
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConceptual similarities between zeolites and artificial enzymes†
Eva M.
Gallego
 ,
Cecilia
Paris
,
Ángel
Cantín
,
Manuel
Moliner
and
Avelino
Corma
*
,
Cecilia
Paris
,
Ángel
Cantín
,
Manuel
Moliner
and
Avelino
Corma
*
Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, 46022 València, Spain. E-mail: acorma@itq.upv.es
First published on 17th July 2019
Abstract
By using a Diels–Alder (DA) reaction as a base case, we show that a pure silica zeolite acting as an entropy-trapping scaffold can be synthesized with an organic structure directing agent (OSDA) analogue of the transition state (TS) of the DA reaction. A cavity stabilization of the TS is observed with the corresponding decrease in the activation energy of the reaction. A lower enthalpy of activation and a larger decrease in entropy are obtained with the zeolite synthesized with the analogue of the DA TS when compared with other zeolitic structures. Those differences are maintained, while catalytic activity is increased, when active sites are introduced in the zeolite. The catalytic zeolitic system synthesized with the OSDA analogue of the TS shows conceptual similarities with “de novo design” of an artificial enzyme to perform DA reactions, in where a suitable scaffold of existing proteins is chosen, and computationally designed active sites able to catalyze the cycloaddition reaction are introduced.
1. Introduction
The Diels–Alder (DA) reaction allows the efficient preparation of complex cyclic and polycyclic organic compounds by reacting electron rich 1,3-dienes with electron-deficient dienophiles. The relevance of the reaction is based on the fact that it can generate two C–C bonds and up to four stereogenic centers in a single concerted step.1 This cycloaddition reaction has played a key role along the years in the chemical industry since its discovery,2 covering from the early applications in the context of total synthesis of natural products,3 to the recent biomass transformation processes for the selective preparation of aromatic molecules.4DA reaction occurs through a highly ordered transition state (TS).5 The reaction can take place by thermal activation,6 photochemical irradiation,7 and homogeneous8 or heterogeneous catalysis,9 among others. It is very interesting that up to recently it was thought that DA reaction does not occur in nature since no enzymes were found to catalyse the cycloaddition reaction. However, it is known there are enzymes that catalyse reactions between dienes and dienophiles in cycloaddition processes.10 For example, the naturally insecticide Spinosyn A is formed through an enzyme catalysed reaction. The reaction mechanism has been explained by assuming that the cyclic molecule can be constrained within the enzyme environment, and the diene and dienophile will be arranged in a geometry closer to the transition state.11 However, since such a constraint will imply a decrease in the entropy of the transition state, this should be compensated by hydrogen bonding with amino acid residues of the active center of the enzyme. If this was so, one could consider that nature may use existing enzymes and make them as the starting point for a general scaffold, which would be evolutionally modified to catalyse a specific reaction, such as cycloaddition reactions.12 This line of thought has inspired the synthesis of artificial enzymes, that could be prepared by combining the proper cavity or scaffold and locate there the necessary groups to achieve the hydrogen bonds that will stabilize the TS.13 In these cases, a protein with a binding pocket that approaches the two substrates and appropriately places hydrogen bond donor and acceptor, can effectively catalyse DA reactions. Following an analogous way of thinking, catalytic antibodies have been generated to a structure that mimics the pericyclic transition state for these types of reactions.14 Even metal ion catalysis has been combined with divers biomolecular scaffolds to promote cycloaddition reactions.15 Taking into account all of the above, a solid catalyst suitably designed for a DA reaction should be formed by a scaffold consisting of a cavity that directs the formation and accommodates the transition state, together with the hydrogen bonding required to minimize the enthalpy of activation of the TS during this reaction. In other words, one would like to design a solid catalyst with cavities that could be used as a molecular trap to decrease the entropic barrier of the reaction, while establishing weak interactions between the transition state and the surface atoms of the solid catalyst to minimize the enthalpy of the TS (ΔH≠).
Thinking about cavities with molecular dimensions on a solid catalyst, zeolites come right away into the game and, in fact, some work on DA reaction with zeolites has already been published.9,16 Probably the first report was made by Dessau,16c who showed that the DA reaction was catalyzed by ZSM-5 zeolite at 250–500 °C. Nevertheless, a high reaction temperature (≥250 °C)4,16c much higher than those needed by artificial enzymes (4–37 °C), was required.13a,15a,17 In general, the zeolite catalysts were selected on the bases of the already existing materials and no specific structures that can act as scaffold to direct the transformation of the DA transition state were searched.
We have recently showed that “ab initio” zeolite structures for a particular reaction can be synthesized by using a TS mimic of the reaction as a molecular “template” or organic structure directing agent (OSDA).18 If a zeolite is synthesized with such a mimic, it should minimize the free energy of the transition state relative to the reactive intermediate. This should be caused by the confinement effect that will stabilize the TS within the pores of the zeolite, decreasing the activation energy of the reaction. In fact, other remarkable catalytic improvements have been described in the literature dealing on confinement effects in zeolites, such as for the dimethylether carbonylation,19 the propane dehydrogenation,20 or the acetone condensation reactions,21 among others. In these cases, a better TS confinement within specific pockets and/or pores has been described, introducing the “right” fit to minimize the free energy barrier of reaction. Such an effect will be more notorious for reactions occurring at lower temperatures, since then the compensating contribution of entropic effects within the zeolite pores will be less important.22
Following this principle, it should be achievable to synthesize a zeolite whose cavities can act as scaffolds for a given DA reaction, using as OSDA a mimic of the DA reaction product, since transition state and reaction product are very close. If zeolite crystallization is successful, then the synthesized zeolite should already correspond to the scaffold for that reaction. Nevertheless, it should be possible to go one-step further if during the synthesis or by post synthesis methods, the required centers could be introduced into the framework structure to enhance the catalytic activity of the solid catalyst.
