
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general concept for the introduction of hydroxamic acids into polymers†
Tobias
Johann
a,
Jennifer
Keth
a,
Matthias
Bros
b and
Holger
Frey
 *a
*a
aInstitute of Organic Chemistry, Johannes Gutenberg University, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de
bDepartment of Dermatology, University Medical Center of the Johannes Gutenberg University Mainz, Langenbeckstrasse 1, 55131 Mainz, Germany
First published on 17th June 2019
Abstract
Hydroxamic acids (HA) form stable complexes with a large variety of metal-ions, affording hydroxamates with high complexation constants. Hydroxamic acid moieties play a crucial role in the natural iron metabolism. In this work, 1,4,2-dioxazoles linked to a hydroxyl group are introduced as key compounds for the installation of hydroxamic acids at synthetic polymers in well-defined positions. A general synthetic scheme is developed that gives access to a series of novel functional key building blocks that can be universally used to obtain hydroxamic acid-based monomers and polymers, for instance as protected HA-functional initiators or for the synthesis of a variety of novel HA-based monomers, such as epoxides or methacrylates. To demonstrate the excellent stability of the dioxazole-protected hydroxamic acids, direct incorporation of the dioxazole-protected hydroxamic acids into polyethers is demonstrated via oxyanionic polymerization. Convenient subsequent deprotection is feasible under mild acidic conditions. α-Functional HA-polyethers, i.e. poly ethylene glycol, polypropylene glycol and polyglycerol based on ethylene oxide, propylene oxide and ethoxy ethyl glycidyl ether, respectively are prepared with low dispersities (<1.2) in the molecular weight range of 1000 to 8500 g mol−1. Water-soluble hydroxamic acid functional poly(ethylene glycol) (HA-PEG) is explored for a variety of biomedical applications and surface coating. Complexation of Fe(III) ions, coating of various metal surfaces, enabling e.g., solubilization of FeOx nanoparticles by HA-PEGs, are presented. No impact of the polyether chain on the chelation properties was observed, while significantly lower anti-proliferative effects were observed than for deferoxamine. HA-PEGs show the same complexation behavior as their low molecular weight counterparts. Hydroxamic acid functional polymers are proposed as an oxidatively stable alternative to the highly established catechol-based systems.
Introduction
In the last two decades, biomimetic materials with chelating and interface-adhesive moieties, such as catechols or their derivatives, have been investigated intensely.1,2 Particularly catechol-bearing polymers that mimic mussel-foot proteins containing dopamine have attracted broad interest, as their outstanding adhesive properties enable strong interaction with almost every surface.1,3,4 Numerous applications like self-healing hydrogels, surface coatings for anti-fouling purposes or the formation of supramolecular networks, especially by combination of chelating moieties with polymers, have been demonstrated.5 In two of our previous works we focused on the direct incorporation of catechols into polyethers via anionic polymerization. Polymers bearing a single catechol moiety were used for the stabilization of MnO nanoparticles in solution. Via a tailored catechol epoxide monomer, multiple units were introduced along the polyether backbone enabling gelation upon iron(III) addition.6 However, most catechols are unstable under aqueous conditions and are known to crosslink via oxidative coupling. This irreversible reaction leads to highly toxic Michael systems. Even though no Michael adduct of amines and the quinone structure was found in studies of Deming et al., the quinone structures lead to cross-linked materials, impeding the reversibility of the formed complexes.4 Nevertheless, catechols are ubiquitous in nature as one of two moieties to form so called siderophores (greek: “iron carrier”). The second prominent class of siderophores relies on hydroxamic acids. These N-hydroxylated amides are capable of forming stable complexes with a large variety of metal-ions, affording hydroxamates with complexation constants in the range of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 28 for tris-complexes to logK = 20 for bis-complexes.7,8 Their binding affinity to metals is generally 7 magnitudes higher compared to carboxylic acids due to the formation of a five membered ring.9 In nature this class of siderophores is essential to solubilize water-insoluble iron oxide, thus increasing the bioavailability of this key element.10 More than 42 different metal-hydroxamate complexes including (but not limited to) Fe(III), Mn(II), Zn(II), Cu(II) and Ni(II) are currently known.7,8,11 In contrast to catechols, hydroxamic acids are stable under physiological conditions and neither undergo oxidation nor crosslinking. They show similar pKs-values in the range of 7–9.12
K = 28 for tris-complexes to logK = 20 for bis-complexes.7,8 Their binding affinity to metals is generally 7 magnitudes higher compared to carboxylic acids due to the formation of a five membered ring.9 In nature this class of siderophores is essential to solubilize water-insoluble iron oxide, thus increasing the bioavailability of this key element.10 More than 42 different metal-hydroxamate complexes including (but not limited to) Fe(III), Mn(II), Zn(II), Cu(II) and Ni(II) are currently known.7,8,11 In contrast to catechols, hydroxamic acids are stable under physiological conditions and neither undergo oxidation nor crosslinking. They show similar pKs-values in the range of 7–9.12
While there are only few studies on hydroxamic acids in the field of synthetic polymers, they are well established in the therapy of iron overload diseases or as potent inhibitors for histone deacetylases that are mostly associated with cancer development.13 Early efforts to combine hydroxamic acids with polymers were patented in 1942 by Du Pont.14 First systematic studies were reported by Winston et al. in the late 1970s. In these works, poly(hydroxamic acids) were synthesized by polymer modification reaction of poly(methacrylate)-based active esters with hydroxylamine derivatives.15,16
A variety of applications was proposed for polymers containing hydroxamic acids. Due to their strong complexation of metal-ions the most prominent application is their use as ion-exchange resins or for waste-water treatment.17–19 In other works, poly(hydroxamic acid)s were used for the separation of rare earth metals.17,19 Furthermore, application of poly(hydroxamic acid)s as starting material for peptide synthesis and as resin-bound acryl transfer reagent was reported.20 We emphasize that in all published reports the hydroxamic acid containing polymers were synthesized via polymer modification (post-polymerization) of carbonyl compounds, such as esters or amides with hydroxylamine. However, this approach inevitably leads to limited conversion due to the harsh reaction conditions, which is an obstacle for many applications, particularly for medical purposes.21 Nome and coworkers showed that the conversion of polyamides to hydroxamic acids by reaction with hydroxylamine leads to an alternating copolymer with both carboxylic and hydroxamic acids due to neighboring effects.22 In recent works Kizhakkedathu et al. reported conjugates of hyperbranched polyethers with deferoxamine (DFO, Desferal), a biologically produced tris-hydroxamic acid used for the therapy of iron overload diseases.23 These modified polyethers improved the plasma half time of deferoxamine 500-fold. This enhancement in pharmacokinetic properties impressively demonstrates the potential of hydroxamic acid functional polyethers. Besides the medical use of hydroxamic acids for the therapy of iron overload diseases, structures analogous to biological siderophores like desferrichrome containing multiple hydroxamic acids have been investigated as potent Zr(IV) chelators for PET imaging.24 Radical polymerization of hydroxamic acid functional methacrylates was shown to proceed with incomplete conversion due to radical transfer to the hydroxamic acid moiety.25 Hence, the attachment of hydroxamic acids to synthetic polymer architectures via polymer modification or direct radical polymerization represents a challenge, and new strategies based on HA-based building blocks are required.
In this work we describe a systematic approach for the introduction of hydroxamic acids to polymers via tailored hydroxyl functional 1,4,2-dioxazoles. The concept presented can be applied to different classes of monomers and initiators that are amenable to both radical and anionic polymerization. Polymers from epoxide and methacrylate based monomers utilizing the 1,4,2-dioxazole group to introduce multiple hydroxamic acids along the polymer backbone are currently in progress and will reported in forthcoming works. At the example of the harsh conditions of the anionic ring opening polymerization, we demonstrate in the second part of this work the introduction of precisely one hydroxamic acid end group in polyethers, in particular in poly(ethylene glycol), aiming at a narrow molecular weight distribution for medical application, combined with a well-defined polymer structure, enabling investigation of the structure-related properties of hydroxamic acid functional polyethers. We aim at combining the chelating properties of hydroxamic acids with PEG as the current “gold standard” of biocompatible, water-soluble polymers.26 Based on this work we propose hydroxamic acids as an oxidation-stable alternative to catechols in the field of materials science.
Results and discussion
A. Molecular construction kit for hydroxamic acid synthons based on 1,4,2-dioxazole protected functional structures
For the introduction of hydroxamic acids to polymers we initially screened various polymer modification approaches. Typically hydroxamic acids are introduced at the end of a synthetic sequence, e.g. at a polymer structure by reaction with hydroxylamine or its protected derivatives, leading to incomplete functionalization with many side products. This strategy also lacks general applicability.22,27 Hence, for the introduction of hydroxamic acids into polymers, we decided to avoid post-polymerization processes with hydroxylamine as a common option for functionalization, as they are restricted to a particular polymer system and thus limit the universal character for further applications. To establish functional groups at well-defined sites during chain-growth polymerization either (i) functional initiators (ii) functional monomers or (iii) functional termination reagents can be employed. Protected hydroxyl functional hydroxamic acids were developed for this approach, featuring both a protected hydroxamic acid to ensure stability during polymerization and a hydroxyl group for further transformation or use as initiators or monomers.The hydroxyl moiety can be conveniently transformed to methacrylate monomers and epoxide monomers (Scheme 2), for instance glycidyl ethers. Additionally, the hydroxyl group can directly be used in anionic ring opening polymerization (AROP) as an initiator for epoxide or lactide polymerization, thus creating a platform for both radical as well as anionic polymerization (Scheme 1). In this work we place the focus on the use of hydroxyl functional initiators for AROP of common epoxides (EO, PO, glycidyl ethers) to combine the hydroxamic acids with medically relevant polymers. Multifunctional polyethers from hydroxamic acid functional epoxides, as well as methacrylate based HA copolymers are currently in progress and will be reported in forthcoming works.
