DOI:
10.1039/C9SC02289A
(Edge Article)
Chem. Sci., 2019,
10, 7641-7648
Total synthesis of griseusins and elucidation of the griseusin mechanism of action†
Received
10th May 2019
, Accepted 26th June 2019
First published on 27th June 2019
Abstract
A divergent modular strategy for the enantioselective total synthesis of 12 naturally-occurring griseusin type pyranonaphthoquinones and 8 structurally-similar analogues is described. Key synthetic highlights include Cu-catalyzed enantioselective boration–hydroxylation and hydroxyl-directed C–H olefination to afford the central pharmacophore followed by epoxidation–cyclization and maturation via diastereoselective reduction and regioselective acetylation. Structural revision of griseusin D and absolute structural assignment of 2a,8a-epoxy-epi-4′-deacetyl griseusin B are also reported. Subsequent mechanistic studies establish, for the first time, griseusins as potent inhibitors of peroxiredoxin 1 (Prx1) and glutaredoxin 3 (Grx3). Biological evaluation, including comparative cancer cell line cytotoxicity and axolotl embryo tail inhibition studies, highlights the potential of griseusins as potent molecular probes and/or early stage leads in cancer and regenerative biology.
1 Introduction
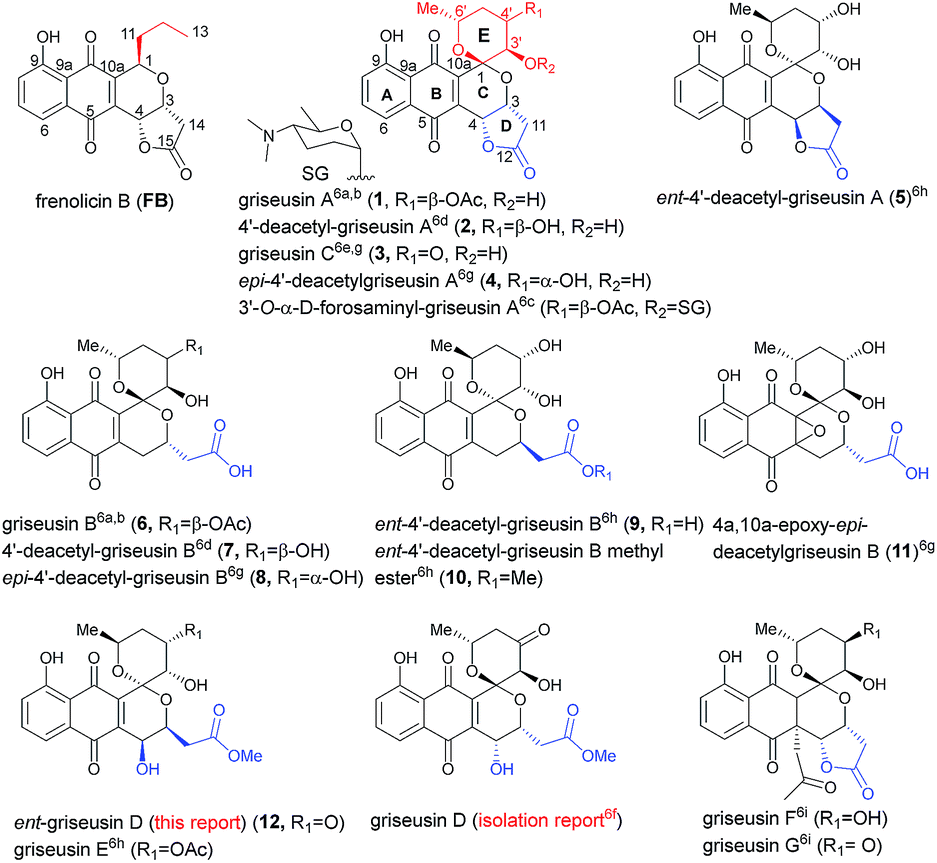
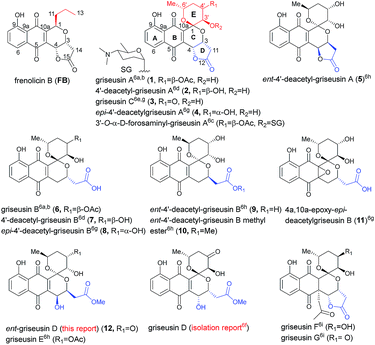
Pyranonaphthoquinone (PNQ) natural products represent an array of structurally and functionally diverse bacterial secondary metabolites.1 Despite extensive advances in PNQ synthetic methodology,2,3 the commercial application of PNQs as feedstock additives and the demonstrated anticoccidial and antimalarial efficacy of PNQs,4 the mechanism of action for PNQs remained unresolved. Enabled by our efficient divergent strategy for the synthesis of frenolicin B (FB, a prototypical PNQ-based natural product; Fig. 1),2f systematic SAR and mechanistic studies revealed FB as the most potent and selective peroxiredoxin 1 (Prx1) and glutaredoxin 3 (Grx3) inhibitors reported to date.5 These studies demonstrated Prx1/Grx3 inhibition by FB led to increased intracellular ROS, the consequence of which was inhibition of mTORC1-mediated 4E-BP1 phosphorylation, apoptosis induction and tumor suppression. Whether this fundamental mechanism of action is unique to FB or more broadly attributed to the other diverse PNQ family members remains to be determined.