While it is true that structure flexibility of the cavities of the zeolite is smaller than either the scaffolds of enzymes, artificial enzymes and antibodies, designed accuracy appears to correlate with scaffold rigidity.13a,23 Then, the control over the backbone conformation that we have in the zeolite synthesized with the mimic, could be helpful for the design of a good solid catalyst.
Herein, we demonstrate the enhancement of the catalytic activity for the DA reaction between 1,3-cyclohexadiene and N-methylmaleimide when using a specific zeolite, ITQ-17 (BEC structure), which has been synthesized using an OSDA that mimics the DA product (see Fig. 1). In fact, the pure silica form of the BEC zeolite already shows a ∼2–2.5 fold increase of the initial reaction rate compared to the thermal reaction or the reaction in the presence of other related pure silica microporous materials (i.e. Beta zeolite). Notice that the activity due to the confinement overcomes the activity of the pure silica MCM-41 with a very large surface area, when working at low temperatures. The achieved data provide evidence that while entropic barriers are more positive on MCM-41 (∼50 J mol−1 K−1 more positive), the much lower enthalpic barrier in BEC (∼20 kJ mol−1 lower) overcomes the entropic barrier at low temperatures. These results put in relevance the importance of the stabilization effects achieved by the catalyst structures depending on the reaction temperature, clearly influencing their transition state enthalpies and entropies. Finally, the reaction rate of the DA process can also be remarkably increased by introducing well-defined single metal atom sites, such as Ti, within the framework of the optimized zeolite BEC microreactor, as it occurs with artificial enzymes.
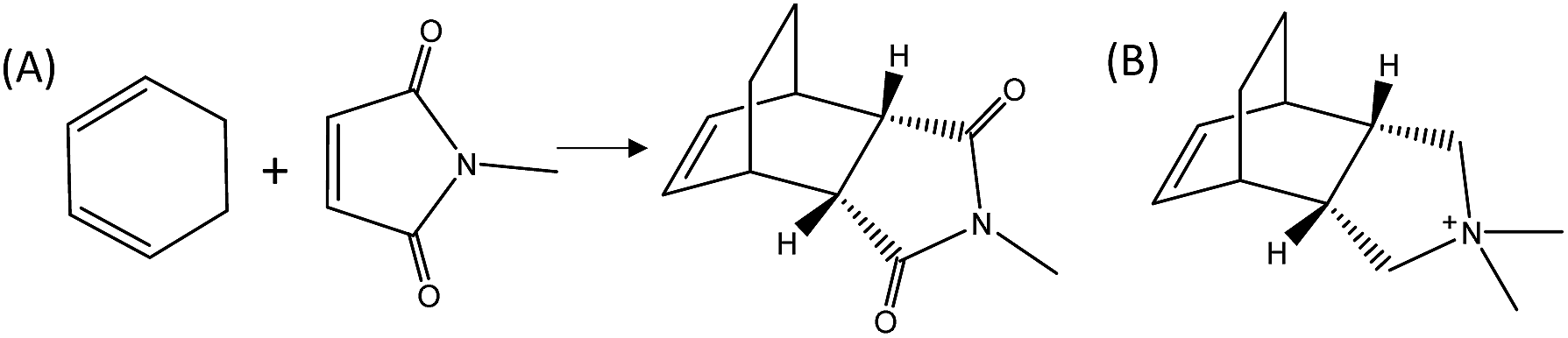 | ||
| Fig. 1 DA reaction between 1,3-cyclohexadiene and N-methylmaleimide (A) and (3aR,4S,7R,7aS)-2,2-dimethyl-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoisoindol-2-ium molecule as OSDA mimic of the DA adduct (B). | ||
2. Experimental section
2.1. Zeolite synthesis
Si-BEC. The synthesis of the pure silica BEC zeolite was carried out using the OSDA mimic of the DA-adduct (see Fig. 1B), according to the recipe described in the literature.24 The final molar composition of the gel was: SiO2:0.54 OSDA(OH):0.25 KOH:0.54 NH4F:7.25 H2O. The gel was heated in Teflon lined stainless steel autoclaves for 14 days at 175 °C, yielding to a white solid after filtration and extensive washing with boiling water and drying at 100 °C overnight. The solid was calcined in air at 580 °C during 6 hours to remove the organic molecules occluded during the hydrothermal synthesis.
The calcined Si-BEC material was ion exchanged with a 1.0 M ammonium chloride solution (1.0 g zeolite in 10 ml solution) at 80 °C for 3 h. The sample was filtered and washed with deionized water. Finally, the calcination was carried out in air at 400 °C for 4 h.