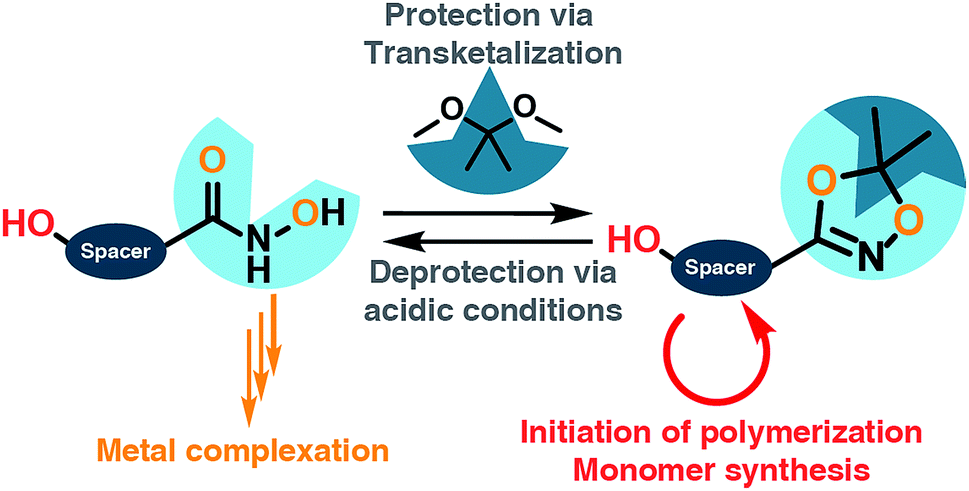 | ||
| Scheme 1 General concept for the introduction of hydroxamic acids at polymers: the hydroxamic acid is protected in the 1,4,2-dioxazole group via transketalization. The remaining free hydroxyl group can be used as a starting point either for monomer synthesis (methacrylates, epoxides) or as an initiator for anionic ring opening polymerization. After acidic treatment the 1,4,2-dioxazole group is cleaved to release the free hydroxamic acid for metal complexation. | ||
 | ||
| Scheme 2 Preparation of hydroxyl functional hydroxamic acids, their corresponding protected 1,4,2-dioxazole form and further transformation to monomers (1d, 1e) developed in this work. See ESI Scheme S1† for additional reactions with C3 and C4 aliphatic spacers. Conditions for the preparation of hydroxamic acids (1b, 2b, 3a, 4a): NH2OH/KOH/MeOH or water, RT; conditions for the preparation of 1,4,2-dioxazoles: 2,2-dimethoxy propane or 2,2-diethoxy propane, CSA, DCM, RT. For detailed synthesis description see experimental section. | ||
To maintain a scalable process, we focused on preparation methods for hydroxamic acids based on esters and hydroxylamine, due to the absence of protecting group chemistry, excellent commercial availability and the preparation in one step. Hence, para-methyl paraben was chosen as a starting material for the synthesis of 2avia nucleophilic cleavage of the ester bond by hydroxylamine (See ESI Fig. S5a–f† for NMR and IR characterization data). Unfortunately, all attempts to synthesize the corresponding hydroxamic acid led to low yields with many side products and thus troublesome purification. This is attributed to the deprotonation of the phenolic hydroxyl group, leading to a delocalized negative charge within the aromatic system. Hence, nucleophilic attack by hydroxylamine to form the hydroxamic acid is highly obstructed. To overcome this issue, we introduced an ethylene glycol C2 spacer, which enabled facile synthesis of the hydroxyl functional hydroxamic acid (“HA-OH”, 1b) in high yields (77%) (ESI Fig. S3a–f†). Aiming at a reduction of the necessary synthetic effort, the aromatic hydroxamic acid (“benzylHA-OH”, 2a) was also prepared from the commercially available ester precursor (ESI Fig. S7a–e†). Additionally lactones were explored for the preparation of aliphatic derivatives (4a, see ESI Scheme S1† for further details), since naturally occurring hydroxamic acid-based siderophores are typically aliphatic structures.10 The preparation of hydroxamic acids from lactones is known in literature.30 The conversion of caprolactone to N,6-dihydroxyhexanamide (4a) (ESI Fig. S9a–f†) and valerolactone to N,5-dihydroxypentanamide (Scheme S1† compound 5) proceeded smoothly in moderate yields, but all attempts to convert butyrolactone to N,4-dihydroxybutanamide led to complex mixtures requiring excessive purification efforts. In summary, the aromatic hydroxamic acids 1b and 3a, as well the aliphatic hydroxamic acids 4a and 5 fulfilled the necessary requirements for facile and large-scale synthesis and were subsequently used for conversion to 1,4,2-dioxazoles.
:1 dioxazole:side product) compared to aromatic derivatives (25–30:1 dioxazole:side product).31 For evaluation purposes, all three aromatic hydroxamic acids (1b, 2a and 3a) were converted to the corresponding 1,4,2-dioxazoles. As expected, the side reaction occurred in only negligible amounts (ESI Fig. S4a–f, S6a–e and S8a–e†).
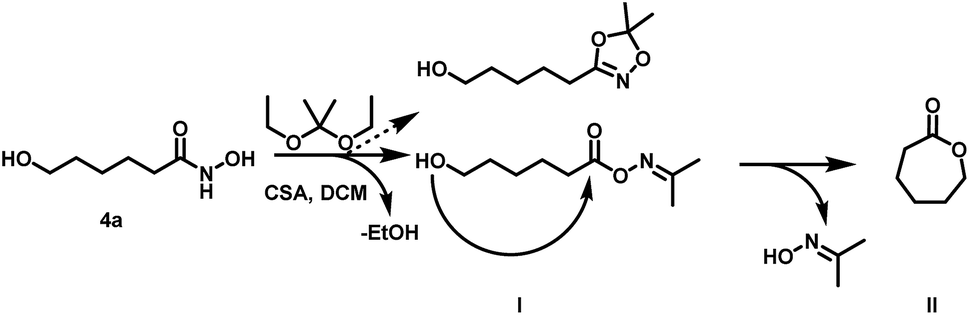 | ||
| Scheme 3 Side reaction during transketalization of 4a leading to caprolactone (II) via formation of O-acyl acetone oxime (I) and subsequent intramolecular ring closure. | ||
Surprisingly, the reaction starting from the aliphatic hydroxamic acid 4a proceeded to form mainly caprolactone by intramolecular ring closure of the O-acyl acetone oxime side product (Scheme 2, see Fig. S10† for 1H NMR of 1,4,2-dioxazole, after purification still some caprolactone is left due to troublesome separation). Hence, intramolecular attack of the hydroxyl group appears to enhance the side reaction, limiting the overall yield (<23%) of the desired aliphatic 1,4,2-dioxazole derivative. Attempts to prepare 4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)butan-1-ol from valerolactone via the hydroxamic acid 5 completely failed and led to this side reaction exclusively (Scheme 4). Considering the synthetic effort and low yields, the aliphatic (4b) as well aromatic (2b) protected hydroxamic acid were not further used for any polymerization. These findings suggest that a rigid spacer like the aromatic ring is beneficial for the preparation of hydroxyl functional 1,4,2-dioxazoles via transketalization.
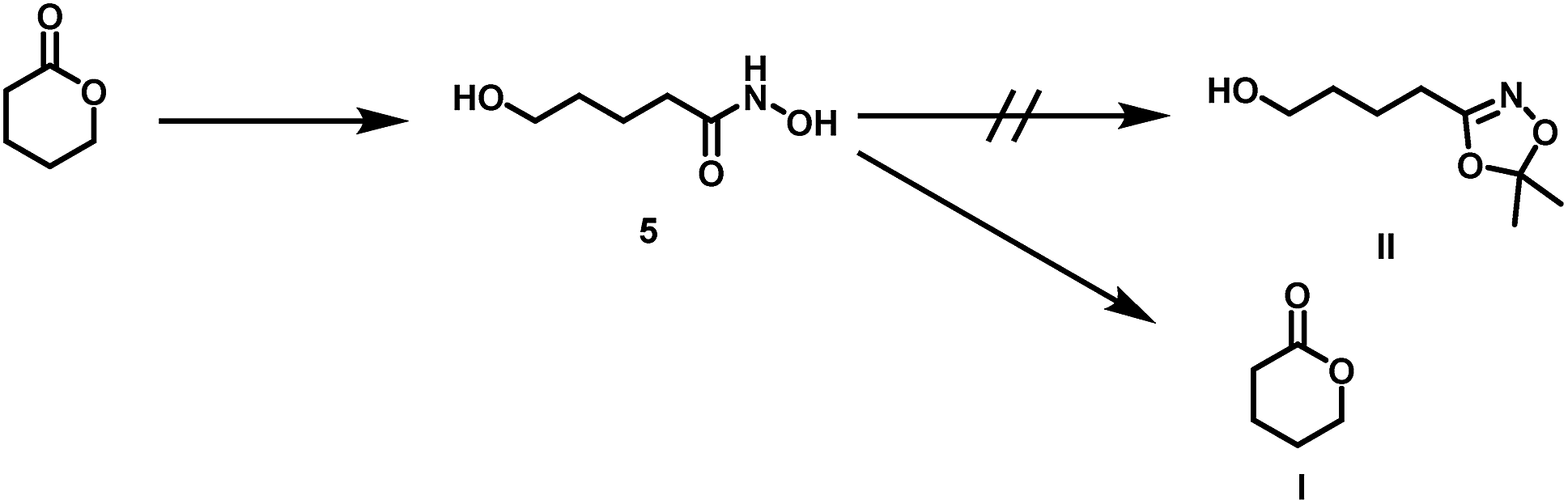 | ||
| Scheme 4 Side reaction during transketalization of 5 leading to valerolactone (I) via formation of O-acyl acetone oxime, subsequent intramolecular ring closure. | ||
Suitable hydroxamic acid initiators for the oxyanionic polymerization have to be both aprotic and base-stable and must possess one hydroxyl group that can be used to form the corresponding alkoxide-initiator salt for the anionic ring opening polymerization. Both aromatic protected hydroxamic acids (1c “HAA-OH”, 3b “benzylHAA-OH”) fulfil these requirements for AROP and have been successfully used for the preparation of functional polyethers (see below). For the different polymer molecular weight series, 1c was selected due to methyl paraben being a cheap and widely commercially available starting material. We emphasize that this synthetic route employs only inexpensive and commercially available chemicals, permitting scale-up to 20 g with 64% overall yield. We have to emphasize that although the aliphatic initiator 4b was not used in this work for further polymerization, aliphatic hydroxamic acids can be installed in polyethers via this approach. Additionally, to further demonstrate the wide applicability of this approach HAA-OH (1b) was used as a starting material for the preparation of an 1,4,2-dioxazol bearing glycidyl ether (1d) and a methacrylate derivative (1e). The hydroxamic acid acetonide glycidyl ether (HAAGE) was synthesized in high yields (81%) from 1bvia phase transfer catalysis in a one-step reaction (See ESI Fig. S11† for NMR analysis). Methacrylate hydroxamic acid acetonide (MAHAA) (1e) was obtained via Steglich esterification of 1b with methacrylic acid (See ESI Fig. S12† for NMR analysis). Polymers based on both monomers are currently in progress and will be reported in a forthcoming work. To sum up, hydroxyl-functional 1,4,2-dioxazoles represent the key structure for the preparation of monomers and polymers.
