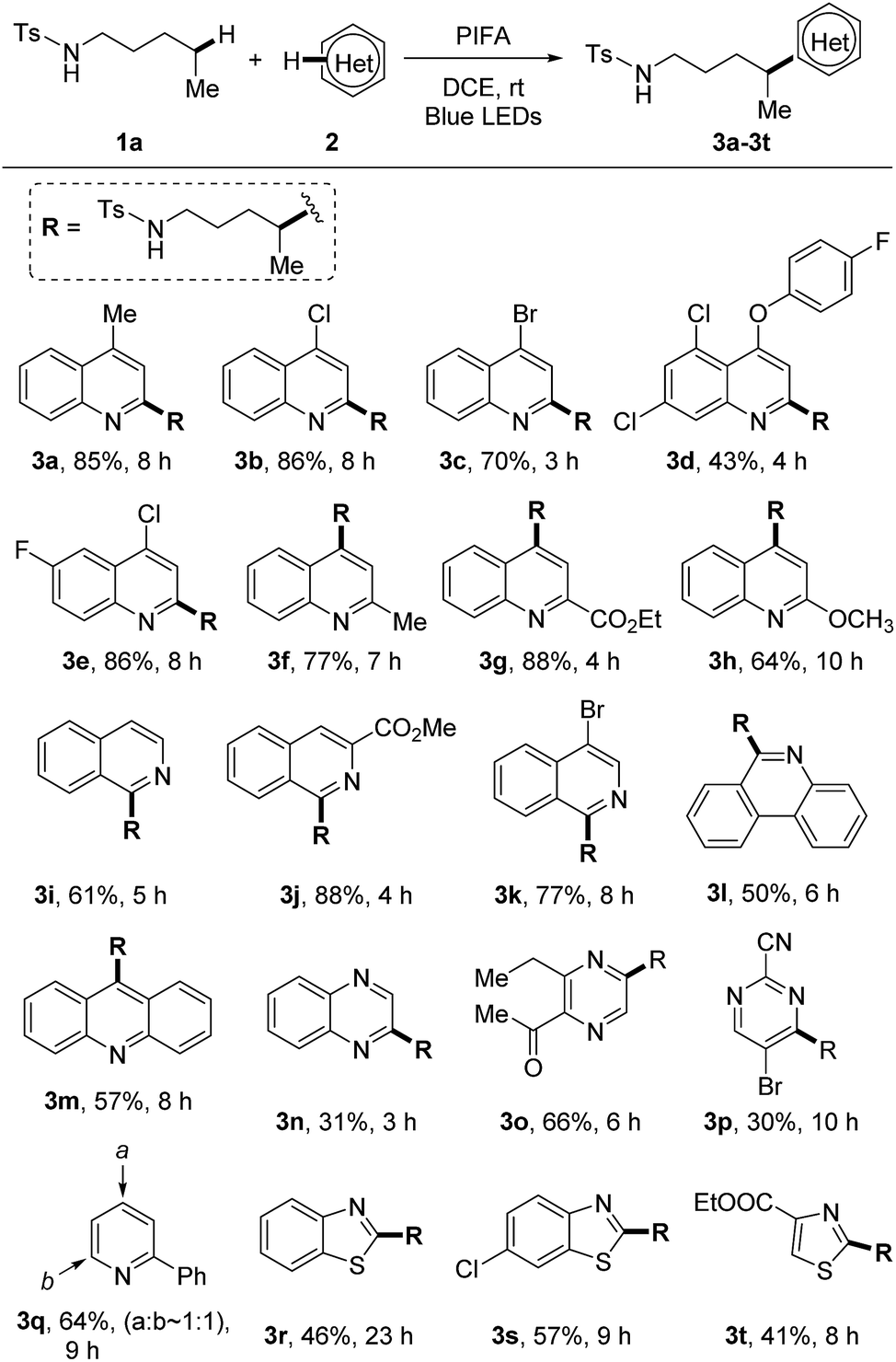 | ||
| Scheme 3 Scope of heteroarenes. Reaction conditions: tosylamide 1a (0.6 mmol), heteroarene 2 (0.2 mmol), and PIFA (0.46 mmol) in DCE (2 mL), irradiated by 2 × 50 W blue LEDs at rt. Yields of isolated products are given. | ||
The scope of sulfonamides was next evaluated (Scheme 4). A number of sulfonamides including both linear and cyclic amides were suitable substrates. While the adduct 3u was obtained in the form of regio-isomers via 1,5- and 1,6-HAT, other products 3v–3ak were generated in unique regioselectivity. In addition to secondary C–H bond, tertiary C–H bond was also readily reacted (3v), albeit resulting in lower yield. Additionally, primary C(sp3)–H bond adjacent to oxygen was also smoothly functionalized (3w). Benzylic C–H bonds were not suitable under the current conditions, the HLF-type byproduct was identified as pyrrolidine obtained from the over-oxidation of benzylic radical. Terminal alkene which is normally sensitive to radical conditions was compatible in this transformation (3z). The reaction directed by secondary amide offered the outcome comparable to that of primary amide (3aa). Site-selective abstraction of the C–H bonds on cycloalkanes provided an efficient approach for the decoration of cycloalkyl skeletons (3ab–3ae). Moreover, tosylamide could be altered to other sulfonamides, such as the aryl (3af and 3ag), heteroaryl (3aj), and alkyl (3ah and 3ak) substituted sulfonamides. Notably, this method could be applied to the modification of complex natural products and drug derivatives (3ai and 3ak). It should be mentioned that while determining the relative configuration of the products with low d.r. values (e.g., 3aa, 3ad, 3ak) was not necessary, for other stereospecific cases (d.r. >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, e.g., 3ab, 3ac, 3ae) the thermodynamically preferred products could be assigned as the major isomers in theory.
:1, e.g., 3ab, 3ac, 3ae) the thermodynamically preferred products could be assigned as the major isomers in theory.
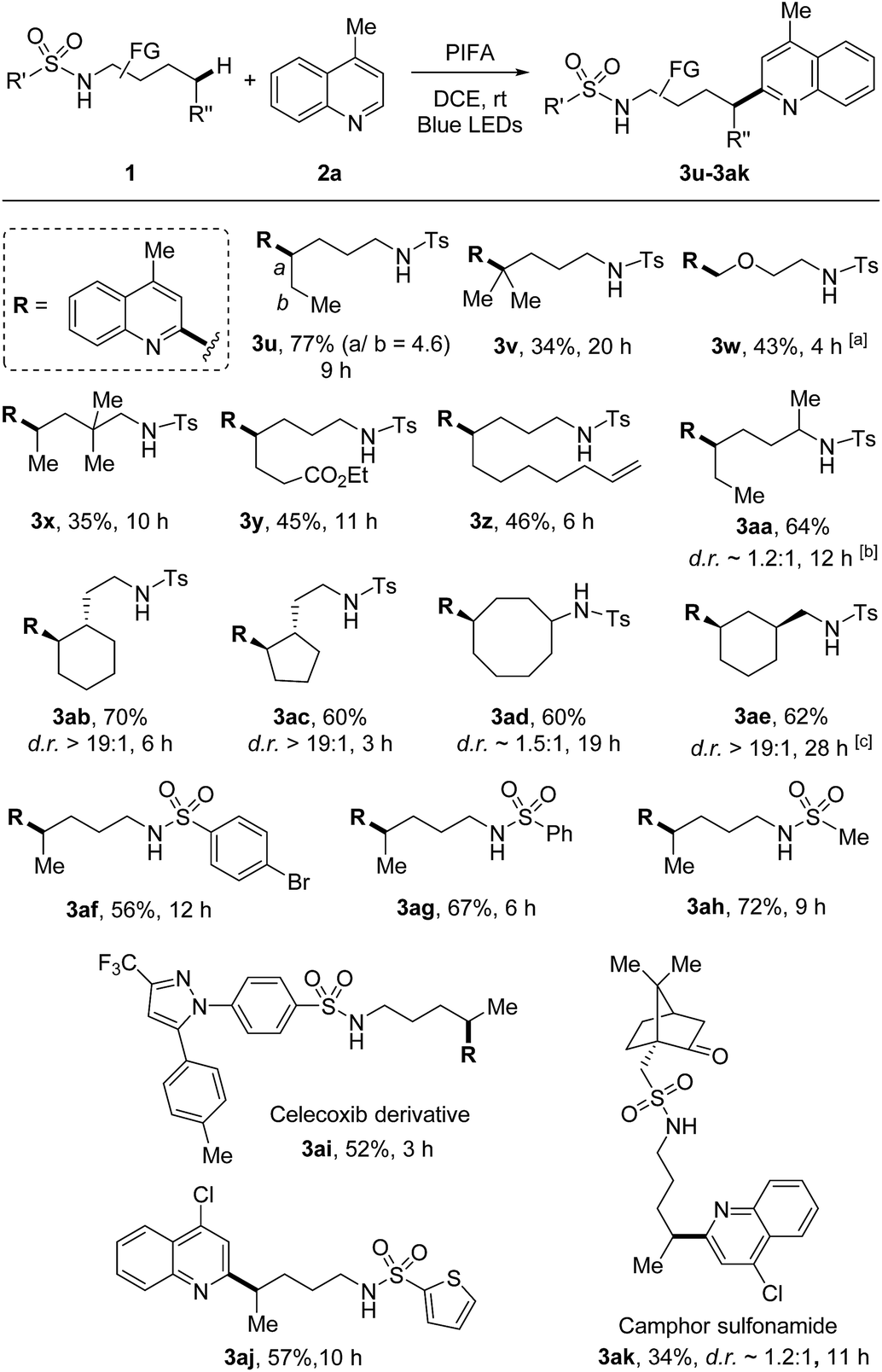 | ||
| Scheme 4 Variation of sulfonamides. Reaction conditions: sulfonamides 1 (0.6 mmol), quinoline 2a (0.2 mmol), and PIFA (0.46 mmol) in DCE (2 mL), irradiated by 2 × 50 W blue LEDs at rt. Yields of isolated products are given. a5.0 equiv. amide was used. bPIFA (0.92 mmol), added in four portions (every 3 h), cPIFA (0.92 mmol), added in two portions (the second portion was added after 12 h). | ||
The generality of this protocol was further illustrated by the heteroarylation of carboxamides and phosphoramides (Scheme 5). A portfolio of electron-rich or deficient benzamides (4a–4f), acetamide (4g), and even the Boc-protected amide (4h) served as competent precursors for the transformation. Only the δ-functionalized products were obtained, no matter whether linear or cyclic amides were used. Likewise, various phosphoramide derivatives such as phosphoramidates (5a–5e) and phosphinamide (5f) also provided the site-specific C–H heteroarylation exclusively at the δ-position, furnishing the desired products in synthetically useful yields.
