
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed enantioselective alkenylation of alkenylbenzene derivatives†
Zhi-Min
Chen‡§
 ,
Jianbo
Liu‡
,
Jing-Yao
Guo
,
Maximillan
Loch
,
Ryan J.
DeLuca
and
Matthew S.
Sigman
*
,
Jianbo
Liu‡
,
Jing-Yao
Guo
,
Maximillan
Loch
,
Ryan J.
DeLuca
and
Matthew S.
Sigman
*
Department of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, Utah 84112, USA. E-mail: sigman@chem.utah.edu
First published on 17th June 2019
Abstract
A regioselective and enantioselective palladium-catalyzed relay Heck alkenylation of alkenylbenzene derivatives to construct remote stereocenters is disclosed. Various β-substituted styrenes were readily obtained in moderate yields with good to excellent levels of enantioselectivity. This strategy provides rapid access to enantioenriched δ, ε, ζ, and η-alkenyl aryl compounds from simple starting materials. Mechanistic studies suggest that termination of the relay reaction is controlled by affinity of the arene for the Pd complex during migration.
Introduction
The migration of a metal through an alkyl chain via iterative β-hydride elimination/migratory insertion processes, often termed chain walking (or running) offers the opportunity to effectively functionalize multiple sites on a substrate.1 Initiation of this process is often accomplished through a metal-catalysed migratory insertion event of an alkene with applications in areas ranging from olefin polymerization2 to asymmetric catalysis.3 After the initial migratory insertion and subsequent chain walking, termination events are often coupled with either a functionalization of a metal-alkyl intermediate or re-establishing the unsaturation.4In this context, our lab has developed various enantioselective Heck-type reactions of di- and tri-substituted alkenes wherein the reaction is terminated by formation of a carbonyl derivative (Scheme 1A).3,5 The formation of the aldehyde facilitates this in two key regards: (1) the penultimate proposed intermediate (C) is thermodynamically stabilized according to computational studies and (2) the resulting aldehyde does not participate in migratory insertion events thus making the process irreversible.6 Coupled together, expanding the scope of chain walking events within a Heck platform will likely require similar tactics.
 | ||
| Scheme 1 Previous work with carbonyl derivatives and proposed work with alkenylbenzene derivatives. | ||
In this regard, our lab disclosed an enantioselective relay Heck arylation of alkenyl carbonyl derivatives, which delivered α,β-unsaturated compounds containing remote stereocenters (Scheme 1B).7 The α,β-unsaturated carbonyls are poor ligands compared to the initial electron rich alkene and do not participate in further Heck reactions. Combined, these results provide a basis for exploring other termination possibilities to expand the scope of these enantioselective processes. As an example, we initially envisaged an aryl group could be employed to deliver a styrenyl product (3) or, with the addition of a transmetallating reagent, a remotely difunctionalized product (I) (Scheme 1C). From a mechanistic perspective, the Pd-alkyl intermediate (D) arising from alkene 1 and alkenyl triflate 2 could undergo β-hydride elimination away from the newly formed stereocenter to deliver intermediate E. We postulated that alkene E would be in equilibrium with alkene F, which would ultimately provide the thermodynamically more stable styrene product (3) after dissociation of the Pd-hydride. However, styrene F could reinsert into the Pd-hydride bond to produce the stabilized π-benzyl intermediate (G) that could be intercepted with a transmetallating reagent to afford intermediate H followed by reductive elimination to deliver product I (in which two new stereocenters were formed). Herein, we demonstrate that the styrene product is favoured and can be produced in high enantioselectivity. Of perhaps more interest, we provide mechanistic evidence to why the reaction terminates in this fashion.