B. Polymer synthesis: hydroxamic acid functional polyethers
α-Functional polyethers were prepared from the two selected functional initiators 1c, 3b to introduce one terminal hydroxamic acid (HA) moiety. Although this limits the binding capacity for metal ions, the use of a single complexing moiety inherently prevents crosslinking via metal chelation, thus providing water-soluble complexes in case of PEG. This requirement is essential to avoid aggregation of coated nanoparticles or gelation.HAA-poly(ethylene glycol)s (HAA-PEGs) P1a, P2a with molecular weights in the range of 1000 to 8500 g mol−1 were prepared via AROP of ethylene oxide (Scheme 5, Table 1) in THF/DMSO. The preparation of polyethylene glycol based polymers was chosen as PEG represents the gold standard polymer for biomedical application. Also polymers based on propylene oxide (PO) P3a, P5a and ethoxy ethyl glycidyl ether (EEGE) P4a, P6a have been prepared to broaden the scope of materials and to demonstrate general application of 1c. Polypropylene oxide (PPO) was chosen as a hydrophobic polyether counterpart to PEG. PPO exhibits a lower critical solution temperature (LCST), enabling temperature dependent aqueous solubility below 20 °C (for PPO50).26,33 One downside of linear polyethers is their limited number of functional groups. In the case of functional initiated polyethers like in this work, only the terminal OH group can be addressed for further functionalization of the polymers. To overcome this issue, poly(ethoxy ethyl glycidyl ether) (PEEGE) polymers were prepared. EEGE is an acetal-protected glycidol monomer. These hydrophobic PEEGE polymers can easily be converted (by acidic treatment) to hydrophilic linear polyglycerol (linPG), which exhibits one hydroxyl group per repeating unit, enabling access to multi-functional polyethers.33
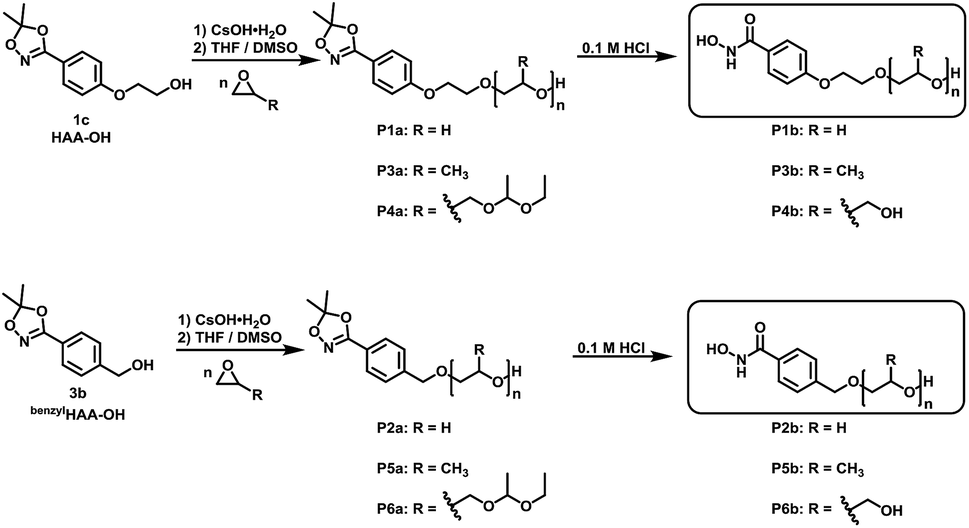 | ||
| Scheme 5 Synthesis of HAA based polyethers (HAA-PEG, HAA-PPO, HAA-PEEGE) and subsequent cleavage of the protecting group to release the free hydroxamic acid functional polyethers (HA-PEG, HA-PPO, HA-linPG). | ||
| Polymer | M n target [g mol−1] | M n (NMR)a [g mol−1] | M n (SEC)b [g mol−1] | Đ |
|---|---|---|---|---|
| a Determined via end group analysis in NMR. b DMF SEC, PEG standard. | ||||
| HAA-PEG21 | 990 | 1160 | 960 | 1.06 |
| HAA-PEG31 | 1470 | 1600 | 1370 | 1.05 |
| HAA-PEG41 | 1950 | 2040 | 1850 | 1.06 |
| HAA-PEG64 | 3010 | 3050 | 2210 | 1.11 |
| HAA-PEG80 | 3800 | 3760 | 3380 | 1.06 |
| HAA-PEG127 | 6180 | 5830 | 4360 | 1.09 |
| HAA-PEG188 | 8080 | 8520 | 6010 | 1.13 |
| benzylHAA-PEG41 | 2200 | 2010 | 1250 | 1.09 |
| benzylHAA-PEG75 | 4220 | 3510 | 1810 | 1.07 |
| HAA-PPO21 | 1400 | 1460 | 1360 | 1.06 |
| benzylHAA-PPO21 | 1370 | 1430 | 1310 | 1.07 |
| HAA-PEEGE7 | 1700 | 1410 | 860 | 1.22 |
| HAA-PEEGE26 | 4470 | 4180 | 1790 | 1.14 |
| benzylHAA-PEEGE6 | 1670 | 1080 | 880 | 1.20 |
In all cases full conversion was achieved within 24 h reaction time. All HAA-PEG, HAA-PPO and HAA-PEEGE polymers exhibit monomodal molecular weight distributions with narrow dispersities below 1.20 (Fig. 1, Table 1, ESI Fig. S1†). The degree of polymerization was kept low (10–20) for both the PPO and PEEGE polymers to limit this side reaction. Higher degrees of polymerization are possible, based on these proof-of-principle studies and further optimization. Purification of the polymers was performed via partitioning between water and DCM to obtain the salt-free, pure polymers in good yields (73–94%).
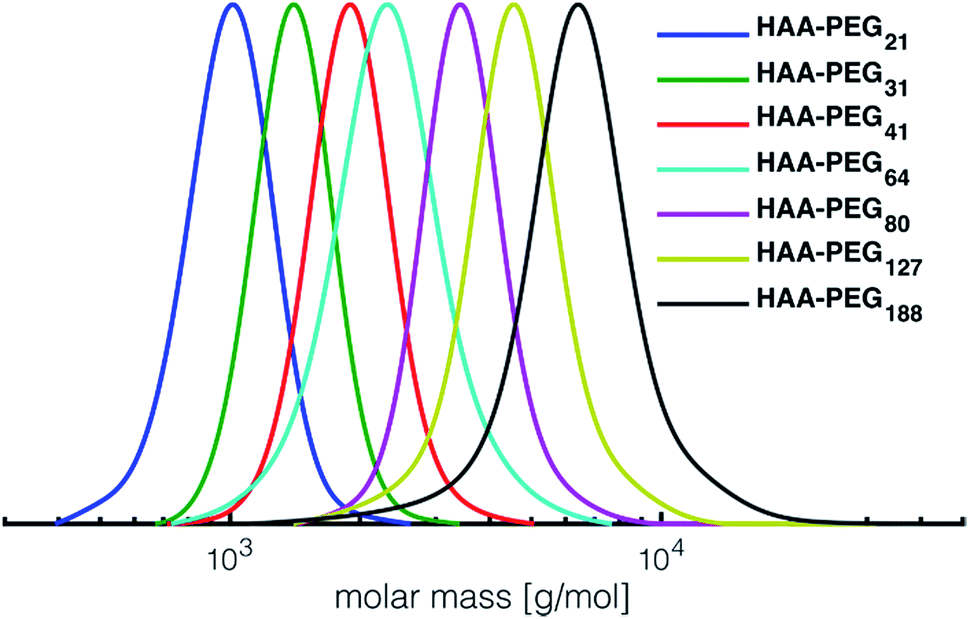 | ||
| Fig. 1 SEC traces of the prepared HAA-PEGs (DMF, PEG-calibration). HA-PEGs after deprotection strongly interact with SEC column material and hence cannot be analyzed via SEC. See Fig. S1† for SEC traces of PPO and PEEGE as well benzylHAA-OH initiated polymers. | ||
For all polymerizations conducted good agreement of target molecular weights with molecular weights determined via NMR end group analysis can be confirmed. Slight differences are accounted to the normalization of the integrals to the initiator, leading to uncertainties in the determination of approx. ±250 g mol−1. The discrepancy between SEC and NMR molecular weight can be explained by the PEG standards used for calibration. Similar underestimation of molecular weights via SEC have been reported for both PPO as well PEEGE polymers.33 In case of PEEGE slight broadening of the molecular weight distribution can be detected. This is attributed to the high polymerization temperature of EEGE (80 °C), increasing known side reactions such as proton abstraction and elimination, leading to overall broadening of the molecular weight distribution.34
1H NMR analysis confirms (ESI Fig. S13–S18†) the successful initiation of the polymers from either HAA-OH or benzylHAA-OH respectively. This can further be verified by the excellent overlap of UV and RI signal in SEC measurements (not shown). The thermo-responsive behaviour of HAA-PPO21 was investigated in deionized water. As expected, a cloud point at 12.9 °C was observable (ESI Fig. S2†), at which the aqueous solution turned turbid and HAA-PPO21 precipitated. In summary, both hydroxyl-functional protected hydroxamic acids have been proven to be a viable initiator for the polymerization of various epoxide monomers, leading to water-soluble hydrophilic PEG, thermo-responsive PPO or multi-functional PEEGE polymers bearing a terminal hydroxamic acid unit.
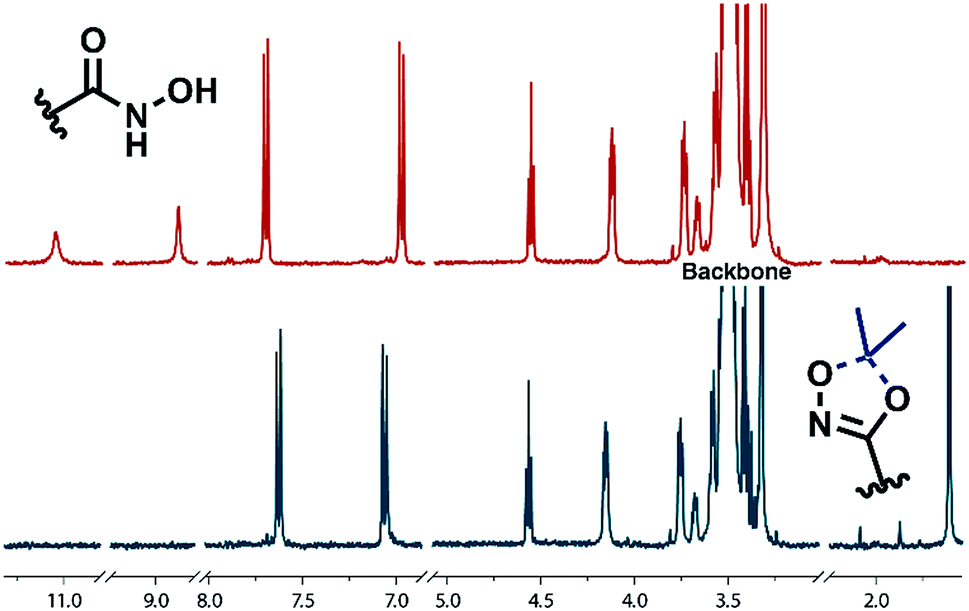 | ||
| Fig. 2 1H NMR analysis of HAA-PEG (bottom) and HA-PEG (top) after cleavage of the protecting group. | ||
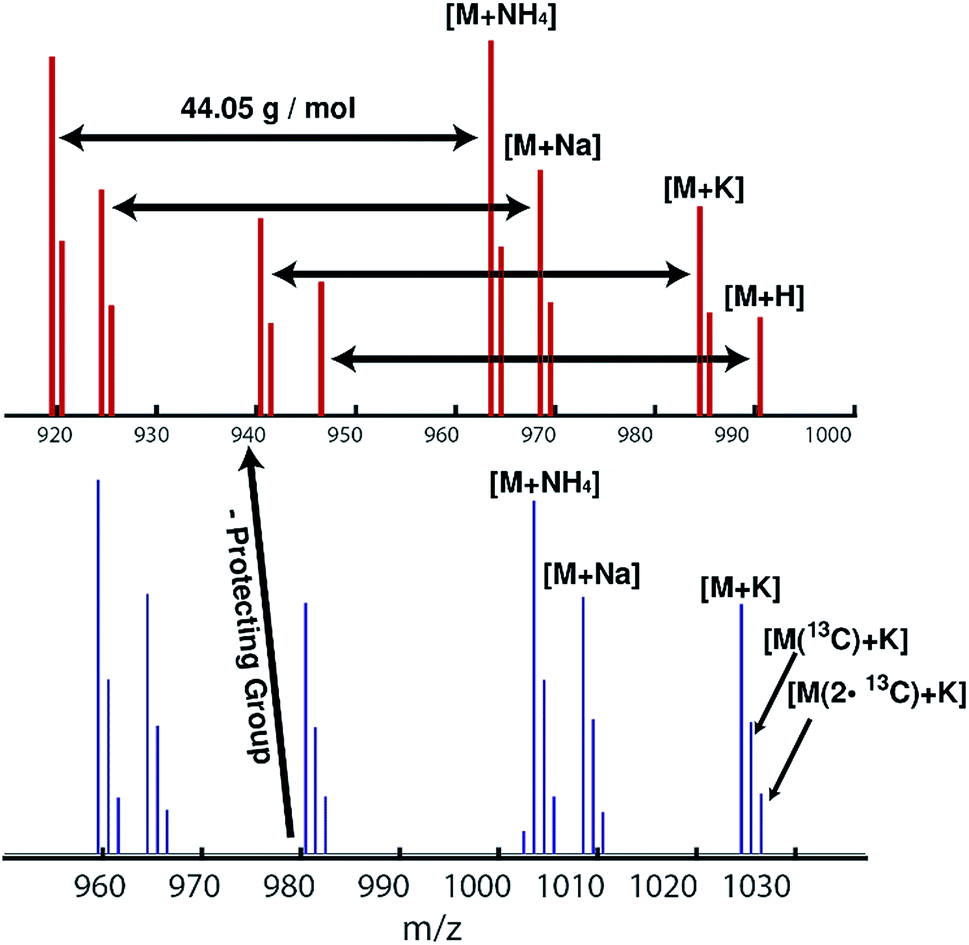 | ||
| Fig. 3 ESI-mass spectra of HAA-PEG31 (bottom) and HA-PEG31 (top). | ||
ESI mass spectra reveal only the desired HAA-PEG and HA-PEG species, respectively. Comparing both polymers, a distinct shift of 40 g mol−1, corresponding to cleavage of the protecting group, can be seen. Each spectrum shows only the distribution of the polymers with 44.05 g mol−1 per repeating unit. No sub-distribution can be detected (see ESI Fig. S19 and S20† for full spectra).