Si-Beta. 6.73 g of tetraethylammonium hydroxide (TEAOH, Sigma-Aldrich, 35% wt) was mixed with 4.8 g of a colloidal silica suspension (Ludox AS-40, Sigma-Aldrich, 40% wt). The resultant mixture was stirred for 1 hour and, afterwards, 2.96 g of an aqueous solution of NH4F (20% wt) was added to the gel and stirred for 4 h. The evaporation of the excess of water was carried out in a vacuum freeze-drying chamber. The final composition of the gel was: SiO2:0.5 TEAOH:0.5 NH4F:1 H2O. The gel was heated at 150 °C in Teflon lined stainless steel autoclaves for 48 hours, and the resultant solid was filtered, washed and dried at 100 °C. The solid was calcined in air at 580 °C during 6 hours to remove the organic molecules occluded during the hydrothermal synthesis.
Si-MCM-41. 4.74 g of an aqueous solution of tetramethylammonium hydroxide (TMAOH, Sigma Aldrich, 25% wt) was mixed with 3.1 g of hexadecyltrimethylammonium bromide (CTMABr, Sigma-Aldrich, 98% wt) and 18.4 g of double-distilled water. The resulting mixture was maintained under stirring at 40 °C until a clear solution was obtained. Finally, 3.0 g of aerosil (Sigma-Aldrich) was added as silica source. The mixture was stirred until evaporation of the excess of water. The composition of the final synthesis gel was: SiO2:0.26 TMAOH:0.17 CTMABr:24.4 H2O. The final gel was charged into a stainless steel autoclave with a Teflon liner, and the crystallization was conducted at 150 °C for 48 h under static conditions. The resultant solid was filtered, washed with water, and dried at 100 °C. Calcination of the sample to remove the organic molecules occluded within the solid particles was performed in air at 580 °C for 8 h.
Ti-BEC. The synthesis of the Ti-containing BEC zeolite was carried out according to the recipe described in the literature.25 The final molar composition of the gel was: SiO2:0.004 TiO2:0.25 OSDA:0.5 NH4F:2 H2O. The gel was heated in Teflon lined stainless steel autoclaves for 14 days at 175 °C, and the resultant solid was filtered, washed and dried at 100 °C overnight. The solid was calcined in air at 580 °C during 6 hours to remove the organic molecules occluded during the hydrothermal synthesis.
Ti-Beta. The synthesis of the Ti-Beta zeolite was carried out according to slight modifications of the recipe described in the literature.26 1.81 g of tetraethylammonium hydroxide (TEAOH, Sigma-Aldrich, 35% wt) was mixed with 36 mg of titanium ethoxide [Ti(C2H5O)4, Alfa Aesar] and 1.2 g of a colloidal silica suspension (Ludox AS-40, Sigma-Aldrich, 40% wt). The resultant mixture was stirred for 1 hour and, afterwards, 1.55 g of an aqueous solution of NH4F (10% wt) was added to the gel, and the mixture was stirred until evaporation of the excess of water. The final composition of the gel was: SiO2:0.02 Ti:0.54 TEAOH:0.54 NH4F:5 H2O. The gel was heated at 175 °C in Teflon lined stainless steel autoclaves for 10 days, and the resultant solid was filtered, washed and dried at 100 °C. The solid was calcined in air at 580 °C during 6 hours to remove the organic molecules occluded during the hydrothermal synthesis.
Ti-MCM-41. 4.74 g of an aqueous solution of tetramethylammonium hydroxide (TMAOH, Sigma Aldrich, 25% wt) was mixed with 3.1 g of hexadecyltrimethylammonium bromide (CTMABr, Sigma-Aldrich, 98% wt), 55 mg of titanium ethoxide [Ti(C2H5O)4, Alfa Aesar] and 18.4 g of double-distilled water. The resulting mixture was maintained under stirring at 40 °C until a clear solution was obtained. Finally, 3.0 g of aerosil (Sigma Aldrich) was added as silica source. The mixture was stirred until evaporation of the excess of water. The composition of the final synthesis gel was: SiO2:0.005 Ti:0.26 TMAOH:0.17 CTMABr:24.4 H2O. The resultant gel was charged into a stainless steel autoclave with a Teflon liner, and the crystallization was conducted at 150 °C for 48 h under static conditions. The achieved solid was filtered, washed with water, and dried at 100 °C. Calcination of the sample to remove the organic molecules occluded within the solid particles was performed in air at 580 °C for 8 h.
2.2. Characterization
Synthesized samples were characterized by powder X-ray diffraction (PXRD) measurements collected on a multisample Philips X'Pert diffractometer equipped with a graphite monochromator, operating at 40 kV and 35 mA, and using Cu Kα radiation (λ = 0.1542 nm).The chemical analysis was determined by a Varian 715-ES ICP-Optical Emission spectrometer, after dissolution of the solids in HNO3/HCl/HF aqueous solution.
The morphology of the samples was studied with a Field-Emission Scanning Electron Microscopy (FESEM) using a ZEISS Ultra-55 microscope.
Nitrogen adsorption isotherms at −196 °C were measured on a Micromeritics ASAP 2020 with a manometric adsorption analyzer to determinate the textural properties of the samples.