Results and discussion
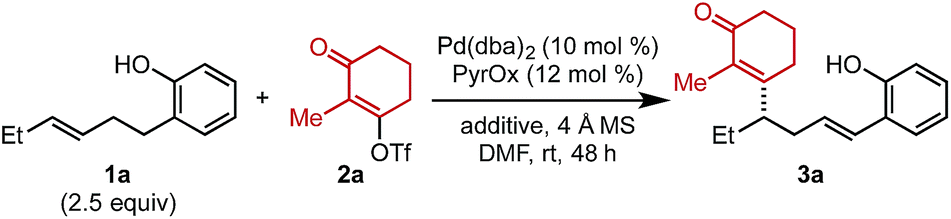
To initiate our investigation, Pd2(dba)3 and a pyridine oxazoline (PyrOx) ligand were combined with alkenylbenzene 1a and alkenyl triflate 2a under previously reported relay Heck conditions for substrates of this type (Table 1).8 Under these conditions, styrenyl product 3a was isolated in 42% yield and 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 er (entry 1). The mass balance was a mixture of the undesired migratory insertion product (insertion at the alkene carbon proximal to the aryl group) and other alkene isomers 4. Switching the precatalyst to Pd(dba)2 led to a slight increase in yield (entry 2) which was further improved to 55% with the addition of 7.5 mol% dba (entry 3).9 Additionally, during the course of our optimization, the effect of phenylboronic acid addition was evaluated (to explore the possibility of a remote difunctionalization reaction), which resulted in an increase in the isolated yield of styrenyl product 3a (entry 4). Lastly, the addition of electron-rich para-methoxyphenylboronic acid improved the yield to 69% (entry 5). It should be noted that no remote difunctionalization product was observed with either arylboronic acid additive (entries 4 and 5); however, the yield of the styrenyl product is increased.
:4 er (entry 1). The mass balance was a mixture of the undesired migratory insertion product (insertion at the alkene carbon proximal to the aryl group) and other alkene isomers 4. Switching the precatalyst to Pd(dba)2 led to a slight