Both protecting groups employed in the synthesis of HAA-PEEGE (P6a) are acetals. Hence, the cleavage of both protecting groups can be achieved simultaneously via acidic treatment. Due to the hydrophobicity of PEEGE the cleavage is very slow in aqueous media. For improved reaction kinetics, the cleavage was performed in isopropanol solution with DOWEX 10WX8 acidic ion exchange resin, leading to HA-linPG (P6b) (Scheme 6).
 | ||
| Scheme 6 Reaction scheme for cleavage of the protecting group in HAA-PEEGE releasing both the hydroxyl groups as well as the hydroxamic acid moiety. | ||
Full cleavage of the protecting groups is confirmed via1H NMR spectroscopy (Fig. 4). After treatment with DOWEX10WX8 in isopropanol all acetal signals of PEEGE (Fig. 4 yellow) as well the methyl signals of the 1,4,2-dioxazole group at 1.6 ppm (Fig. 4 green) have vanished. The free hydroxamic acid can be confirmed via the NH-signal appearing at 11 ppm (Fig. 4 blue, top). In conclusion, the 1,4,2-dioxazole group can be simultaneously cleaved with other protecting groups reacting under the same acidic conditions, like acetals without interference leading to multi-functional linPG bearing one hydroxamic acid (Scheme 6). Considering the high stability of the 1,4,2-dioxazole group under basic conditions, orthogonal protecting group chemistry should also be possible for selective release of the functional moieties.
 | ||
| Fig. 4 Stacked 1H NMR spectra of HAA-PEEGE7 (I) before cleavage of the protecting groups and (II, top) HA-linPG7 as the reaction product. | ||
Chelation properties. Fundamental chelation properties of the HA-PEGs were investigated, relying on UV-Vis spectroscopy of the highly colored iron(III) complexes. As already reported by Winston et al.16, at low iron(III) concentration the tris(hydroxamato)iron(III) complex is favored. When increasing the iron(III) to HA-PEG ([Fe]/[HA-PEG]) ratio the tris-complex is consecutively cleaved to form bis- and finally mono(hydroxamato)iron(III) complexes (Scheme 7). Simultaneously, the color changes from the red tris-complex (λmax = 500 nm) to the purple mono-complex (λmax = 550 nm) (Fig. 5, ESI Fig. S21). This distinct shift cannot be detected in case of chelating tris-hydroxamic acids such as deferoxamine due to their highly favored formation of tris-complexes.16
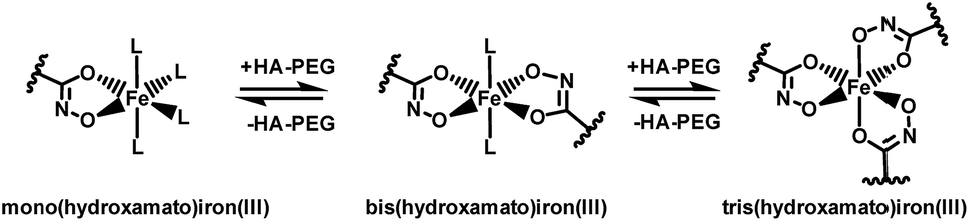 | ||
| Scheme 7 Equilibria of the formation of mono-, bis-, or tris(PEG-hydroxamato)iron(III) complexes. | ||
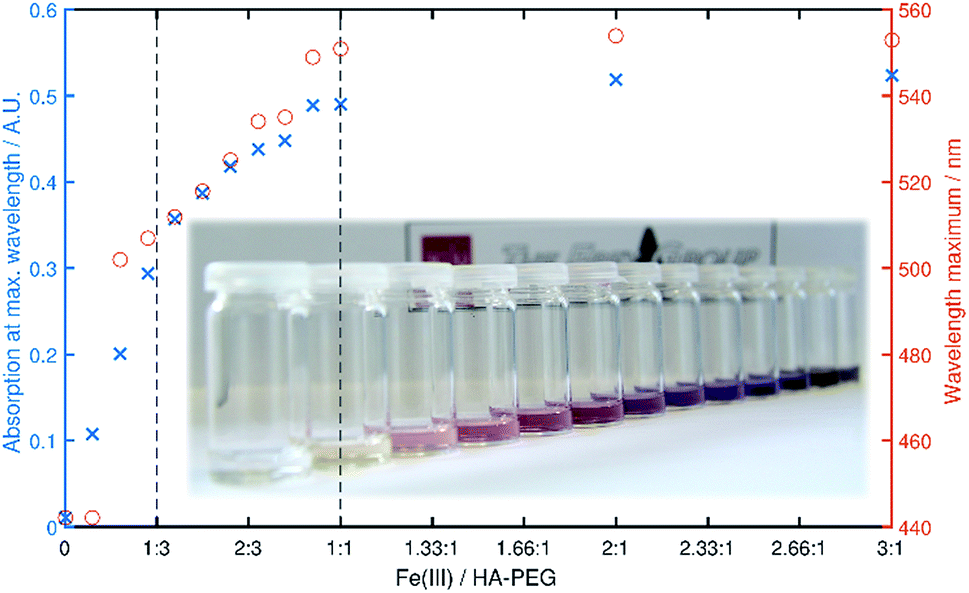 | ||
| Fig. 5 Maximum absorbance (blue) and λmax (red) in dependency of the [Fe]/[HA-PEG188] ratio. The vertical lines denote a ratio of 1:3 (tris complex) and 1:1 (mono complex) respectively. | ||
This behavior can also be verified by a steep increase in absorbance in the course of iron(III) chloride addition, until a ratio of 1:3 ([Fe]/[HA-PEG]) is achieved. With further addition of iron(III) the absorbance increases until equimolarity is obtained. At this ratio all hydroxamic acid moieties are saturated as mono-complexes, and thus no increase in absorbance can be induced by further addition of iron(III). This behavior suggests that there is no influence of the polymer chain on the chelation behavior of the hydroxamic acid end group connected to the polyether backbone. Hence, the chelation properties of HA-PEGs are comparable to low molecular weight hydroxamic acids and are not altered due to the conjugation with PEG.
Nanoparticle and surface modification. Besides binding of low molecular iron, the coating of macroscopic surfaces or metal oxide nanoparticles is another prominent application of polymeric chelators.1 As a proof of principle for the coating of surfaces with HA-PEGs, solubilization of iron oxide nanoparticles (FeOx-NP) in water as well as enhancement of the hydrophilicity of different metal surfaces were demonstrated (ESI Fig. S22 and S23†). In all cases, the hydrophilicity of the surfaces increased due to coating with HA-PEGs. By coating FeOx-NP with HA-PEGs with a molecular weight of 1000, 3800 and 8500 g mol−1 the NP diameter was systematically increased from 20 nm for the oleate-coated NP up to 50 nm in case of HA-PEG with a molecular weight of 8500 g mol−1. Control experiments with non-functionalized PEG resulted only in minor changes in size within the error range of the uncoated NP distribution (Fig. 6, see Table S1†).
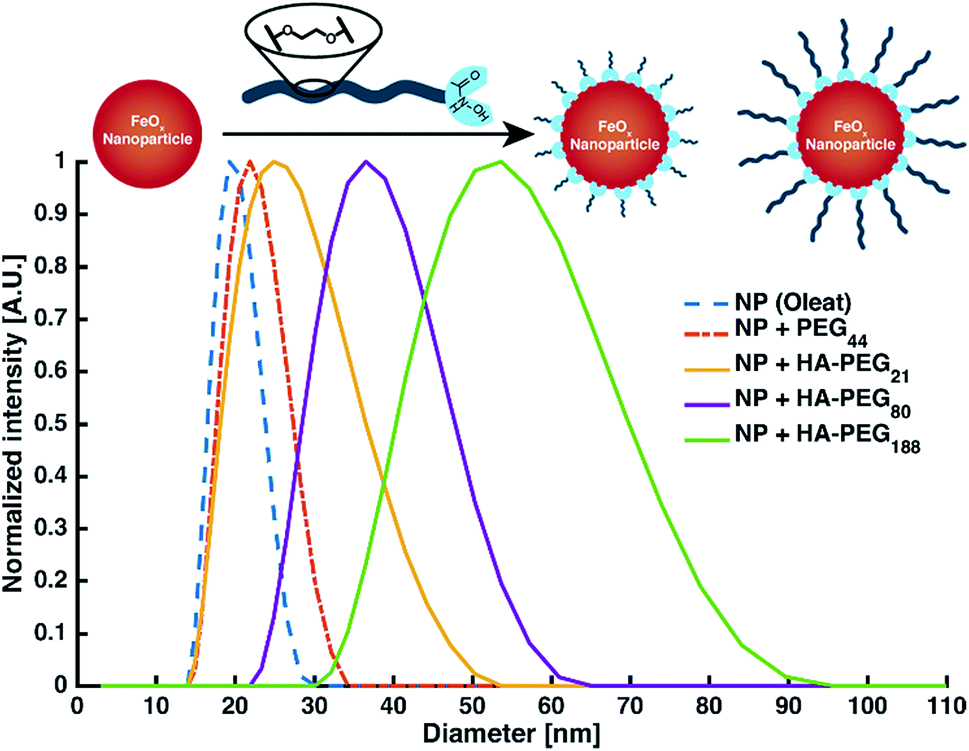 | ||
| Fig. 6 Intensity distribution of the FeOx nanoparticles by DLS. | ||
After coating, the initially water-insoluble NPs were freely soluble in water or methanol (ESI Fig. S21†). TEM images prepared from water show the coated, non-agglomerated NPs (Fig. 7). As expected, no differences in size or distribution in comparison to the NP prior to coating can be detected.
 | ||
| Fig. 7 TEM image of non-modified (oleate coated) FeOx nanoparticles (left, blue histogram, prepared from hexane) and FeOx nanoparticles after coating with HA-PEG188 (right, red histogram, obtained from aqueous solution). | ||
Cell toxicity and proliferation assays. First, biocompatibility tests were performed using primary human blood cells to explore applicability of the polymers for the transport of Fe(III) or other metal ions in medicine. In all cases HA-PEG as well as their protected counterpart HAA-PEG showed no significant impact on the metabolic cell activity within the range of tested concentrations, thus low to no toxicity can be expected (ESI Fig. S24 and S26†). To further investigate the shielding effect of the PEG chain, T-Cell proliferation studies were performed.