 |
| | Fig. 1 Naturally-occurring frenolicin prototype (FB) and griseusins. | |
First reported in 1976, the griseusin-based PNQ metabolites are structurally distinguished by their fused spiro-ring C/E system (Fig. 1) in lieu of the FB C1-alkyl side chain of FB.6 Griseusin members are further differentiated via ring D forms (open or closed), relative stereochemistry, oxidation, glycosylation, and/or O- or C-acetylation. Like their frenolicin counterparts, many of the griseusins display potent cancer cell line cytotoxicity. Representative griseusins have also been reported as COMPARE negative (indicating a novel mechanism of activity)6g and potentially synergistic with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in TRAIL-resistant gastric adenocarcinoma cell lines.6h Yet, the molecular target and mechanism of action for griseusin remains unknown.
Enabled by our recently reported synthetic approach to griseusin A (1), 4′-deacetyl-griseusin A (2), and griseusin C (3),3m herein we report the further development and implementation of the first concise divergent synthetic approach to a broad set of griseusin A–E analogs (twelve naturally-occurring griseusins and eight additional synthetic analogues). Methodological highlights include mechanistic investigation of C–H olefination en route to the griseusin C-ring, enantioselective Cu-catalyzed boration–hydroxylation to afford open D-ring griseusin B members and a series of stereoselective and regioselective transformations for E-ring diversification. This enabling chemistry also facilitated revision of the previous reported griseusin D structure, assignment of stereocenters in 4a,10a-epoxy-epi-4′-deacetyl griseusin B and the broadest griseusin comparative cancer cell line cytotoxicity SAR analysis to date. Subsequent biochemical and cell-based studies revealed representative griseusins to inhibit Prx1/Grx3 and 4E-BP1 phosphorylation in manner similar to FB and to inhibit tail regeneration in our recently developed axolotl tail regeneration assay.7 Given the notable demonstrated in vivo efficacy of optimized FB analogs,5 the current study considerably expands the range PNQ pharmacophores available for further optimization of Prx1/Grx3 selectivity and/or ADMET.