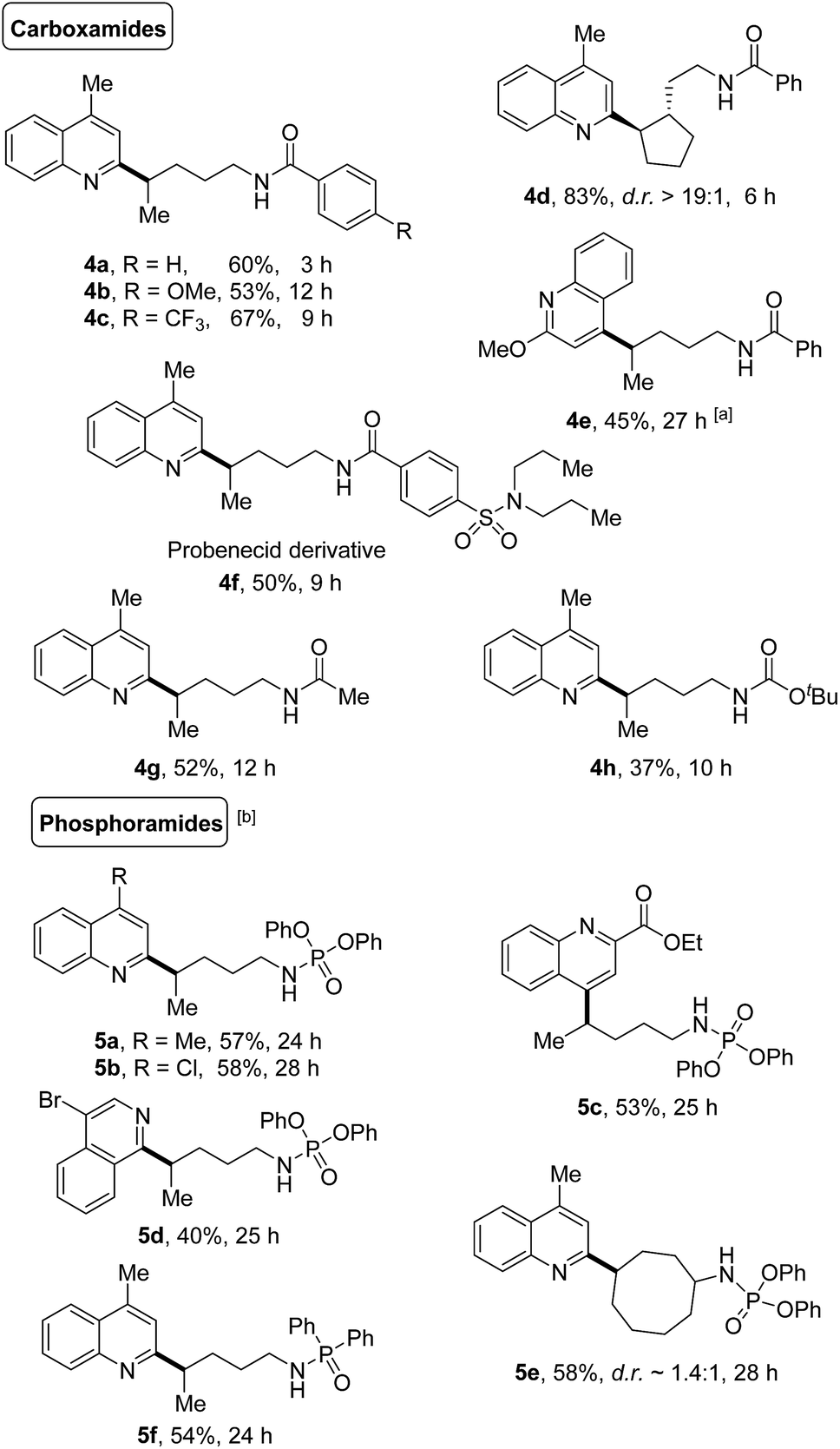 | ||
| Scheme 5 Transformation of carboxamides and phosphoramides. Reaction conditions: amides (0.6 mmol), quinoline (0.2 mmol), and PIFA (0.46 mmol) in DCE (2 mL), irradiated by 2 × 50 W blue LEDs at rt. Yields of isolated products are given. aPIFA (0.46 mmol), added in two portions (the second portion was added after 12 h). bPIFA (0.92 mmol), added in two portions (the second portion was added after 12 h). | ||
A set of experiments were performed to shine light on the mechanism. The free N–H bond of amides was requisite to the transformation as the heteroarylation did not proceed with the methylated tosylamide 6 (Scheme 6a), supporting the hypothesis that HAT process was enabled by amidyl radical generated from N–H bond. The mixture of 1a and PIFA showed a weak absorption in the field ranging from 410 to 460 nm that overlapped with the emission of blue LEDs (Scheme 6b), indicating that homolysis of the N–I intermediate might be triggered from light absorption.