The UV-Vis spectra were collected using a Varian Cary 5 spectrometer equipped with a diffuse reflectance accessory.
2.3. Diels–Alder reaction
30 mg of each catalyst was activated under vacuum at 150 °C for 3 hours. After cooling down the solid at room temperature under N2 atmosphere, 22.9 mg of N-methylmaleimide (0.20 mmol; Sigma-Aldrich, 97%) and 2 ml of CDCl3 were added. The mixture was stirred and the temperature fixed at 60, 80 and 100 °C. Then, 25 μl of freshly distilled 1,3-cyclohexadiene (0.26 mmol, Sigma-Aldrich, 96%) was added via syringe. 1,4-Dinitrobenzene (Sigma-Aldrich, 98%) was used as internal standard. The samples were taken at different reaction times, and they were analyzed by liquid 1H NMR to obtain initial rates and their respective rate constants (see 1H NMR spectra in Fig. S1†).The DA reaction between 1,3-cyclohexadiene and N-methylmaleimide follows a second-order kinetics (first-order in both Diels–Alder reactants). According to this, it is possible to obtain the reaction rate constant (k) and the initial rates for each catalyst at given temperatures.
The activation energy, Ea, can be calculated from the Arrhenius equation:
| k = Ae−Ea/RT | (1) |
The eqn (1) can be linearized by the logarithmic transformation of the dependent variable:
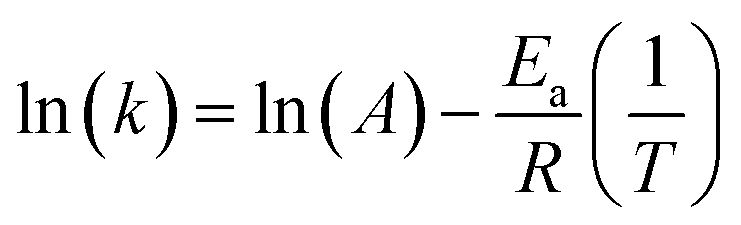 | (2) |
Thus, a plot of ln(k) versus 1/T allows the calculation of the pre-exponential A factor and the activation energy (Ea).
The enthalpy, ΔH≠, and the entropy, ΔS≠, can be calculated from the Eyring–Polanyi equation:
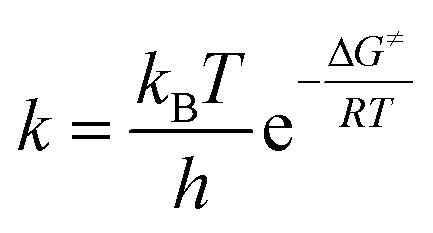 | (3) |
The Gibbs energy of activation during a reaction is equal to the change in the enthalpy minus the product of the temperature and the change in the entropy of the system.
| ΔG≠ = ΔH≠ − TΔS≠ | (4) |
Therefore, the combination of eqn (3) and (4) can be linearized as follows:
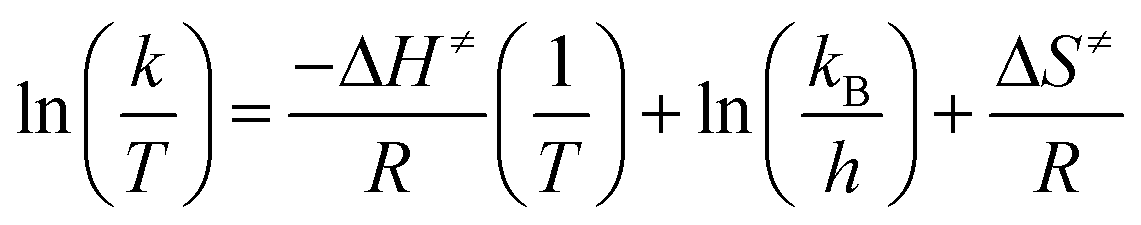 | (5) |
Thus, a plot of ln(k/T) versus 1/T produces a straight line with gradient equal to −ΔH≠/R and with a y-intercept of ln(kB/h) + ΔS≠/R.
3. Results and discussion
To prove the concept, we have used a mimic of the product formed during the DA reaction between 1,3-cyclohexadiene and N-methylmaleimide (see Fig. 1A). The chosen OSDA, (3aR,4S,7R,7aS)-2,2-dimethyl-2,3,3a,4,7,7a-hexahydro-1H-4,7-ethanoisoindol-2-ium molecule (see Fig. 1B), corresponds to the DA product and, it is well-known that the TS and the final product in a DA reaction are very similar.27 The use of this organic molecule as OSDA has allowed the efficient crystallization of the pure silica BEC structure (Si-BEC).24 This material presents a three-directional large pore system, in where the three channels cross in a larger cavity-like void space. The zeolite BEC synthesized is a polymorph of the commercially manufactured Beta zeolite (BEA). They are polymorphs composed by the same crystallographic layer, which has been arranged under different stacking order for each material, resulting in related but different crystalline structures (see Fig. S2†).28 Though both polymorphs are closely related, the specific difference in the crystalline structure should have an impact on the stabilization of the DA TS and the desired scaffold specify could be well tested with these two zeolites. Therefore, we have first synthesized pure silica BEC (Si-BEC) and pure silica BEA (Si-BEA) in where no framework specific potentially active sites have been introduced. In this case, the DA TS stabilization should be only accomplished by confinement and dispersion forces, as mastered by using the mimic of DA as organic template that results in the synthesis of the BEC structure.As said before, both zeolites have been obtained in the pure silica form, with very good crystallinities (see PXRD patterns in Fig. 2a) and with similar crystallite size (see Table 1). Indeed, Si-BEA shows small crystallites of 0.2–0.3 μm that are agglomerated in the form of 1–2 μm nanoparticles, while Si-BEC is formed by the aggregation of tetragonal crystals with size of 0.2–1 μm (see Fig. S3†). When the activity for the DA condensation of 1,3-cyclohexadiene and N-methylmaleimide is evaluated at 60 °C, the pure silica form of the BEC zeolite already shows a ∼2.5 fold increase of the initial reaction rate compared to the thermal reaction or when using the related pure Beta zeolite. The results show that even if a confinement by the nanopores of the solid catalyst already enhances the rate of the reaction, the larger structural specificity achieved by the scaffold synthesized with the mimic OSDA, has an impact on the rate of reaction.