increase in yield (entry 2) which was further improved to 55% with the addition of 7.5 mol% dba (entry 3).9 Additionally, during the course of our optimization, the effect of phenylboronic acid addition was evaluated (to explore the possibility of a remote difunctionalization reaction), which resulted in an increase in the isolated yield of styrenyl product 3a (entry 4). Lastly, the addition of electron-rich para-methoxyphenylboronic acid improved the yield to 69% (entry 5). It should be noted that no remote difunctionalization product was observed with either arylboronic acid additive (entries 4 and 5); however, the yield of the styrenyl product is increased.
After optimized conditions were identified (entry 5), the scope of the alkenylbenzene substrate was explored (Table 2). In general, the desired styrenyl products were obtained with moderate yields and excellent enantiomeric ratios. Again, alkenyl triflate 2a was completely converted and the mass balance was mainly composed of alkene isomers 4 (a putative explanation is discussed below). A phenyl group on the substrate (1b) gave the desired styrenyl product in 64% and 96.5:3.5 er. A variety of electron-rich aryl groups were well-tolerated including p-methyl (3c), p-methoxy (3d), o-methoxy (3e), p-hydoxy (3f), and p-dimethylamino (3g).
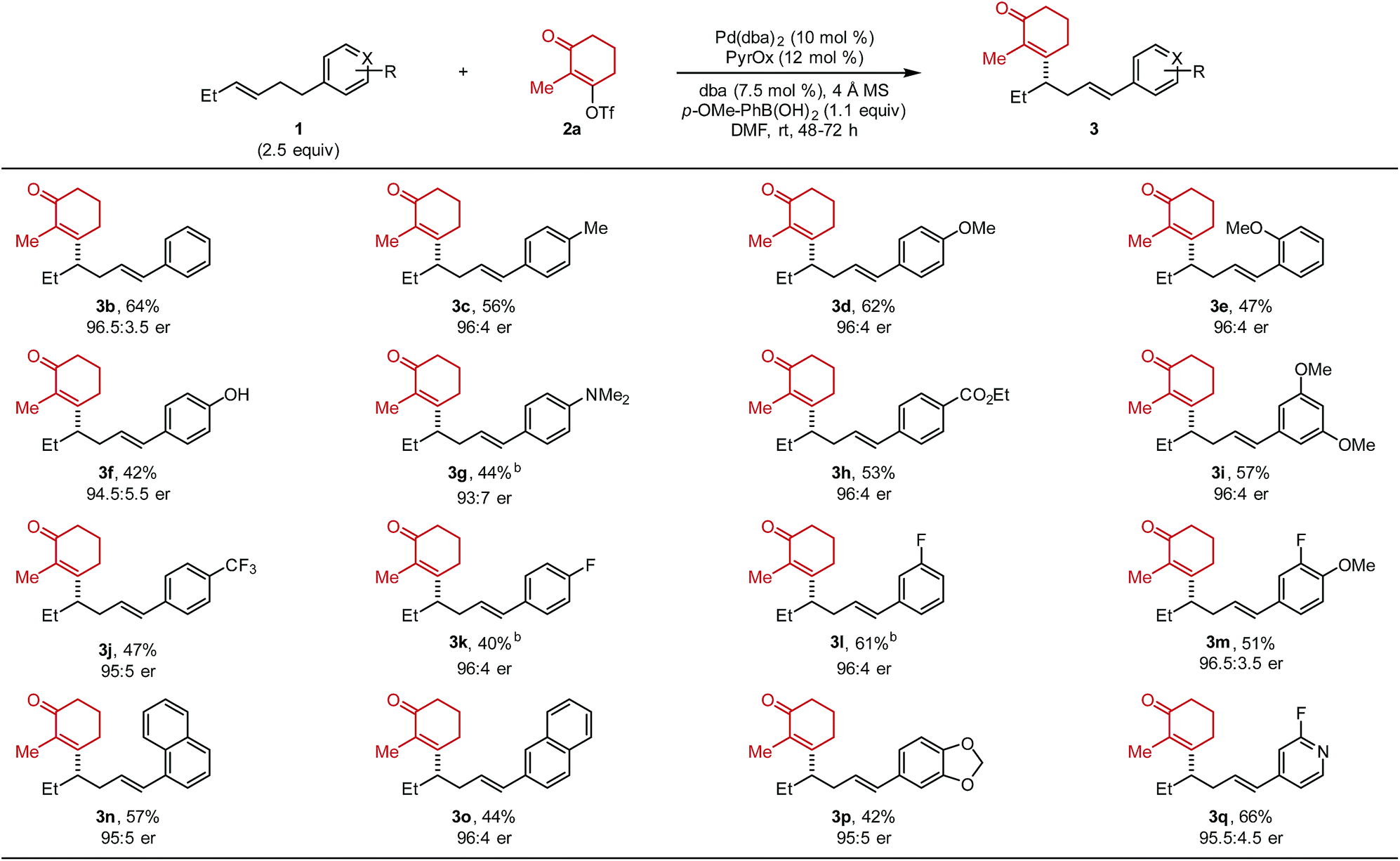