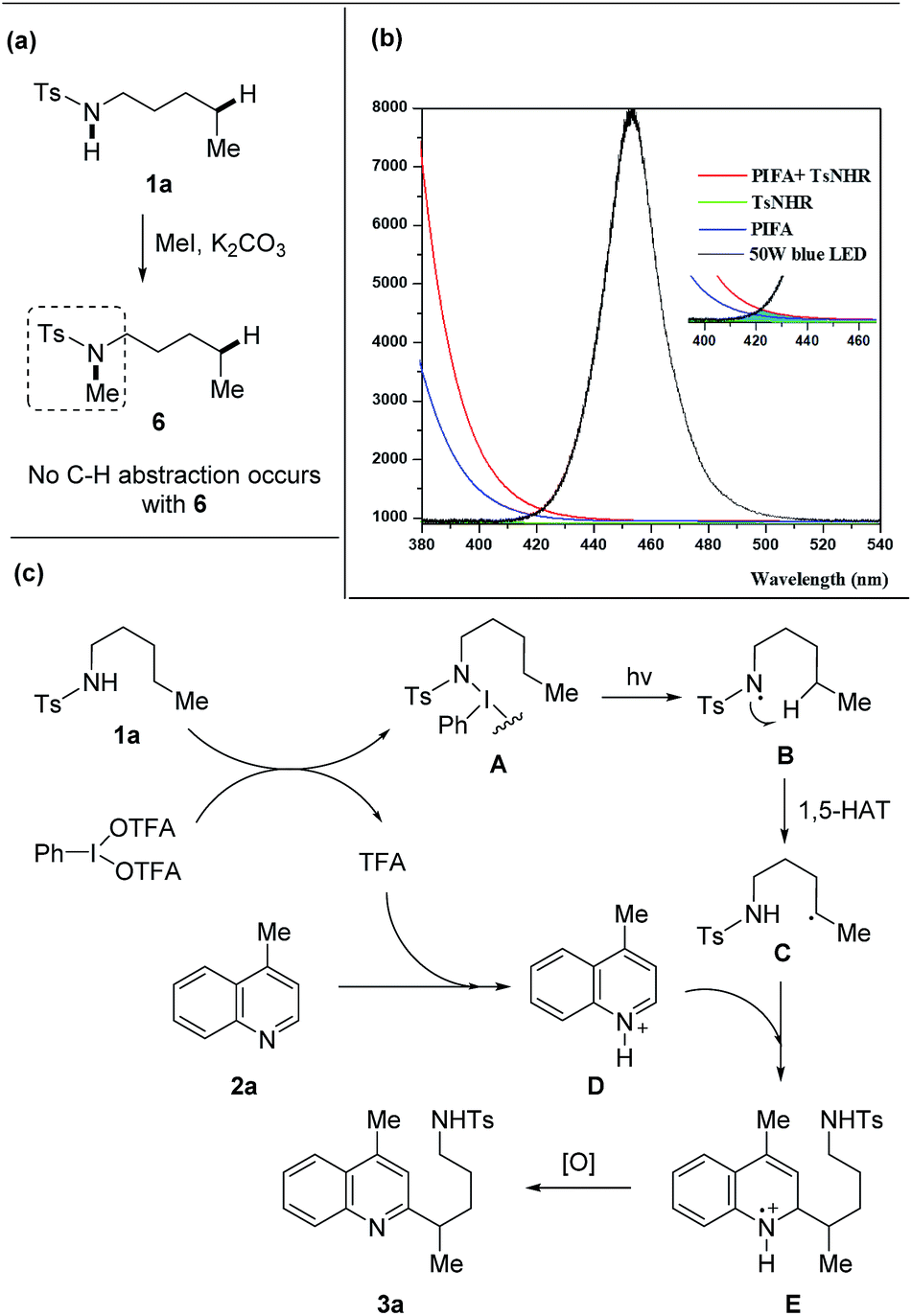 | ||
| Scheme 6 Mechanistic investigation and proposed mechanism. (a) Reaction of tosylamide without N–H bond. (b) Absorption spectra (1a + PIFA) and emission spectra (blue LEDs). (c) Proposed mechanism. | ||
A proposed mechanism is depicted (Scheme 6c). Initially, the interaction of 1a with PIFA generates the N–I(III) complex A and TFA.16 According to the DFT calculations,16b the N–I(III) complex A is probably generated from amide and PIFA via the ligand exchange on hypervalent iodine. The light-induced homolysis of N–I bond of A affords the amidyl radical B that undergoes 1,5-HAT to form the alkyl radical C. Meanwhile, heteroarene 2a is pre-activated through the protonation with in situ generated TFA and then reacts with the nucleophilic alkyl radical C, leading to the intermediate E. Further single-electron oxidation of E furnishes the final product 3a. The quantum yield (4.8%) of this reaction also suggested a photochemical process (for details, see ESI†).