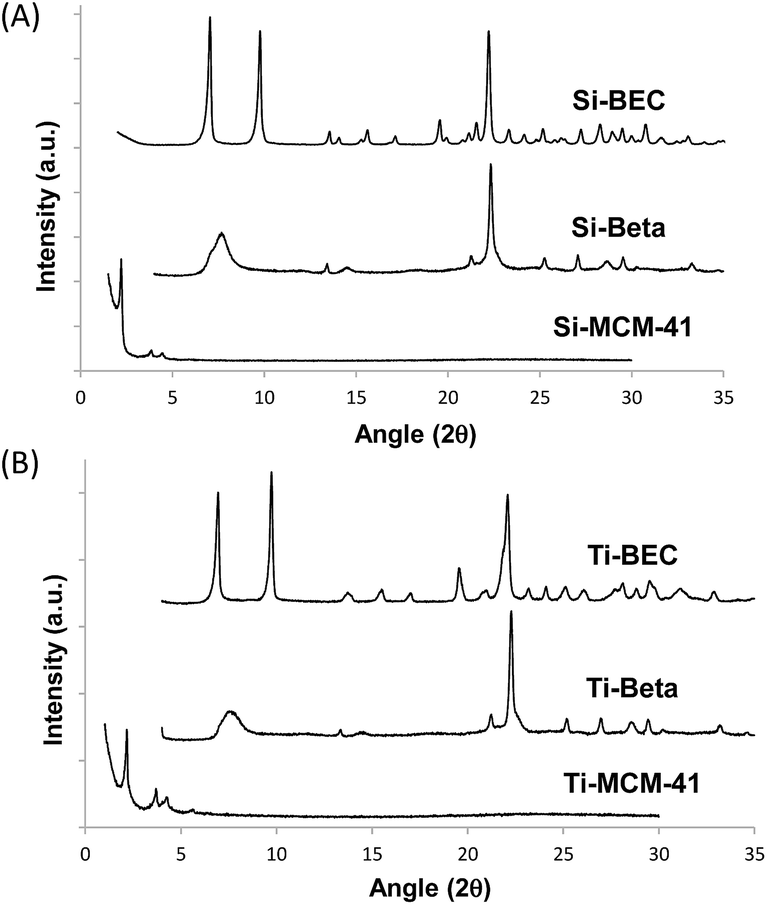 | ||
| Fig. 2 PXRD patterns of the calcined pure silica (A) and Ti-containing microporous and mesoporous materials (B). | ||
| Sample | Crystal size (μm) | Si/Ti | BET surface area (m2 g−1) | Ext. surface area (m2 g−1) | Micropore area (m2 g−1) | r 0/r0thermal (60 °C) | r 0/r0thermal (100 °C) |
|---|---|---|---|---|---|---|---|
| Si-MCM-41 | 0.05–0.10 | — | 731 | — | — | 1.6 | 1.7 |
| Si-Beta | 0.2–0.3 | — | 504 | 116 | 388 | 1.3 | 1.1 |
| Si-BEC | 0.2 × 1 | — | 389 | 24 | 365 | 2.5 | 1.4 |
| Ti-MCM-41 | 0.05–0.10 | 180 | 968 | — | — | 1.7 | 1.6 |
| Ti-Beta | 0.5 | 50 | 494 | 25 | 469 | 1.7 | — |
| Ti-BEC | 0.4–0.5 | 240 | 500 | 40 | 460 | 3.7 | 1.9 |
At this point, it is worth noting that some authors have described that periodically ordered mesoporous materials, i.e. MCM-41 or FSM-16, perform better than some crystalline silicate zeolites for different DA reactions, fact that was explained by the promoter effect of the weak acidity of external silanols.9b These ordered mesoporous materials show extended pores compared to those of zeolites, in general being ranged between 20–30 Å. Thus, for comparison purposes, a well-ordered mesoporous pure silica MCM-41 material was prepared (see details in Experimental section). The obtained Si-MCM-41 material presents the standard PXRD pattern of a well-ordered mesoporous MCM-41, as revealed by the presence of an intense peak centered at ∼2 degrees and two smaller peaks between ∼4–5 degrees (see Si-MCM-41 in Fig. 2A). In addition, the Si-MCM-41 shows a higher BET surface area (∼730 m2 g−1, see Table 1) than the previously synthesized zeolites. According to this, it was asserted that the Si-MCM-41 shows an improvement in the catalytic activity for the DA reaction at 60 °C compared to the Beta zeolite (see Fig. 3A).