Commercially available hydroxamic acid-based drugs like deferoxamine mesylate (Desferal, Novartis) are known to strongly inhibit T-cell proliferation.35 In contrast HAA-PEG, as well as HA-PEG with a MW of 8500 g mol−1 did not significantly influence the proliferation of T-cells, while the low molecular weight HA 1b showed dose-dependent inhibition of the proliferation (Fig. 8, ESI Fig. S25†).
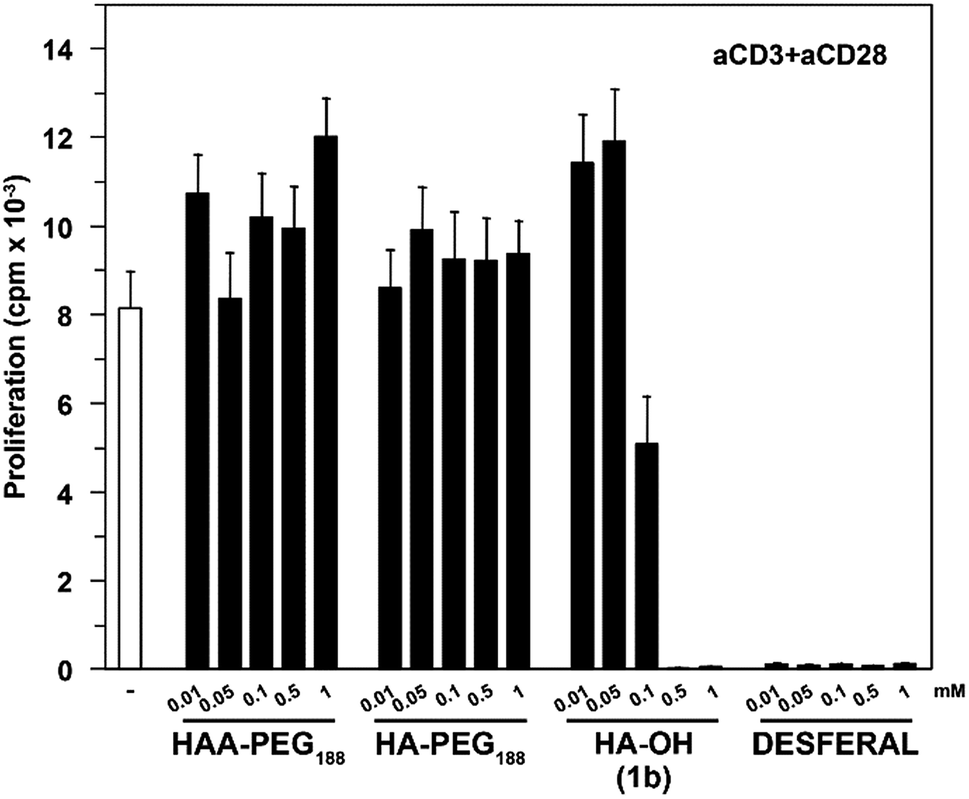 | ||
| Fig. 8 T-Cell proliferation assay. Human PBMC (4 donors) were stimulated with aCD3 and aCD28 antibodies to induce T-cell proliferation in the presence of different test compounds. | ||
Conclusions
1,4,2-Dioxazoles with an adjacent hydroxyl group have been demonstrated to represent key structures for the direct introduction of hydroxamic acids into polymers. These structures permit to install hydroxamic acids at synthetic polymers in a well-defined manner at the α-position. A general synthetic scheme was developed that gives access to a large variety of novel functional key building blocks that can be universally used to obtain hydroxamic acid-based monomers and polymers, for instance as protected HA-functional initiators or for the synthesis of a variety of novel HA-based monomers, exemplified by epoxides and methacrylates in this work.Relying on these novel synthons, hydroxamic acid functional polyethers based on ethylene oxide, propylene oxide and ethoxy ethyl glycidyl ether, respectively, were prepared to show the general applicability of this approach, even under the harsh conditions of the AROP. After mild acidic treatment of the resulting polyethers, the free hydroxamic acid can be recovered. Consequently, hydroxyl-functional 1,4,2-dioxazoles are broadly applicable for the initiation of AROP or for preparation of HA-based monomers by modification of the hydroxyl group with polymerizable structures.
The HA-initiated polyethers were studied both with respect to their future use in materials science and for medical purposes, for instance in the treatment of diseases related to the iron metabolism. No measureable difference in terms of chelation properties of the hydroxamic acid functional polyethers was detected, compared to low molecular weight hydroxamic acids, while anti-proliferative effects of low molecular weight hydroxamic acid therapeutics like deferoxamine were highly reduced. The complexation of Fe(III) ions, the coating of various metal surfaces as well as FeOx nanoparticles confirm outstanding chelation properties. From these proof-of-principle studies and the basic synthons established in this work, a vast variety of typical applications known for other polymeric chelators, such as catechol-bearing or terpyridine-bearing polymers become viable. The combination of hydroxamic acids with polymers is a promising alternative to catechols with their low oxidative stability. Hydroxamic acid based polyethers offer promise for iron-depletion therapies, for medical purposes related to metal ion transport in general as well as in materials science and for surface coating.
Based on the general concept for protected hydroxamic acids introduced in this work, a variety of novel monomers based on acrylates, methacrylates and epoxides has been prepared and polymerized. These structures and their use for both material science and medicine will be reported in due course.
Experimental section
Materials
Unless otherwise stated, all reagents were used without further purification. All chemicals and solvents were purchased from Acros, Aldrich, Fisher Scientific, Fluka, Riedel-de-Haën or Roth. Deuterated solvents were purchased from Deutero GmbH. THF was dried via distillation over sodium/benzophenone. DMSO was dried using molecular sieve 4 Å. Hydrophobic FeOx nanoparticles coated with oleic acid were provided by Jan Hilgert (Group of Prof. Tremel, University of Mainz).Abbreviations
Ethyl acetate (EA), petrol ether (PE), dichloromethane (DCM), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), ethylene oxide (EO), camphor sulfonic acid (CSA), methanol (MeOH), correlation spectroscopy (COSY), heteronuclear single-quantum correlation spectroscopy (HSQC), heteronuclear multiple-bond correlation spectroscopy (HMBC).Characterization techniques
000; nominal underfocus: 0.5–1.5 mm). For sample preparation, carbon grids for TEM applications were negatively glow discharged at 25 mA for 30 s in an Emitech K100X glow discharge system. A droplet of the sample (5 μl) was placed on the grid and incubated for 30 s. The grid was dried with Whatman filter paper no. 4 by side-blotting.
Synthesis procedures of hydroxamic acids and 1,4,2-dioxazoles
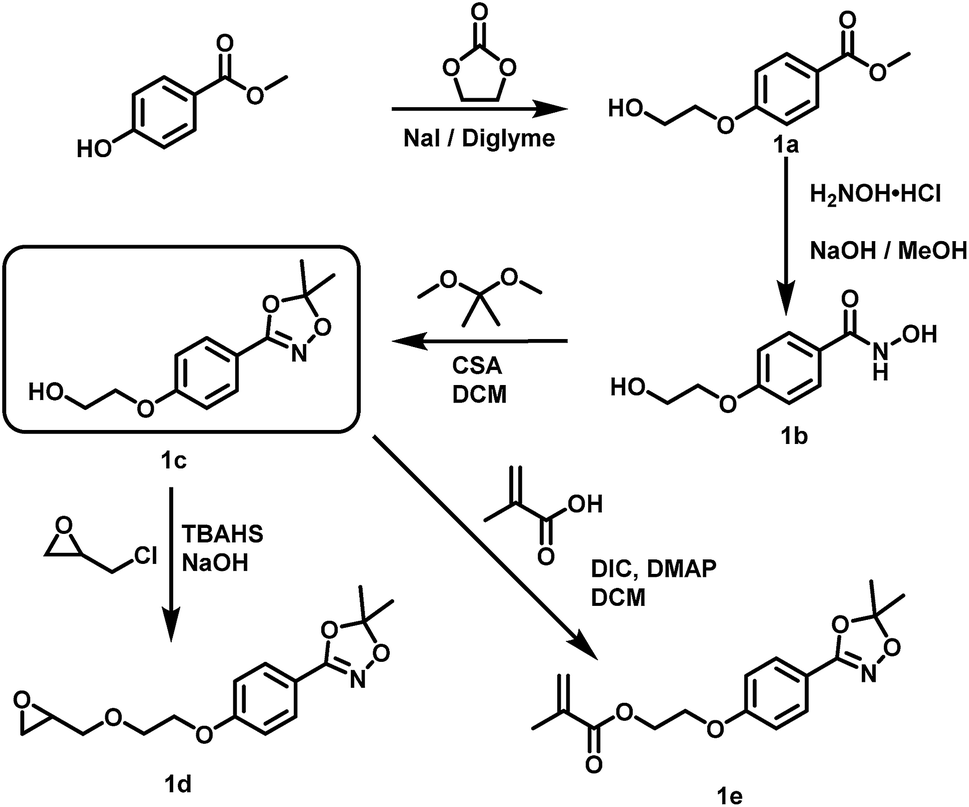
(1a) Methyl 4-(2-hydroxyethoxy)benzoate. (1a) was prepared in a modified procedure described elsewhere.36 A 250 mL flask equipped with a condenser and gas bubbler was charged with 76.07 g (0.500 mol, 1.00 eq.) methyl 4-hydroxybenzoate, 46.25 g (0.525 mol, 1.05 eq.) ethylene carbonate, 7.50 g (0.050 mol, 0.10 eq.) NaI and 100 mL diglyme and heated under vigorous stirring for 16 h at 140 °C oil bath temperature. Finally, the temperature was increased to 160 °C for two hours. The solution was cooled to room temperature and the solvent were removed until dryness in vacuo. The residue was dissolved in 300 mL EA and washed with 50 mL water, 50 mL saturated NaHCO3 and 50 mL brine and dried using Na2SO4. After evaporation of the solvent 93.6 g (0.477 mol, 95%) of methyl 4-(2-hydroxyethoxy)benzoate were obtained.