Conclusions
In summary, we have described a simple and practical approach for the regioselective heteroarylation of amides via unactivated C(sp3)–H bond functionalization. The transformation is promoted by visible-light irradiation which leads to the mild generation of amidyl radicals directly from the amide N–H bonds. A vast array of heteroarenes as well as amides including carboxamides, sulfonamides, and phosphoramides are readily functionalized. It is noteworthy that while the classic Minisci reaction usually requires the addition of extrinsic acid, this protocol takes place under neutral conditions. This metal-free process may find the potential use in medicinal chemistry in near future.Experimental section
General procedure for heteroarylation of remote C(sp3)–H bonds
Heteroarene 2 (0.2 mmol) and amide 1 (0.6 mmol) were loaded in a reaction vial without N2 atmosphere. Then DCE (2.0 mL) followed by PIFA (0.46 mmol) was added to the mixture. The reaction was irradiated with 2 × 50 W blue LEDs from 5 cm away and kept at 25 °C under fan cooling. After the reaction completion monitored by TLC, the mixture was neutralized by aq. KOH until pH > 8 and then extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were washed by brine, dried over Na2SO4, filtered, concentrated, and purified by flash column chromatography on silica gel (eluent: ethyl acetate/petroleum ether) to give the desired products 3–5.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. Z. is grateful for the financial support from Soochow University, the National Natural Science Foundation of China (21722205), the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201708), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- (a) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed; (b) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443 CrossRef CAS PubMed.
- For selected reviews on C(sp3)–H activation, see: (a) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (e) O. Daugulis, J. Roane and L. Trane, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (f) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- (a) A. W. Hofmann, Chem. Ber., 1883, 16, 558 CrossRef; (b) K. Löffler and C. Freytag, Chem. Ber., 1909, 42, 3427 CrossRef.
- For selected reviews on HAT, see: (a) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94 RSC; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (c) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2017, 2056 CrossRef CAS PubMed; (d) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569 CrossRef CAS PubMed; (e) X.-Q. Hu, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960 CrossRef CAS PubMed.
- (a) F. G. Bordwell and G.-Z. Ji, J. Am. Chem. Soc., 1991, 113, 8398 CrossRef CAS; (b) F. G. Bordwell, S. Zhang, X.-M. Zhang and W.-Z. Liu, J. Am. Chem. Soc., 1995, 117, 7092 CrossRef CAS.
- For selected reviews on the N-radical chemistry, see: (a) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603 RSC; (b) B. Quiclet-Sire and S. Z. Zard, Beilstein J. Org. Chem., 2013, 9, 557 CrossRef CAS PubMed; (c) H. Song, X. Liu and Y. Qin, Acta Chim. Sin., 2017, 75, 1137 CrossRef; (d) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem.–Eur. J., 2018, 24, 12154 CrossRef CAS PubMed.