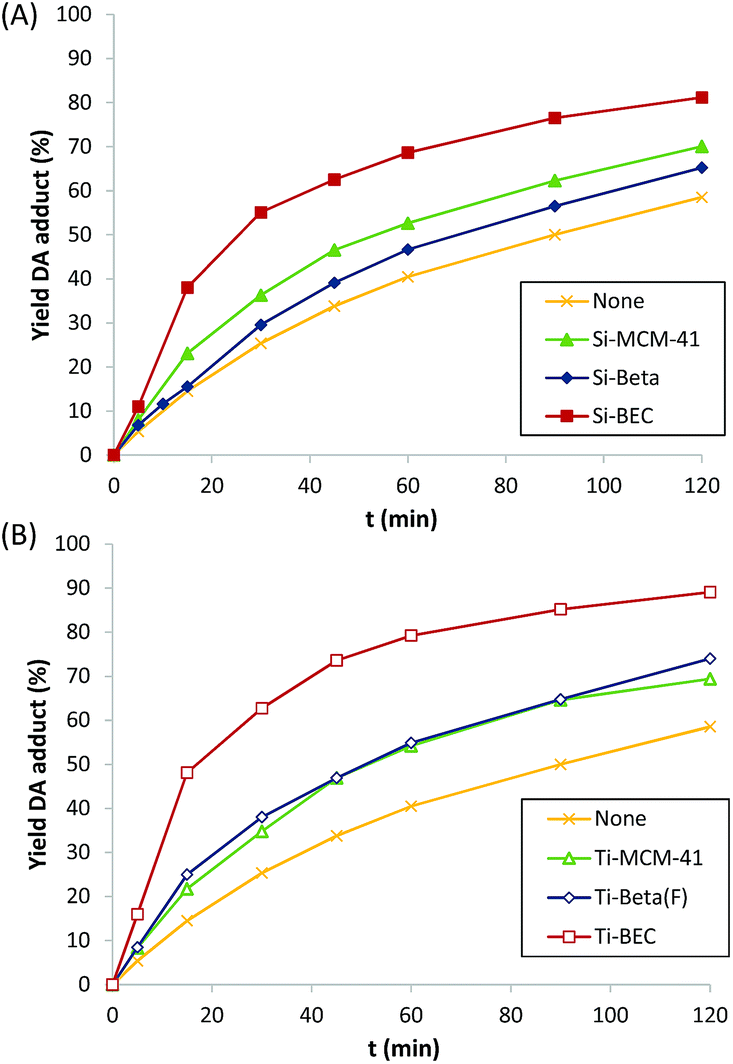 | ||
| Fig. 3 Catalytic activity for the Diels–Alder reaction using different silicate (A) and titanosilicate materials (B). Reaction conditions: 0.26 mmol 1,3-cyclohexadiene, 0.20 mmol N-methylmaleimide, 2 ml CDCl3, 30 mg of catalyst, and T = 60 °C. | ||
However, the Si-BEC catalyst, which was synthesized using the mimic of the DA adduct as OSDA, performs clearly better than the mesoporous Si-MCM-41 (see Fig. 3A), presenting a ∼1.7-fold increase of the initial reaction rate when the DA is carried out at 60 °C (see Table 1). The results obtained from the synthesis and catalytic evaluation of the different pure silica materials, shows the catalytic benefit of using an “imprinted” scaffold to facilitate the DA reaction in an analogous way as catalytic antibodies do for these processes.
As stated above, the differences in the macroscopic catalytic behavior observed should be related with the larger stabilization of the TS of the reaction within the scaffold of the Si-BEC zeolite, with the corresponding decrease of the activation energy of the reaction. Then, the calculated activation energies (Ea) give 18, 27 and 36 kJ mol−1 for the reaction carried out with Si-BEC, Si-BEA and Si-MCM-41, respectively (see linearized Arrhenius plots in Fig. S4A† and calculated Ea values in Table 2). Moreover, the Eyring–Polanyi approximation (see Fig. 4A) gives enthalpies of activation of 15, 24 and 33 kJ mol−1, respectively (see Table 2), while the decrease in entropy is remarkably higher for the Si-BEC zeolite (−247 kJ mol−1 K−1, see Table 2).