1H NMR (300 MHz, CDCl3) δ [ppm] = 7.99 (AA′BB′, 2H, HC2,6), 6.93 (AA′BB′, 2H, HC3,5), 4.17 (t, J = 4.1 Hz, 2H, CH2–O–Ar), 3.99 (t, J = 4.1 Hz, 2H, HO–CH2–), 3.88 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ [ppm] = 166.92 Ar–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 162.48 C4, 131.78 C2,6, 123.13 C3,5, 114.24 C1, 69.45 Ar–O–C, 61.42 HO–C, 52.06 CH3.
O, 162.48 C4, 131.78 C2,6, 123.13 C3,5, 114.24 C1, 69.45 Ar–O–C, 61.42 HO–C, 52.06 CH3.
(1b) N-hydroxy-4-(2-hydroxyethoxy)benzamide. (1b) was prepared in a modified procedure based on the work of Hauser and Renfrow.37 42.9 g (0.62 mol, 2.00 eq.) H2NOH·HCl were dissolved in 240 mL methanol and neutralized by addition of a solution of 52.1 g (0.93 mol, 3.00 eq.) KOH in 140 mL methanol. After cooling in an ice-bath for 5 min the solution was filtered to remove precipitated KCl. To this solution 60.6 g (0.31 mol, 1.00 eq.) methyl 4-(2-hydroxyethoxy)benzoate (1a) were added in one step, and the solution was stirred until homogenous. After standing for 24 h at room temperature, the basic solution was acidified with 2 M HCl to pH 4 and all solvents were removed until dryness in vacuo. The residue was transferred into a soxhlet apparatus and extracted using THF (1000 mL) overnight. After cooling at −20 °C and vacuum filtration 47.5 g (0.24 mol 77%) N-hydroxy-4-(2-hydroxyethoxy)benzamide (1b) were obtained as a colourless solid.
1H NMR (300 MHz, DMSO-d6) δ [ppm] = 11.06 (s, 1H, NH), 8.90 (s, 1H, NH–OH), 7.71 (AA′BB′, 2H, HC2,6), 6.98 (AA′BB′, 2H, HC3,5), 4.89 (t, J = 5.5 Hz, 1H, HO–CH2), 4.03 (t, J = 4.9 HZ, 2H, CH2–O–Ar), 3.72 (dt, J = 5.5, 4.9 Hz, 2H, HO–CH2–).
13C NMR (75 MHz, DMSO-d6) δ [ppm] = 164.02 C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O, 160.95 C4, 128.62 C2,6, 124.82 C1, 123.13 C3,5, 69.70 Ar–O–C, 59.48 HO–C.
O, 160.95 C4, 128.62 C2,6, 124.82 C1, 123.13 C3,5, 69.70 Ar–O–C, 59.48 HO–C.
IR-ATR ν [cm−1] = 3279 m, 2947 w, 2750 m br, 1638 m, 1606 vs. (CO), 1564 s, 1503 s, 1445 m, 1413 m, 1378 m, 1335 w, 1304 m, 1253 vs. (C–O), 1185 m, 1162 m, 1115 w, 1088 s, 1050 s, 1031 m, 1010 m, 919 m, 894 vs., 847 vs., 817 m.
R
f (EA:PE = 2:1) = 0.05.
Elemental analysis. Found C: 53.68, H: 6.09, N: 6.66 – calculated 54.82, H: 5.62, N: 7.10.
FD-MS (m/z) = 197.2532 (100%), 198.2448 (8.1%).
(1c) 2-(4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenoxy)ethan-1-ol. (1c) was prepared in a modified procedure based on the work of Couturier et al.31 19.72 g (0.10 mol, 1.00 eq.) N-hydroxy-4-(2-hydroxyethoxy)benzamide (1b) were suspended in 2000 mL DCM in a dry 3 L flask. To this suspension 31.25 g (0.30 mol, 3.00 eq.) 2,2-dimethoxypropane and 23.23 g (0.10 mol, 1.00 eq.) camphor sulfonic acid were added. The suspension was vigorously stirred for 15 h and then neutralized by addition of 250 mL 4 M aqueous NaOH solution. After stirring over night the organic layer was separated and washed twice with 100 mL 2 M NaOH, dried using Na2SO4 and evaporated to dryness in vacuo. The pure product was obtained by column chromatography (SiO2, EA
:PE = 1:1 as eluent). Yield: 20.89 g (0.088 mol, 88%) as colourless liquid that solidified at −20 °C.
1H NMR (300 MHz, DMSO-d6) δ [ppm] = 7.64 (AA′BB′, 2H, HC3,5), 6.94 (AA′BB′, 2H, HC2,6) 4.12 (t, J = 5.2 Hz, 2H, Ar–O–CH2–), 3.98 (t, J = 5.2 Hz, 2H, HO–CH2–), 1.66 (s, 6H, CH3).
13C NMR (75 MHz, DMSO-d6) δ [ppm] = 161.12 C1, 158.21 CN, 128.58 C3,5, 116.50 C4, 115.34 C(CH3)2, 114.71 C2,6, 69.45 Ar–O–CH2–, 61.43 HO–CH2–, 24.97 CH3.
IR-ATR ν [cm−1] = 3396 vw br (OH), 2935 vw, 1710 vw, 1606 m (CN), 1514 m, 1454 w, 1423 m, 1363 m, 1306 m, 1251 vs., 1215 s, 1166 s, 1115 m, 1078 s, 1044 m, 1010 m, 978 m, 945 w, 916 m, 884 m, 836 s, 816 m, 771 m.
R
f (EA:PE = 1:1) = 0.38.
FD-MS (m/z) = 237.2485 (100%), 238.2470 (11.5%), 239.2456 (1.2%).
(1d) 5,5-dimethyl-3-(4-(2-(oxiran-2-ylmethoxy)ethoxy)phenyl)-1,4,2-dioxazole. A 250 mL three-necked flask equipped with a dropping funnel, thermometer and mechanical stirrer was charged with 56 mL 50% (w/w) aqueous NaOH solution, 35.0 mL (0.45 mol, 5.3 eq.) epichlorhydrin and 1.18 g (3.5 mmol, 0.04 eq.) tetrabutylammonium hydrogen sulfate (TBAHS). 20.0 g (84.3 mol, 1.0 eq.) 2-(4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenoxy) ethan-1-ol were added within 30 minutes while cooling in an ice bath. After addition, the ice bath was removed and the suspension stirred vigorously overnight. After complete conversion of the alcohol was verified via TLC the reaction was quenched by addition of 100 mL of an ice-water mixture. The organic phase was separated, and the aqueous phase was extracted three times each with 60 mL dichloromethane. The combined organic phase was washed with brine until a pH of 8 was obtained, then dried using Na2SO4, filtered and all solvents were removed in vacuo. After column chromatography (SiO2, eluent: PE
:EA = 4:1) 20.0 g (68.2 mmol, 81%) 5,5-dimethyl-3-(4-(2-(oxiran-2-ylmethoxy)ethoxy)phenyl)-1,4,2-dioxazole were obtained as a colourless liquid.
1H NMR (400 MHz, DMSO-d6) δ = 7.68–7.59 (AA′BB′, 2H, HC3,5), 7.11–7.02 (AA′BB′, 2H, HC2,6), 4.21–4.14 (m, 2H, Ar–O–CH2–CH2–), 3.85–3.72 (m, 3H, Ar–O–CH2–CH2, epoxide-CH2–O), 3.35–3.27 (m, 1H, epoxide-CH2–O), 3.15–3.09 (m, CH), 2.73 (dd, J = 5.1, 4.2 Hz, 1H, CH2 in epoxide), 2.56 (dd, J = 5.1, 2.7 Hz, 1H, CH2 in epoxide), 1.61 (s, 6H, acetonide CH3).
(1e) 2-(4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenoxy)ethyl methacrylate. In a 50 mL Schlenk flask 6.06 g (0.026 mol, 1.10 eq.) 2-(4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenoxy)ethan-1-ol, 2.00 g (0.023 mol, 1.00 eq.) methacrylic acid and 0.23 g (1.9 mmol, 0.08 eq.) 4-dimethylaminopyridine (DMAP) were dissolved in 15 mL dichloromethane. Under ice cooling it was added 3.23 g (0.026 mol, 1.10 eq.) N,N′-diisopropylcarbodiimide (DIC) within 5 minutes. After stirring for 15 minutes under ice cooling, the mixture was stirred for 5 h at rt. The mixture was filtered through a frit, and the filtrate was washed twice with 100 mL of a saturated sodium hydrogen carbonate solution. The organic phase was dried over MgSO4 and the solvent was removed under reduced pressure. The solid was purified by column chromatography in a solvent mixture of PE
:EA (6:1). Methacrylate hydroxamic acid acetonide (MAHAA, 1e) was obtained in yields of 5.35 g (0.018 mol, 76%).
1H NMR (300 MHz, CDCl3): δ [ppm] = 7.73 (AA′BB′, 2H, HC3,5), 6.93 (AA′BB′, 2H, HC2,6), 6.15 (s, 1H, HCC), 5.60 (s, 1H, HCC), 4.51 (m, 2H, MA–O–CH2–), 4.26 (m, 2H, Ar–O–CH2), 1.96 (s, 3H, CH3–CC), 1.67 (s, 6H, acetonide CH3).
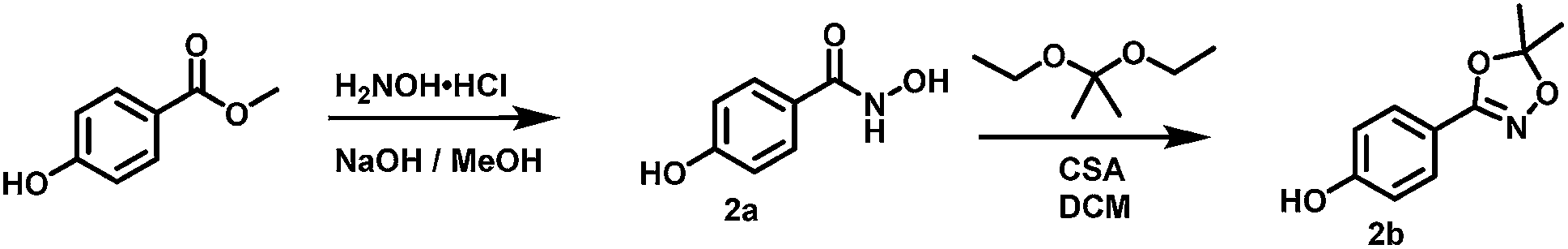
(2a) N,4-dihydroxybenzamide. (2a) was prepared in a modified procedure based on the work of Jeanrenaud et al.38 A 250 mL round bottom flask was charged with 3.80 g (25 mmol, 1 eq.) methyl paraben and 3.47 g (50 mmol, 2 eq.) H2NOH·HCl in 87.5 mL water. To this suspension 3.5 g (88 mmol, 3.5 eq.) NaOH as a 2 M solution was added. After 4 hours of stirring at room temperature the solution was acidified using 110 mL 1 M HCl and cooled overnight at 4 °C. Unreacted reagents were filtered off and the residue reduced in vacuo to few mL residual volume. The residual aqueous phase was extracted four times with 50 mL EA each. The combined organic phase was dried using Na2SO4 and freed from any solvents in vacuo. The crude product was purified using column chromatography (SiO2, PE
:EA = 1:1 as starting eluent, increasing elution strength gradually up to EA:MeOH 19:1). Yield 820 mg (5.4 mmol, 21%) of N,4-dihydroxybenzamide (2a) as a colourless solid.