Additionally, electron-withdrawing substituents such as an ethyl ester (3h), two methoxy groups at the meta positions (3i), and a trifluoromethyl group (3j) were all competent. A fluorine atom was readily incorporated on the aromatic ring (3k–3m). Substrates containing naphthyl groups (1n and 1o) produced the desired products 3n and 3o in 57% and 44% yield, respectively. A benzodioxole group (1p) was successfully incorporated delivering styrenyl product 3p in 42% yield and 95:5 er. Lastly, a fluoropyridyl group (1q) delivered the corresponding product (3q) in 66% yield. Trisubstituted alkene substrates were also evaluated under the reaction conditions but no desired product was observed. It should be noted that the absolute configuration of 3d was determined to be (S) and thus the rest of the products were assigned as (S) by analogy.10
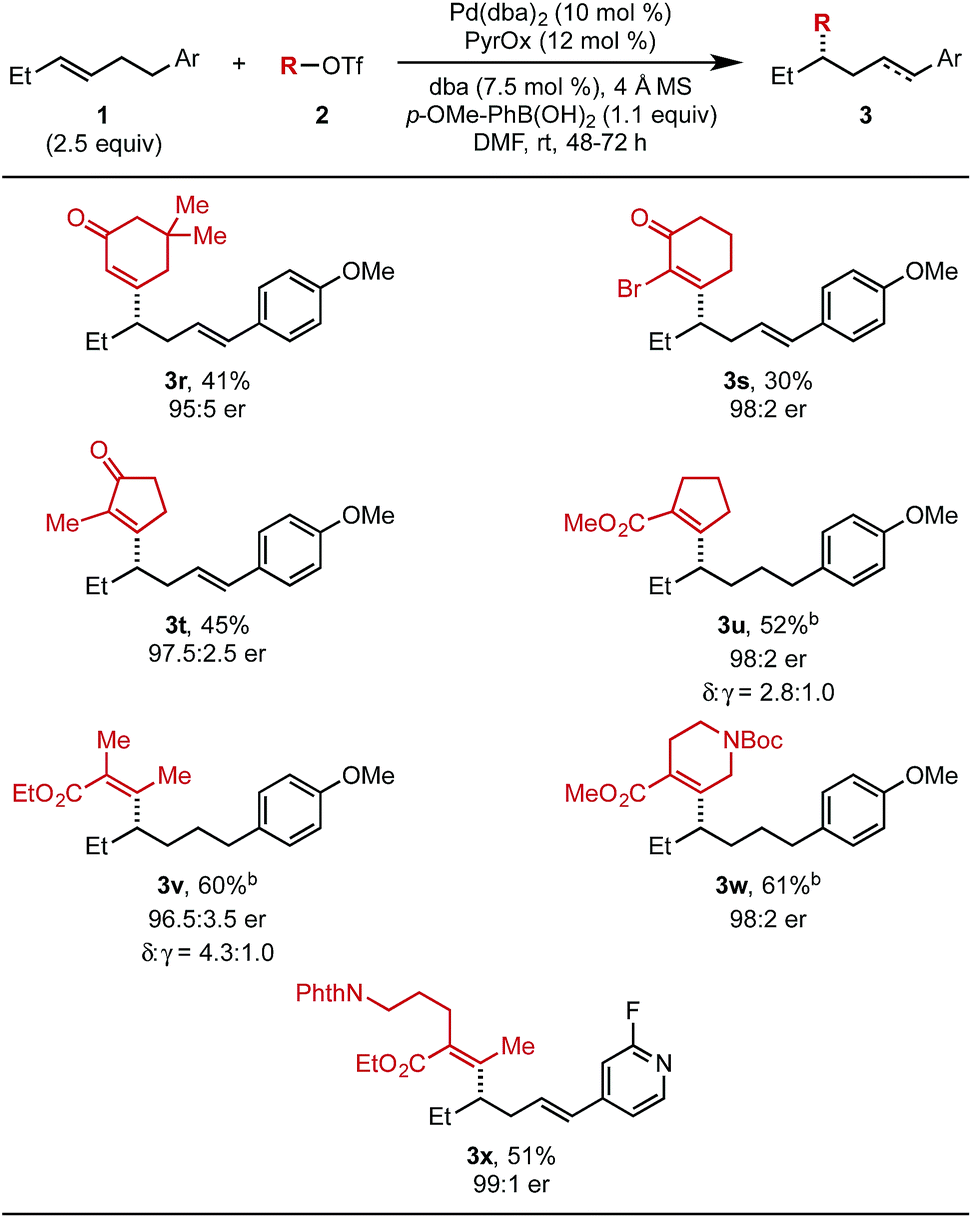
Next, the scope of alkenyl triflates was investigated (Table 3). For example, 5,5-dimethylcyclohexenone triflate (2r) and 2-bromo-substituted triflate 2s gave the corresponding products (3r and 3s) in 41% and 30% yield, respectively. An enol triflate containing a methyl-substituted cyclopentenone (2t) delivered product 3t in 45% yield and 97.5:2.5 er. Reaction with β-keto ester derived triflates 1u and 1v yielded the desired products (3u, 3v) in 52% and 60% yield, respectively. A β-keto ester derived triflate containing a Boc-protected amine (1w) produced 3w in 61% yield and 98:2 er. Additionally, an enol triflate containing a phthalimide (1x) furnished the desired styrenyl product in 51% yield and 99:1 er. The isolated yields of products 3u, 3v, and 3w were determined after hydrogenation, as a mixture of alkene isomers were produced and complicated purification.