2 Results and discussion
2.1 Retrosynthetic analysis and rationale
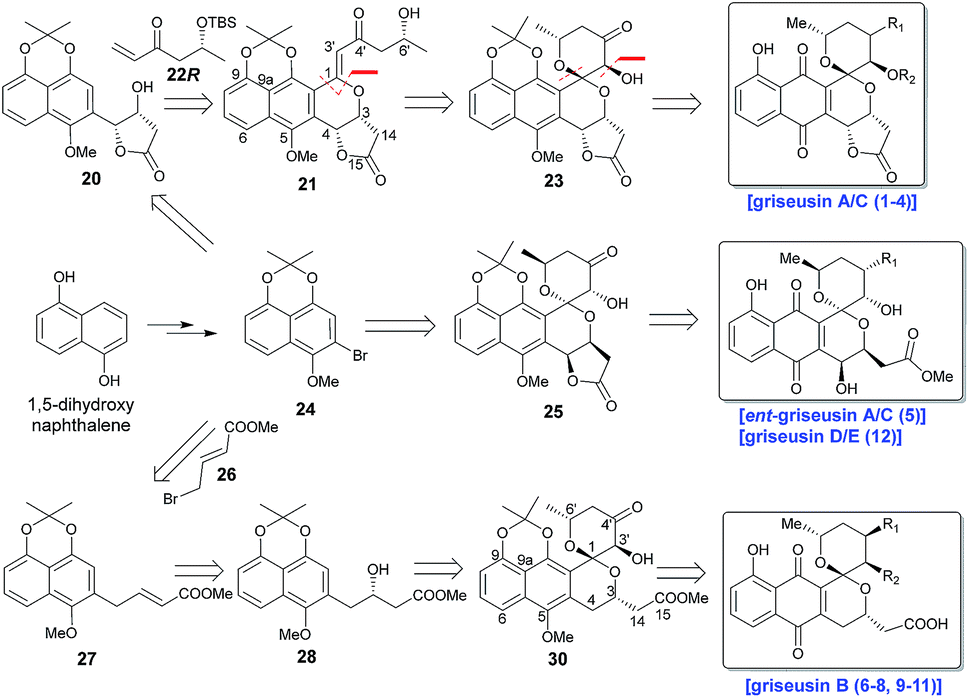
Scheme 1 highlights a conceptual overview of the envisioned synthetic route to griseusin-type PNQs. C1 epimerization within the context of spiropyran construction and/or subsequent scaffold maturation presents a primary hurdle to griseusin total synthesis. We postulated that diastereoselective C1–C3′ epoxidation and subsequent C6′–OH intramolecular cyclization could provide the key spiro-pyrano core 23, the C1 stereocenter of which should be stable to late-stage deprotection of the acetonide.8 For this approach, the critical 21 C1–C3′-enone would derive from simple convergent C–H olefination using 20 and 22R, the latter of which would result from Sharpless asymmetric dihydroxylation9 of the Heck-coupled product of methyl 3-butenoate and bromine 24. Following the same strategy, ent-griseusin C (and the griseusin D/E subclass) may derive from enantiomer 25 and AD-mix-α. For the griseusin B series, alcohol 28 would stem from copper-catalyzed enantioselective boration–hydroxylation10 of 27, accessible via a palladium coupling between bromine 26 and a 24-derived boronic acid. Final maturation/tailoring reactions would also leverage the C4′ ketone of precursors 23, 24 and 30 where applicable.