1H NMR (300 MHz, DMSO-d6) δ [ppm] = 10.97 (s, 1H, NH), 9.96 (s, 1H, Ar–OH), 8.84 (s, 1H, NH–OH), 7.62 (AA′BB′, 2H, HC2,6), 6.78 (AA′BB′, 2H, HC3,5)
13C NMR (75 MHz, DMSO-d6) δ [ppm] = 164.48 (CO), 160.12 C1, 128.81 C2,6, 123.40 C4, 114.98 C3,5.
IR-ATR ν [cm−1] = 3314 m, 3191 m, br, 2923 w, 1606 s (CO), 1570 s, 1534 m, 1493 m, 1457 m, 1457 m, 1350 m, 1316 m, 1263 s, 1128 vs. (C–O), 1177 m, 1157 vs., 1028 m, 971 m, 893 s, 848 vs., 768 s.
R
f (EA:PE = 2:1) = 0.09.
Mp: 146 °C.
(2b) 4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenol. (2b) was prepared in a modified procedure based on the work of Couturier et al..31 A dried 100 mL Schlenk flask was charged with 337 mg (2.2 mmol, 1 eq.) N,4-dihydroxybenzamide (2b) and dried with 4 mL benzene under high vacuum. The solid residue was suspended in 50 mL of dry DCM (dried over P2O5, followed by distillation) and 872 mg (6.6 mmol, 3 eq.) 2,2-diethoxypropane were added. To this suspension 511 mg (2.2 mmol, 1 eq.) camphor sulfonic acid was added at once under argon atmosphere. After 105 minutes of stirring at room temperature the reaction was quenched by the addition of 10 mL saturated NaHCO3 solution. The mixture was stirred over night at room temperature and then the organic phase separated. The aqueous phase was extracted three times with 50 mL ether, and the combined organic phase dried using Na2SO4. After evaporation of all solvents in vacuo the residue was dissolved in THF and diethyl ether (to a final ratio of 1
:1) was added. 4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenol (2b) crystallized after days as colourless needles. Yield: 160 mg (0.8 mmol, 38%).
1H NMR (300 MHz, DMSO-d6) δ [ppm] = 7.65 (AA′BB′, 2H, HC3,5), 6.89 (AA′BB′, 2H, HC2,6), 1.68 (s, 6H, CH3).
13C NMR (75 MHz, DMSO-d6) δ [ppm] = 159.27 C1, 158.75 (CN), 128.90 C3,5, 115.94 C2,6, 115.58 CCH3, 115.24 C4, 24.90 CH3.
R f (EA) = 0.92.
(3a) N-hydroxy-4-(hydroxymethyl)benzamide. The synthesis was carried out in a modified procedure based on the work of Hauser and Renfrow analogous to the synthesis of (1b).37 Starting from 14.95 g (0.09 mol, 1 eq.) methyl 4-(hydroxymethyl)benzoate 11.28 g (0.068 mol, 75%) of N-hydroxy-4-(hydroxymethyl)benzamide (3a) were obtained as a colourless solid.
1H NMR (400 MHz, DMSO-d6) δ [ppm] = 11.08 (s, 1H, NH), 8.98 (s, 1H, NH–OH), 7.77–7.64 (AA′BB′, 2H, HC2,4), 7.46–7.30 (AA′BB′, 2H, HC3,5), 5.28 (t, J = 5.7 Hz, 1H, HO–CH2), 4.53 (d, J = 5.7 Hz, 2H, CH2).
13C NMR (100 MHz, DMSO-d6) δ [ppm] = 164.54 CO, 145.83 C4, 131.10 C1, 126.70 C2,4, 126.10 C3,5, 62.4S4 CH2.
(3b) (4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenyl)methanol. The synthesis was carried out in a modified procedure based on the work of Couturier et al. analogous to the synthesis of 1c.31 Starting from 5.03 g (0.03 mol, 1 eq.) N-hydroxy-4-(hydroxymethyl)benzamide 3.43 g (0.016 mol, 55%) (4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenyl)methanol (3b) was obtained as a colourless liquid which solidified after one week at −20 °C.
1H NMR (400 MHz, DMSO-d6) δ [ppm] = 7.72–7.59 (AA′BB′, 2H, HC3,5), 7.50–7.39 (AA′BB′, 2H, HC2,4), 5.36 (t, J = 5.7 Hz, 1H, CH2OH), 4.56 (d, J = 5.7 Hz, 2H, CH2), 1.63 (s, 6H, CH3).
13C NMR (100 MHz, DMSO-d6) δ [ppm] = 157.49 CN, 146.58 C1, 126.68 C2,4, 126.11 C3,5, 115.36 C(CH3)2, 62.36 CH2, 24.41 CH3.
R
f (EA:PE = 1:4) = 0.35.
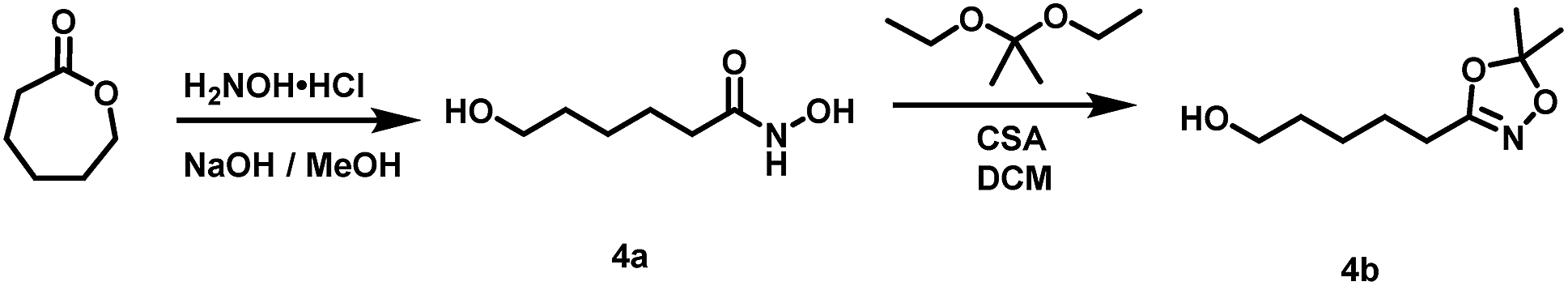
(4a) N,6-dihydroxyhexanamide. The synthesis was carried out in an modified procedure of Hauser and Renfrow.37 A 250 mL round bottom flask was charged with 17.12 g (150 mmol, 1.00 eq.) freshly distilled ε-caprolactone. In a separated flask 11.47 g (165 mmol, 1.1 eq.) H2NOH·HCl is dissolved in 65 mL methanol and neutralized with a solution of 12.62 g (225 mmol, 1.5 eq.) KOH in 32 mL methanol under shaking and cooling in a water bath. After 15 minutes the solution was filtered from precipitated KCl and added to the round bottom flask containing the caprolactone and stirred for 90 minutes. After the reaction had proceeded, the solution was acidified using conc. HCl to pH 4 and reduced under vacuum until most of the product solidified. The residue was recrystallized twice from THF (approx. 200 mL). 11.24 g (76 mmol, 51%) N,6-dihydroxyhexanamide (4a) were recovered after crystallization for 72 h at −20 °C as a colourless solid.
1H NMR (300 MHz, DMSO-d6) δ [ppm] = 10.37 (s, 0.9H, NH), 9.72 s (s, 0.1H, NH), 9.03 (s, 0.1H, NH–OH), 8.68 (s, 0.9H, NH–OH), 4.37 (s, 1H, CH2–OH), 3.35 (t, J = 6.4 Hz, 2H, C6H2), 1.93 (t, J = 7.5 Hz, 2H, C2H2), 1.52–1.42 (m, 2H, C3H2), 1.42–1.32 (m, 2H, C5H2), 1.30–1.16 (m, 2H, C4H2).
13C NMR (75 MHz, DMSO-d6) δ [ppm] = 169.25 C1, 60.67 C6, 32.42 C2, 32.30 C5, 25.23 C4, 25.16 C3.
IR-ATR ν [cm−1] = 3311 m, 3205 m, 3029 m, 2932 m, 2858 m br, 1616 vs. (CO), 1544 m, 1449 m, 1466 m, 1362 m, 1274 m, 1116 m, 1078 m, 1055 vs., 1012 vs., 977 vs., 785 m.
R
f (EA:MeOH = 9:1) = 0.42.
Mp: 84 °C.
(4b) 5-(5,5-dimethyl-1,4,2-dioxazol-3-yl)pentan-1-ol. The synthesis of 4b was carried out analogous to the preparation of 1 on a scale of 4.27 g (29 mmol) N,6-dihydroxyhexanamide (4a). Purification was performed via column chromatography (SiO2, EA
:PE = 1:1 as eluent). Yield: 1.25 g (7 mmol, 23%) 5-(5,5-dimethyl-1,4,2-dioxazol-3-yl)pentan-1-ol (4b) as a colourless liquid.
1H NMR (300 MHz, CDCl3) δ [ppm] = 3.64 (t, J = 6.3 Hz, 2H, C1H2), 2.31 (t, J = 7.3 Hz, C5H2), 1.76 (s, 1H, OH), 1.69–1.56 (m, 4H, C2H2, C4H2), 1.55 (s, 6H, CH3), 1.50–1.39 (m, 2H, C3H2).
13C NMR (75 MHz, CDCl3) δ [ppm] = 160.39 CN, 114.54 C(CH3)2, 62.65 C1, 32.26 C2, 25.22 C3, 25.18 C4, 24.91 CH3, 23.95 C5.
R
f (EA:PE = 1:1) = 0.46.
Polymerization procedures
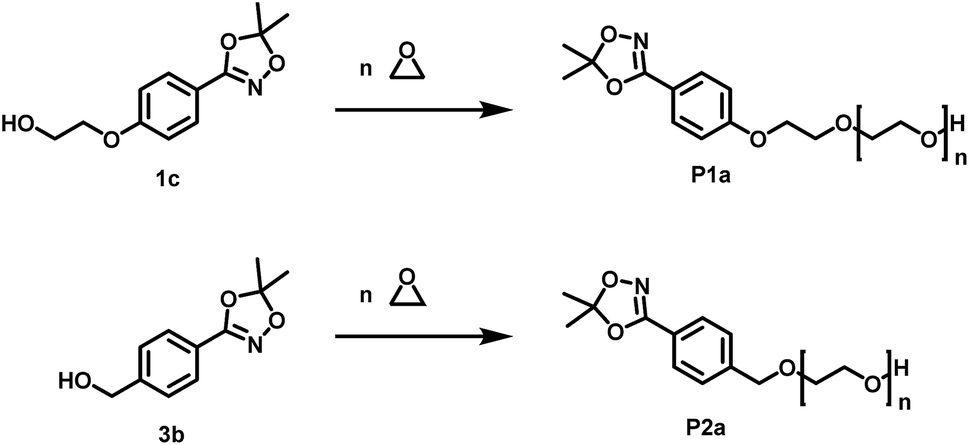
(P1a/P2a) synthesis of HAA-PEG and benzylHAA-PEG. Preparation in example of HAA-OH as initiator. In a typical procedure, 2-(4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenoxy)ethan-1-ol (1 eq.) (1d) was deprotonated by 90 mol% cesium hydroxide monohydrate in 10 mL benzene in a 100 mL Schlenk flask. After stirring for 60 minutes at 50 °C under vacuum the solvents were removed with high vacuum (10−3 mbar) and subsequently the flask was heated to 60 °C overnight under high vacuum. The formed alkoxide salt was dissolved in 15 mL dry THF and 2 mL dry DMSO. EO was first distilled in the cold into a graduated ampoule and subsequently into the reaction flask containing the dissolved initiator. The mixture was heated to 50 °C and stirred for 2 days. Afterwards, 1 mL methanol was added to quench the reaction. Except for high boiling DMSO all residual solvents were removed in vacuo. The residue was diluted with 40 mL DCM and washed three times with 10 mL water. The organic phase was separated and dried using Na2SO4. The volume was reduced in vacuo to approx. 5 mL and the polymer precipitated in cold diethyl ether (45 mL, −20 °C, recovery through centrifugation at 4500 RPM for 10 minutes). Yields: 73–94%.