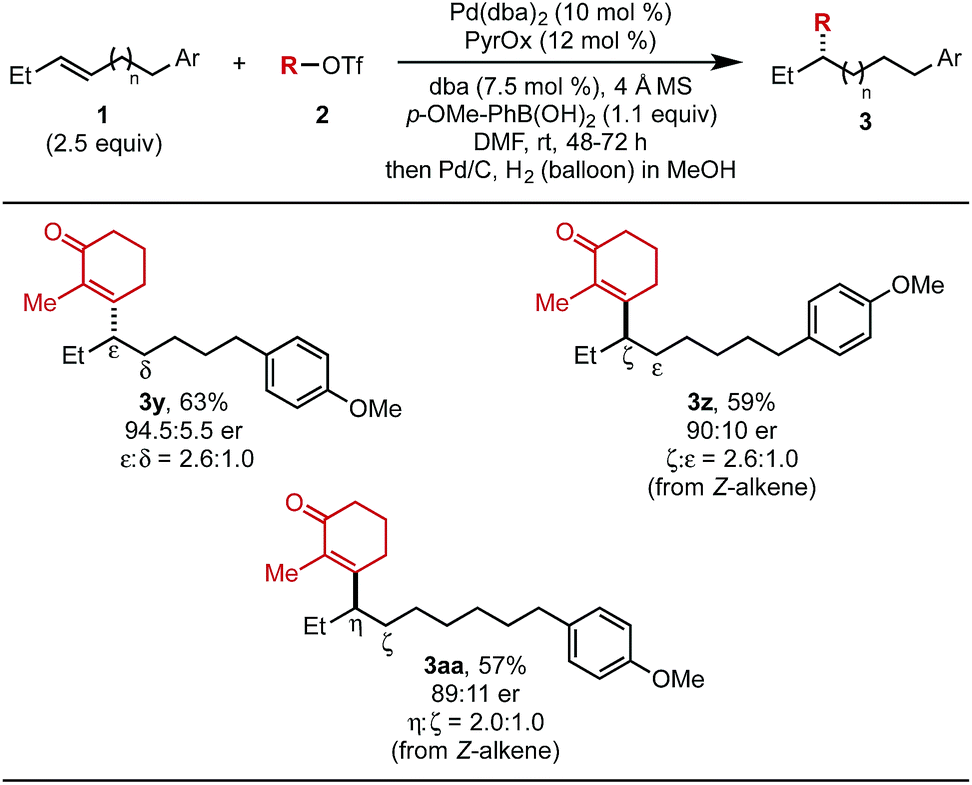
The effect of chain length on yield and selectivity was determined by comparing three additional substrates containing additional methylene units between the alkene and the aryl moiety (1y–1aa) (Table 4). When bishomoallylic aryl compound 1y was subjected to the standard reaction conditions followed by hydrogenation, product 3y was obtained in 63% (2.6:1.0 ratio of insertion products). Trishomoallylic aryl compound 1z delivered product 3z in similar yield and insertion selectivity, albeit with a slightly diminished er. Lastly, when the substrate contained an additional methylene unit (1aa), product 3aa was observed in 57% yield and reduced site selectivity (2.0:1.0 mixture of migratory insertion products). On the basis of our previous work,5b using (Z)-alkene substrates, the opposite enantiomer would be produced compared to reaction of the (E)-alkene. Thus, the absolute configuration of 3z and 3aa were assigned as (R).
As alluded to above, interesting features of the reaction include the observation of alkene isomers using several of the differentiated aryl substrates as well as the inability to capture Pd-alkyl intermediates through a subsequent cross-coupling. Therefore, we set out to interrogate the reaction mechanism with a focus on why the formation of the styrene product generally terminates the chain walking event(s). To initiate this investigation, deuterated alkenylbenzene 1y–D2 was subjected to the optimized reaction conditions with alkenyl triflate 2a (Table 5). The goal of this study was to determine the dynamics of the chain walking process. In the event, mono-deuterated products 3y(H–D) and 3y(D–H) were observed in 23% and 12% yield, respectively. In addition, di-deuterated product 3y(D–D) was also observed in 12% yield. This mixture suggests the palladium catalyst undergoes reversible β-hydride elimination/migratory insertion at the benzylic position of the substrate, which results in the scrambling of the deuterium atoms.