| Sample | E a (J mol−1) (±3.5) | ΔH≠ (kJ mol−1) (±3.5) | ΔS≠ (J mol−1 K−1) (±9) |
|---|---|---|---|
| Thermal | 34.4 | 31.5 | −206 |
| Si-MCM-41 | 36.0 | 33.1 | −197 |
| Si-Beta | 27.2 | 24.3 | −225 |
| Si-BEC | 18.4 | 15.5 | −247 |
| Ti-MCM-41 | 32.1 | 29.1 | −209 |
| Ti-BEC | 16.4 | 13.5 | −249 |
 | ||
| Fig. 4 Linearized Eyring–Polanyi plots of the Diels–Alder reaction for silicates (A) and Ti-silicates (B). | ||
The kinetic parameters strongly support the benefit of using a zeolitic scaffold synthesized by using the TS mimic template as OSDA. According to the previous results, it could be argued that the catalytic improvement for the DA reaction was already achieved only by confinement, in particular when the reaction is carried out at low temperatures. Now, we can proceed with the zeolite as in the case of synthetic enzymes, by introducing potentially active groups within the scaffold. This has been done here introducing Lewis acid sites (i.e. Ti) within the zeolite framework by direct synthesis. This metal was already observed to be an active site in zeolites for DA reactions catalysis.9c If this is so, the preparation of the BEC zeolite including isolated Lewis acid sites within its crystalline framework could also enhance the catalytic activity for the DA reaction. Having that in mind, the titanosilicate form of the BEC zeolite has been prepared,25 together with the titanosilicate forms of Beta and MCM-41 zeolites for comparison purposes (see Experimental details †).26 The physico-chemical characteristics of Ti-BEC, Ti-BEA and Ti-MCM-41 are summarized in Table 1. Interestingly, UV-Visible spectra for these materials show that most of the Ti is tetrahedrally-coordinated in framework positions (see bands centered at ∼220–230 nm in Fig. S6†).29
The catalytic results for the DA reaction show further improvement with all catalysts when Ti is incorporated (see Fig. 3B), being this increase relatively higher for the Ti zeolites (see r0/r0thermal at 60 °C in Table 1). Moreover, the activation energy is still lower for the Ti-BEC (see linearized Arrhenius plots in Fig. S4B† and calculated Ea values in Table 2). Notice that enthalpies of activation are slightly lower with the Ti containing samples (15.5 and 13.5 kJ mol−1 for BEC and Ti-BEC, respectively, see Table 2) while entropies of activation remain practically unaltered (−247 and −249 kJ mol−1 K−1 for BEC and Ti-BEC, respectively, see Table 2).
4. Conclusions
We have shown the catalytically benefit achieved by synthesizing a zeolite with an OSDA analogue to the TS of a DA reaction in where the crystallized zeolites provides transition state cavity stabilization, acting as the entropy trapping scaffold, in where active sites have been introduced. This is conceptually, at least, not too different from what it is called “de novo design” of an artificial enzyme to perform DA reactions in where a suitable scaffold of existing proteins is chosen and computationally designed active sites able to catalyze the cycloaddition are introduced. If we take into account what it has been said by Preiswerk et al.13a when working on the impact of scaffold rigidity on the design and evolution of artificial Diels–Alderase: “Because design accuracy appears to correlate with scaffold rigidity, improved control over backbone conformation will likely be the key to future efforts to design more efficient enzymes for divers chemical reactions”, it would appear that the smaller framework flexibility of the zeolite would be acceptable for DA reactions, provided that the right scaffold is used as shown in the present work. This approach brings again conceptual parallelism between enzymes and zeolites.30Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the European Union through ERC-AdG-2014-671093 (SynCatMatch), by the Spanish Government-MINECO through ‘‘Severo Ochoa” (SEV-2016-0683) and RTI2018-101033, and by La Caixa Foundation through MIT-SPAIN SEED FUND (LCF/PR/MIT17/11820002). Eva M. Gallego acknowledges “La Caixa – Severo Ochoa” International PhD Fellowships (call 2015). The Electron Microscopy Service of the UPV is acknowledged for their help in sample characterization.Notes and references
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- (a) R. B. Woodward, F. Sondheimer, D. Taub, K. Heusler and W. M. McLamore, J. Am. Chem. Soc., 1952, 74, 4223–4251 CrossRef CAS; (b) R. B. Woodward, F. E. Bader, H. Bickel, A. J. Frey and R. W. Kierstead, J. Am. Chem. Soc., 1956, 78, 2023–2025 CrossRef CAS.
- (a) J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS; (b) A. Settle, L. Berstis, N. Rorrer, Y. Roman-Leshkov, G. Beckham, R. Richards and D. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- M. I. Page and W. P. Jencks, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1678–1683 CrossRef CAS.
- (a) G. M. Ho, C. J. Huang, E. Y. Li, S. K. Hsu, T. Wu, M. M. L. Zulueta, K. B. Wu and S. C. Hung, Sci. Rep., 2016, 6, 35147 CrossRef PubMed; (b) A. Marrocchi, L. Minuti, A. Taticchi and H. W. Scheeren, Tetrahedron, 2001, 57, 4959–4965 CrossRef CAS.
- (a) D. A. Seeley, J. Am. Chem. Soc., 1972, 94, 4378–4380 CrossRef CAS; (b) F. Xu, X. Xiao and T. R. Hoye, J. Am. Chem. Soc., 2017, 139, 8400–8403 CrossRef CAS.
- (a) S. Yamabe, T. Dai and T. Minato, J. Am. Chem. Soc., 1995, 117, 10994–10997 CrossRef CAS; (b) O. Yilmaz, N. S. Kus, T. Tunç and E. Sahin, J. Mol. Struct., 2015, 1098, 72–75 CrossRef CAS; (c) S. Song, G. Wu, W. Dai, N. Guan and L. Li, J. Mol. Catal. A: Chem., 2016, 420, 134–141 CrossRef CAS.