- For selected examples on the amide-mediated HAT, see: (a) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640 CrossRef CAS PubMed; (b) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771 CrossRef CAS PubMed; (c) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692 CrossRef CAS PubMed; (d) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940 CrossRef CAS PubMed; (e) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 CrossRef CAS PubMed; (f) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945 CrossRef CAS PubMed; (g) Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288 CrossRef CAS PubMed; (h) X. Shen, J.-J. Zhao and S. Yu, Org. Lett., 2018, 20, 5523 CrossRef CAS PubMed; (i) H. Chen, L. Guo and S. Yu, Org. Lett., 2018, 20, 6255 CrossRef CAS PubMed; (j) C. G. Na and E. J. Alexanian, Angew. Chem., Int. Ed., 2018, 57, 13106 CrossRef CAS PubMed; (k) Y. Tang, Y. Qin, D. Meng, C. Li, J. Wei and M. Yang, Chem. Sci., 2018, 9, 6374 RSC; (l) Z. Zhang, L. M. Statemana and D. A. Nagib, Chem. Sci., 2019, 10, 1207 RSC; (m) K. Wu, L. Wang, S. Colón-Rodríguez, G.-U. Flechsig and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 1774 CrossRef CAS PubMed; (n) Z. Liu, H. Xiao, B. Zhang, H. Shen, L. Zhu and C. Li, Angew. Chem., Int. Ed., 2019, 58, 2510 CrossRef CAS PubMed; (o) F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385 CrossRef CAS PubMed.
- (a) P. De Armas, R. Carrau, J. L. Concepción, C. G. Francisco, R. Hernández and E. Suárez, Tetrahedron Lett., 1985, 26, 2493 CrossRef CAS; (b) R. Carrau, R. Hernández, E. Suárez and C. Betancor, J. Chem. Soc., Perkin Trans. 1, 1987, 1, 937 RSC; (c) R. Hernández, M. C. Medina, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1987, 28, 2533 CrossRef; (d) R. Freire, A. Martín, I. Pérez-Martín and E. Suárez, Tetrahedron Lett., 2002, 43, 5113 CrossRef CAS; (e) A. Martín, I. Pérez-Martín and E. Suárez, Org. Lett., 2005, 7, 2027 CrossRef PubMed.
- (a) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef PubMed; (b) P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muñiz, Angew. Chem., Int. Ed., 2017, 56, 8004 CrossRef CAS PubMed; (c) P. Becker, T. Duhamel, C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2018, 57, 5166 CrossRef CAS PubMed; (d) T. Duhamel, C. J. Stein, C. Martínez, M. Reiher and K. Muñiz, ACS Catal., 2018, 8, 3918 CrossRef CAS.
- (a) E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974 CrossRef CAS PubMed; (b) E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204 CrossRef CAS PubMed; (c) L. M. Stateman, E. A. Wappes, K. M. Nakafuku, K. M. Edwards and D. A. Nagib, Chem. Sci., 2019, 10, 2693 RSC.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS PubMed.
- (a) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef PubMed; (b) D.-F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897 CrossRef CAS PubMed.
- W. Yuan, Z. Zhou, L. Gong and E. Meggers, Chem. Commun., 2017, 53, 8964 RSC.
- (a) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- (a) X. Wu, H. Zhang, N. Tang, Z. Wu, D. Wang, M. Ji, Y. Xu, M. Wang and C. Zhu, Nat. Commun., 2018, 9, 3343 CrossRef PubMed; (b) X. Wu, M. Wang, L. Huan, D. Wang, J. Wang and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 1640 CrossRef CAS PubMed.
- (a) X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712 CrossRef CAS PubMed; (b) C. Zhu, Y. Liang, X. Hong, H. Sun, W.-Y. Sun, K. N. Houk and Z. Shi, J. Am. Chem. Soc., 2015, 137, 7564 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02564b |
| ‡ N. T. and X. W. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2019 |