| a Each entry represents the isolated yield on 0.2 mmol scale. |
|---|

|
To further probe the mechanism, we investigated the effect of various features of the terminating aryl group on the selective formation of styrenes. Specifically, a ratio of alkene products 3 and 4 was observed as a function of the aryl group (Fig. 1A). To probe this relationship, systematic changes of the terminal aryl on this ratio were evaluated while conserving the remainder of the reaction features (Fig. 1B), and the results were compared to a number of DFT-derived physical organic parameters. These were calculated using a simple aryl model system depicted in Fig. 1C. A correlation was identified between the observed product selectivity (represented as ΔΔG‡) and the energy of an empty π* orbital as calculated by natural bond orbital (NBO) analysis (Fig. 1C). To validate this correlation, two extrapolations were tested that provide modestly improved selectivity. However, a substrate containing a para-Ph substituent performed in an unpredictable manner.
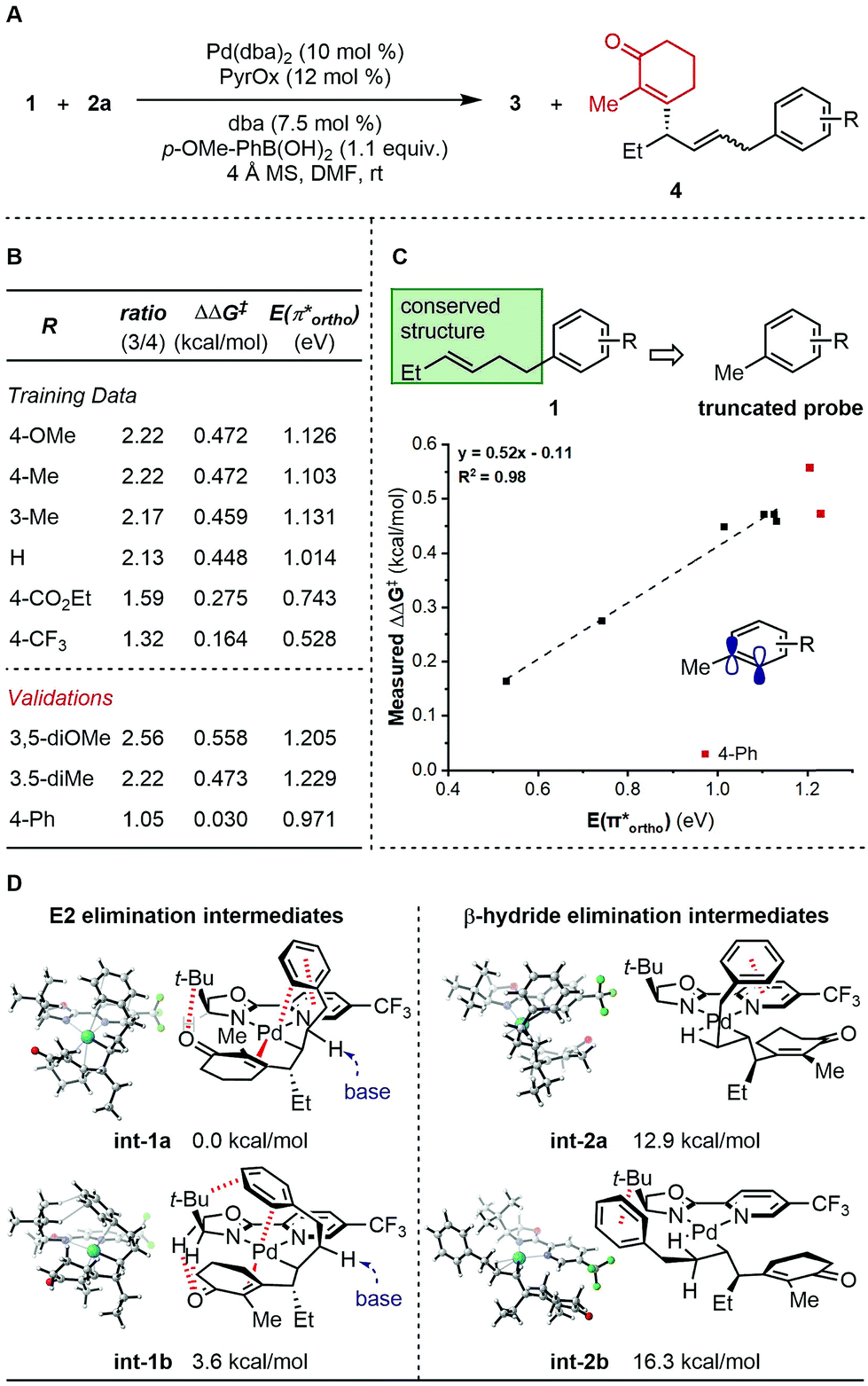 | ||
| Fig. 1 Analysis of side product formation. (A) Reaction scheme. (B) Experimental data. (C) Quantitative analysis. (D) Proposed original of side products. | ||
The discovery that relative π* orbital energy is correlative with the selectivity of alkene isomers formed, suggested that Pd may coordinate with the arene while chain walking. As the arene becomes less donating, the back-bonding interaction of the π-system with Pd, presumably favourable for the formation of the side product 4, is enhanced.11 To further inspect this hypothesis, computational optimization of intermediate structures was performed. The cationic organopalladium intermediate stabilized by interactions with both the alkenyl and the terminal aryl group is calculated to be >10 kcal mol−1 lower in energy than the conformer leading up to β-hydride elimination, thus significantly raising the energy barrier of the chain walking process (Fig. 1D). Additionally, computational results support that, through the proposed palladium–Ar bonding interaction, the γ-palladium intermediates are stabilized in conformations prone to E2 elimination, with the a H–C–C–Pd dihedral angle of int-1a (φint-1a) being 160.8°, and φint-1b being 178.5° (see ESI† for further