1H NMR (400 MHz, DMSO-d6) δ [ppm] = 7.63 (AA′BB′, 2H, ArC3,5H), 7.06 (AA′BB′, 2H, ArC2,6H), 4.56 (t, J = 5.5 Hz, 1H, terminal OH), 4.19–4.11 (m, 2H, Ar–O–CH2–), 3.79–3.71 (m, 2H, Ar–O–CH2–CH2–O–), 3.71–3.28 (m, polyether backbone), 1.61 (s, 6H, CH3).
(P1b/P2b) removal of the acetal protecting group of HAA-PEG (2a) to form HA-PEG.

Preparation in example of HAA-PEG (P1a). In a typical procedure, 500 mg HAA-PEG (P1a) were dissolved in 20 mL 0.1 M HCl and shaken for 24 h. The polymer was recovered by adding 40 mL DCM and partitioning between the organic and aqueous phase. The organic phase was separated and dried over Na2SO4 and reduced in vacuo to a volume of approx. 5 mL. After precipitation in ice cold diethyl ether (45 mL, −20 °C, recovery using centrifugation at 4500 RPM for 10 minutes), HA-PEG (P1b) was obtained in near quantitative (>98%) yields.
1H NMR (400 MHz, DMSO-d6) δ [ppm] = 11.06 (s, 1H, NH), 8.90 (s, 1H, NH–OH), 7.72 (AA′BB′, 2H, ArC2,6H), 6.99 (AA′BB′, 2H, ArC3,5H), 4.57 (t, J = 5.5 Hz, 1H, terminal OH), 4.19–4.08 (m, 2H, Ar–O–CH2–), 3.79–3.72 (m, 2H, Ar–O–CH2–CH2–O–), 3.73–3.37 (m, polyether backbone).
(P3a, P4a, P5a, P6a) synthesis of HAA-PPO, HAA-PEEGE, benzylHAA-PPO, benzylHAA-PEEGE. Preparation example of HAA-PPO with HAA-OH as an initiator. In a typical procedure 2-(4-(5,5-dimethyl-1,4,2-dioxazol-3-yl)phenoxy)ethan-1-ol (1 eq.) (1d) was deprotonated by 90 mol% cesium hydroxide monohydrate in 10 mL in a 25 mL Schlenk flask. After stirring for 60 minutes at 50 °C under vacuum the solvents were removed with high vacuum (10−3 mbar) and subsequently the flask heated to 60 °C overnight under high vacuum. 1 mL dry propylene oxide (dried via cryo-distillation over CaH2) were added to the formed alkoxide using a syringe and the polymerization was performed at 40 °C under vacuum overnight. Subsequently, 1 mL methanol was added to quench the reaction and the polymer was dissolved in 40 mL DCM and washed three times with 10 mL. The organic phase was separated, dried using Na2SO4 and all solvents were evaporated in vacuo. Typical yield: 78-95% as highly viscous amorphous polymer.
Preparation of PEEGE polymers was carried out analogously.
1H NMR (300 MHz, CDCl3) δ [ppm] = 7.70 (AA′BB′, 2H, ArC3,5H), 6.94 (AA′BB′, 2H, ArC2,6H), 4.19–4.11 (m, 2H, Ar–O–CH2–), 4.16 (m, 2H, Ar–O–CH2–CH2–O–), 3.85 (m, 2H, Ar–O–CH2–CH2–O–), 3.69–3.28 (m, polyether backbone), 1.66 (s, 6H, C(CH3)2), 1.34-1.08 (m, propylene oxide CH3).
(P6b) cleavage of the protection groups of HAA-PEEGE to obtain HA-linPG (linear polyglycerol). 50 mg of HAA-PEEGE (P6a) were dissolved in 10 mL isopropanol and 200 mg DOWEX 50WX8 acidic ion exchange resin were added. The mixture was shaken for 2 days and filtered afterwards. The resin was washed with 5 mL isopropanol and 5 mL methanol, and the combined organic phases were evaporated in vacuo to obtain 41 mg (81%) of HA-linPG (P6b) as yellowish amorphous polymer.
Cleavage of the protecting group of P3a, P4a and P5a was carried out analogous.
1H NMR (300 MHz, DMSO-d6) δ [ppm] = 11.07 (s, 1H, NH), 7.70 (AA′BB′, 2H, ArC2,6H), 6.99 (AA′BB′, 2H, ArC3,5H), 4.19–4.04 (m, 2H, Ar–O–CH2–), 3.72–3.13 (m, polyether backbone).
Nanoparticle modification
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. J. acknowledges a fellowship by the Max Planck Graduate Center. The authors thank Monika Schmelzer for SEC measurements, Tatjana Dänzer for TEM measurement, Thi Dinh for experimental assistance and Jan Hilgert for providing FeOx nanoparticles.Notes and references
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236 CrossRef CAS
.
-
(a) E. J. Werner, J. Kozhukh, M. Botta, E. G. Moore, S. Avedano, S. Aime and K. N. Raymond, Inorg. Chem., 2009, 48, 277 CrossRef CAS PubMed
- N. Patil, C. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2018, 82, 34 CrossRef CAS
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825 CrossRef CAS
-
(a) B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038 CrossRef CAS PubMed
-
(a) K. Niederer, C. Schüll, D. Leibig, T. Johann and H. Frey, Macromolecules, 2016, 49, 1655 CrossRef CAS
- G. Schwarzenbach and K. Schwarzenbach, Helv. Chim. Acta, 1963, 46, 1390 CrossRef CAS
- Y. K. Agrawal and S. G. Tandon, J. Inorg. Nucl. Chem., 1972, 34, 1291 CrossRef CAS
- J. P. Folkers, C. B. Gorman, P. E. Laibinis, S. Buchholz, G. M. Whitesides and R. G. Nuzzo, Langmuir, 1995, 11, 813 CrossRef CAS
- J. B. Neilands, Science, 1967, 156, 1443 CrossRef CAS PubMed
-
(a) R. Codd, Coord. Chem. Rev., 2008, 252, 1387 CrossRef CAS
- L. Bauer and O. Exner, Angew. Chem., Int. Ed. Engl., 1974, 13, 376 CrossRef
-
(a) A. Vannini, C. Volpari, G. Filocamo, E. C. Casavola, M. Brunetti, D. Renzoni, P. Chakravarty, C. Paolini, R. de Francesco, P. Gallinari, C. Steinkühler and S. Di Marco, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15064 CrossRef CAS PubMed
- US2402604A, 1946
-
(a) J. W. Rosthauser and A. Winston, Macromolecules, 1981, 14, 538 CrossRef CAS
- A. Winston and E. T. Mazza, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2019 CrossRef CAS
- Y. K. Agrawal, H. Kaur and S. K. Menon, React. Funct. Polym., 1999, 39, 155 CrossRef CAS
- T. Hirotsu, S. Katoh, K. Sugasaka, M. Sakuragi, K. Ichimura, Y. Suda, M. Fujishima, Y. Abe and T. Misonoo, J. Polym. Sci., Polym. Chem. Ed., 1986, 24, 1953 CrossRef CAS
- F. A. Alakhras, K. A. Dari and M. S. Mubarak, J. Appl. Polym. Sci., 2005, 97, 691 CrossRef CAS
-
(a) P. N. Sophiamma and K. Sreekumar, React. Funct. Polym., 1997, 35, 169 CrossRef CAS
-
(a) W. Kern and R. C. Schulz, Angew. Chem., 1957, 69, 153 CrossRef CAS
- R. S. Mello, E. S. Orth, W. Loh, H. D. Fiedler and F. Nome, Langmuir, 2011, 27, 15112 CrossRef CAS PubMed
-
(a) M. Imran ul-haq, J. L. Hamilton, B. F. L. Lai, R. A. Shenoi, S. A. Horte, I. Constantinescu, H. A. Leitch and J. N. Kizhakkedathu, ACS Nano, 2013, 7, 10704 CrossRef CAS PubMed
- C. J. Adams, J. J. Wilson and E. Boros, Mol. Pharmaceutics, 2017, 14, 2831 CrossRef CAS PubMed
- G. M. Iskander, H. M. Kapfenstein, T. P. Davis and D. E. Wiley, J. Appl. Polym. Sci., 2000, 78, 751 CrossRef CAS
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170 CrossRef CAS PubMed
-
S. R. Sandler and W. Karo, Organic functional group preparations, Acad. Press, Orlando, 1989, vol. 12 Search PubMed
- D. Geffken and J. Froböse, J. Prakt. Chem., 1994, 336, 550 CrossRef CAS
-
(a) J. Lim, Y. Song, J.-H. Jang, C.-H. Jeong, S. Lee, B. Park and Y. H. Seo, Arch. Pharmacal Res., 2018, 41, 967 CrossRef CAS PubMed
- D. Cerniauskaite, J. Rousseau, A. Sackus, P. Rollin and A. Tatibouët, Eur. J. Org. Chem., 2011, 2011, 2293 CrossRef
- M. Couturier, J. L. Tucker, C. Proulx, G. Boucher, P. Dubé, B. M. Andresen and A. Ghosh, J. Org. Chem., 2002, 67, 4833 CrossRef CAS
- D. Geffken and J. Froböse, J. Prakt. Chem., 1993, 335, 555 CrossRef CAS
- M. Schömer and H. Frey, Macromolecules, 2012, 45, 3039 CrossRef
-
(a) M. Hans, H. Keul and M. Moeller, Polymer, 2009, 50, 1103 CrossRef CAS
- B. E. Bierer and D. G. Nathan, Blood, 1990, 76, 2052 CAS
- T. Yoshino, S. Inaba and Y. Ishido, Bull. Chem. Soc. Jpn., 1973, 46, 553 CrossRef CAS
- C. R. Hauser and W. B. Renfrow Jr, Org. Synth., 1939, 19, 15 CrossRef CAS
- A. Jeanrenaud, Ber. Dtsch. Chem. Ges., 1889, 22, 1270 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR, SEC, LCST and mass spectroscopy characterization, cell toxicity and proliferation assay data. See DOI: 10.1039/c9sc02557j |
| This journal is © The Royal Society of Chemistry 2019 |