- (a) Y. Narayana Murthy and C. Pillai, Synth. Commun., 1991, 21, 783–791 CrossRef; (b) Y. Kubota, H. Ishida, R. Nakamura and Y. Sugi, Stud. Surf. Sci. Catal., 2003, 146, 749–752 CrossRef CAS; (c) M. V. Gomez, A. Cantin, A. Corma and A. de la Hoz, J. Mol. Catal. A: Chem., 2005, 240, 16–21 CrossRef CAS.
- (a) H. Oikawa and T. Tokiwano, Nat. Prod. Rep., 2004, 21, 321–352 RSC; (b) H. Oikawa, Comprehensive Natural Products II, Chem. And Bio., ed. B.-L. Mander and H.-W. Liu, Oxford Elsevier, 2010, pp. 277–314 Search PubMed.
- E. G. Gordeev and V. P. Ananikov, PLoS One, 2015, 10, e0119984 CrossRef.
- H. Oikawa, Cell Chem. Biol., 2016, 23, 429–430 CrossRef CAS.
- (a) N. Preiswerk, T. Beck, J. D. Schulz, P. Milovník, C. Mayer, J. B. Siegel, D. Baker and D. Hilvert, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8013–8018 CrossRef CAS PubMed; (b) J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. R. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS.
- A. C. Braisted and P. G. Schultz, J. Am. Chem. Soc., 1990, 112, 7430–7431 CrossRef CAS.
- (a) G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230–3232 CrossRef CAS PubMed; (b) M. T. Reetz, Chem. Rec., 2012, 12, 391–406 CrossRef CAS.
- (a) M. Onaka, R. Yamasaki and T. Ookoshi, Stud. Surf. Sci. Catal., 1999, 121, 139–142 CrossRef CAS; (b) A. Cantín, M. V. Gómez and A. De la Hoz, Beilstein J. Org. Chem., 2016, 12, 2181–2188 CrossRef; (c) R. M. Dessau, J. Chem. Soc., Chem. Commun., 1986, 1167–1168 RSC.
- (a) A. Palma, M. Artelsmair, G. Wu, X. Lu, S. Barrow, N. Uddin, E. Rosta, E. Masson and O. Scherman, Angew. Chem., Int. Ed., 2017, 56, 15688–15692 CrossRef CAS; (b) P. Deuss, G. Popa, A. M. Z. Slawin, W. Laan and P. C. J. Kamer, ChemCatChem, 2013, 5, 1184–1191 CrossRef CAS.
- (a) E. M. Gallego, M. T. Portilla, C. Paris, A. León-Escamilla, M. Boronat, M. Moliner and A. Corma, Science, 2017, 355, 1051–1054 CrossRef CAS; (b) C. Li, C. Paris, J. Martinez-Triguero, M. Boronat, M. Moliner and A. Corma, Nat. Catal., 2018, 1, 547–554 CrossRef CAS.
- (a) A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS; (b) M. Boronat, C. Martínez and A. Corma, Phys. Chem. Chem. Phys., 2011, 13, 2603–2612 RSC.
- R. Gounder and E. Iglesia, J. Am. Chem. Soc., 2009, 131, 1958–1971 CrossRef CAS.
- S. Herrmann and E. Iglesia, J. Catal., 2017, 346, 134–153 CrossRef CAS.
- R. Gounder and E. Iglesia, Acc. Chem. Res., 2012, 45, 229–238 CrossRef CAS PubMed.
- M. E. Davis, CATTECH, 1997, 1, 19–26 CAS.
- A. Cantin, A. Corma, M. J. Diaz-Cabañas, J. L. Jordá, M. Moliner and F. Rey, Angew. Chem., Int. Ed., 2006, 45, 8013–8015 CrossRef CAS.
- M. Moliner, P. Serna, A. Cantín, G. Sastre, M. Díaz-Cabañas and A. Corma, J. Phys. Chem. C, 2008, 112, 19547–19554 CrossRef CAS.
- (a) T. Blasco, M. A. Camblor, A. Corma, P. Esteve, J. M. Guil, A. Martínez, J. A. Perdigón-Melón and S. Valencia, J. Phys. Chem. B, 1998, 102, 75–88 CrossRef CAS; (b) A. Corma, M. T. Navarro and J. Pérez-Pariente, J. Chem. Soc., Chem. Commun., 1994, 147–148 RSC.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS.
- J. B. Higgins, R. B. LaPierre, J. L. Schlenker, A. C. Rohrman, J. D. Wood, G. T. Kerr and W. J. Rohrbaugh, Zeolites, 1988, 8, 446–452 CrossRef CAS.
- M. Moliner and A. Corma, Microporous Mesoporous Mater., 2014, 189, 31–40 CrossRef CAS.
- (a) M. Boronat, C. Martínez-Sánchez, D. Law and A. Corma, J. Am. Chem. Soc., 2008, 130, 16316–16323 CrossRef CAS PubMed; (b) A. Corma, Angew. Chem., Int. Ed., 2016, 55, 6112–6113 CrossRef CAS; (c) R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S. J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02477h |
| This journal is © The Royal Society of Chemistry 2019 |