details), which through solvent mediated deprotonation will lead to minor products 4. A low-energy aryl π* orbital, which allows for strong back-bonding, can lead to both enhanced stabilizing effect, and an early transition state of the proposed E2 elimination process. The unexpected low selectivity observed for the para-Ph-C6H4 substrate can be explained by the additional non-covalent interactions of the para-position phenyl group with the ligand backbone, which further stabilizes the organopalladium intermediate for elimination.12,5d
Conclusions
In summary, we have successfully developed an intermolecular enantioselective alkenylative relay Heck reaction of alkenylbenzene derivatives to synthesize enantiomerically enriched β-substituted styrenes in good to high enantioselectivity. Preliminary mechanistic studies suggest reversible β-hydride elimination/migratory insertion to arrive at the thermodynamic styrenyl product. This information will provide the foundation for the future development of new termination strategies using a metal-chain walking reaction process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by National Institute of Health (NIGMS R01GM063540). The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged.References
- For selected reviews, see: (a) R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27 CrossRef CAS PubMed; (b) N. Kuznik and S. Krompiec, Coord. Chem. Rev., 2007, 251, 222 CrossRef CAS; (c) S. Krompiec, M. Krompiec, R. Penczek and H. Ignasiak, Coord. Chem. Rev., 2008, 252, 1819 CrossRef CAS; (d) E. Larionov, H. Li and C. Mazet, Chem. Commun., 2014, 50, 9816 RSC; (e) M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. van Otterlo, Chem. Rev., 2015, 115, 5462 CrossRef CAS PubMed; (f) M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1706 CrossRef CAS; (g) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209 CrossRef CAS PubMed; (h) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153 CrossRef CAS PubMed.
- Selected examples and reviews on olefin polymerization, see: (a) M.-H. Prosenc and H.-H. Brintzinger, Organometallics, 1997, 16, 3889 CrossRef CAS; (b) Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059 CrossRef CAS PubMed; (c) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS.
- E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed.
- Selected examples: for Rh-catalyzed chain walking, see (a) K. Yoshida and T. Hayashi, J. Am. Chem. Soc., 2003, 125, 2872 CrossRef CAS PubMed; (b) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062 CrossRef CAS PubMed; for Pd-catalyzed chain walking, see (c) C. C. Oliveira, R. A. Angnes and C. R. D. Correia, J. Org. Chem., 2013, 78, 4373 CrossRef CAS PubMed; (d) E. Larionov, L. Lin, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2014, 136, 16882 CrossRef CAS PubMed; (e) T. Hamasaki, Y. Aoyama, J. Kawasaki, F. Kakiuchi and T. Kochi, J. Am. Chem. Soc., 2015, 137, 16163 CrossRef CAS PubMed; (f) S. Singh, J. Bruffaerts, A. Vasseur and I. Marek, Nat. Commun., 2017, 8, 14200 CrossRef PubMed; (g) D. G. Kohler, S. N. Gockel, J. L. Kennemur, P. J. Waller and K. L. Hull, Nat. Chem., 2018, 10, 333 CrossRef CAS PubMed; for Co-catalyzed chain walking, see (h) J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107 CrossRef CAS PubMed; for Ni-catalyzed chain walking, see (i) W.-C. Lee, C.-H. Wang, Y.-H. Lin, W.-C. Shih and T.-G. Ong, Org. Lett., 2013, 15, 5358 CrossRef CAS PubMed; (j) Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061 CrossRef CAS PubMed; (k) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (l) L. Peng, Y. Li, Y. Li, W. Wang, H. Pang and G. Yin, ACS Catal., 2018, 8, 310 CrossRef CAS; (m) Z. Wang, H. Yin and G. C. Fu, Nature, 2018, 563, 379 CrossRef CAS PubMed.
- Selected examples: (a) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (b) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (c) C. Zhang, C. B. Santiago, J. M. Crawford and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 15668 CrossRef CAS PubMed; (d) Z.-M. Chen, M. J. Hilton and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 11461 CrossRef CAS PubMed; (e) N. J. Race, C. S. Schwalm, T. Nakamuro and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 15881 CrossRef CAS PubMed; (f) Z.-M. Chen, C. S. Nervig, R. J. Deluca and M. S. Sigman, Angew. Chem., Int. Ed., 2017, 56, 6651 CrossRef CAS PubMed; (g) Q. Yuan and M. S. Sigman, J. Am. Chem. Soc., 2018, 140, 6527 CrossRef CAS PubMed.
- (a) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960 CrossRef CAS PubMed; (b) M. J. Hilton, L.-P. Xu, P.-O. Norrby, Y.-D. Wu, O. Wiest and M. S. Sigman, J. Org. Chem., 2014, 79, 11841 CrossRef CAS PubMed.
- C. Zhang, C. B. Santiago, L. Kou and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 7290 CrossRef CAS PubMed.
- (a) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462 CrossRef CAS PubMed; (b) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 14226 CrossRef CAS PubMed.
- H. M. Nelson, B. D. Williams, J. Miró and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3213 CrossRef CAS PubMed.
- For details on the assignment of absolute configuration, please see the ESI.†.
- (a) A. Falceto, E. Carmona and S. Alvare, Organometallics, 2014, 33, 6660 CrossRef CAS; (b) S. M. Reid, R. C. Boyle, J. T. Mague and M. J. Fink, J. Am. Chem. Soc., 2003, 125, 7816 CrossRef CAS PubMed; (c) B. W. Smucker, J. M. Hudson, M. A. Omary and K. R. Dunbar, Inorg. Chem., 2003, 42, 4714 CrossRef CAS PubMed; (d) B. Inés, R. SanMartin, F. Churruca, E. Domínguez, M. K. Urtiaga and M. I. Arriortua, Organometallics, 2008, 27, 2833 CrossRef; (e) K. Wieghardt, H. J. Küppers, E. Raabe and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 1101 CrossRef.
- Selected examples on non-covalent interactions application in selective catalysis, see: (a) C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020 CrossRef CAS PubMed; (b) F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609 CrossRef CAS PubMed; (c) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637 CrossRef CAS PubMed; (d) M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803 CrossRef CAS PubMed; (e) R. A. Angnes, L. M. Thompson, M. S. Mashuta, C. R. D. Correia and G. B. Hammond, Adv. Synth. Catal., 2018, 360, 3760 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02380a |
| ‡ Z.-M. C. and J. L. contributed equally to this work. |
| § Present address: Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P. R. China. |
| This journal is © The Royal Society of Chemistry 2019 |