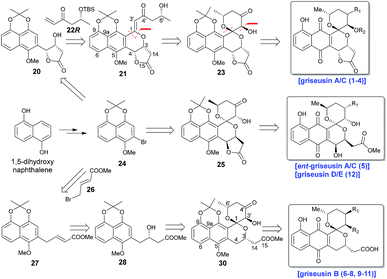 |
| | Scheme 1 Retrosynthetic analysis for griseusin subclasses A–E. | |
2.2 Synthesis of key griseusin A/C precursors
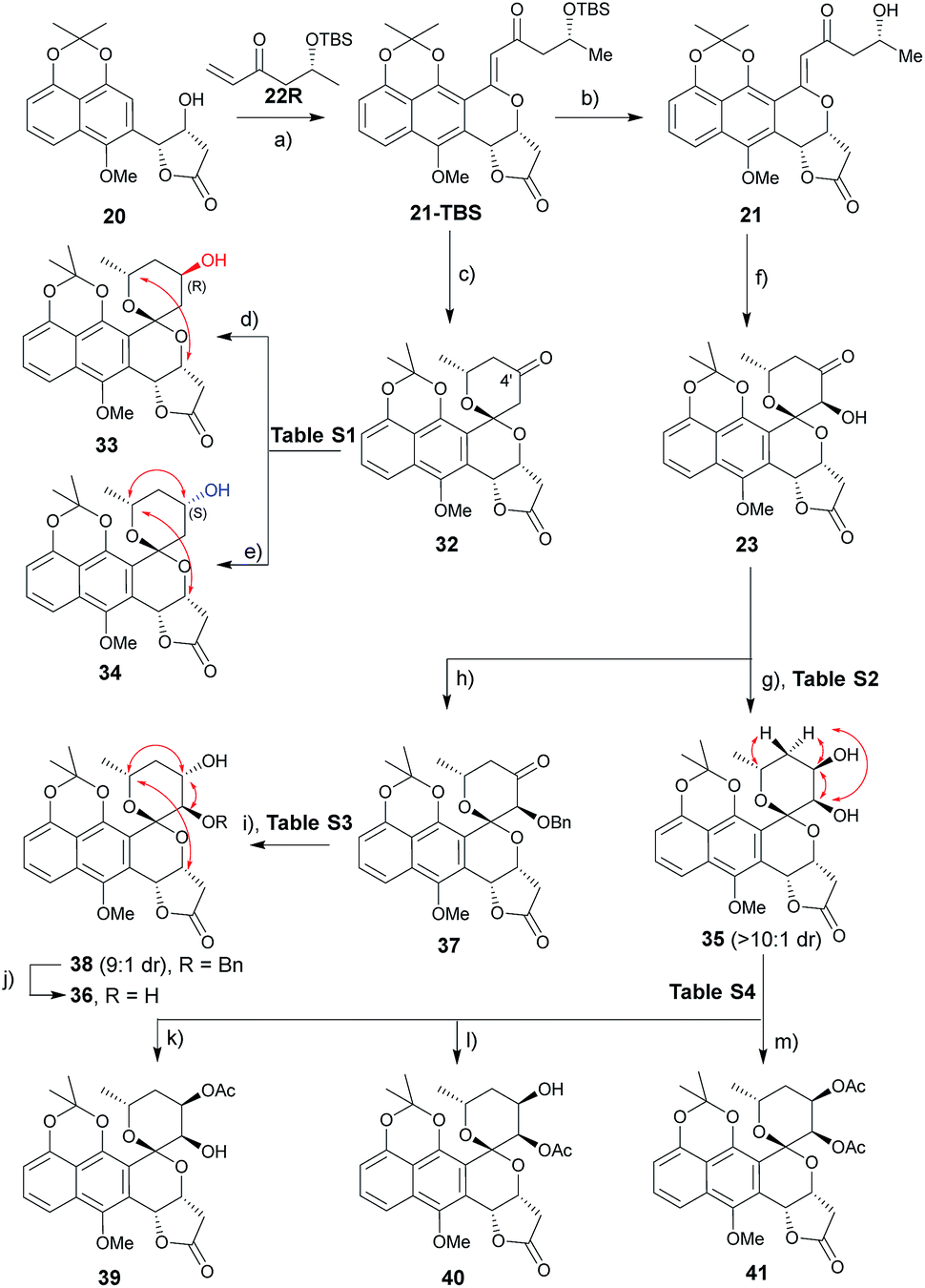
A range of conditions were explored for reduction of the 32 (ref. 3m) C4′ ketone, the primary goal of which was to establish a general reductive method with bilateral C4′ diastereoselectivity (Table S1† and Scheme 2). Among the common borohydride reagents (Table S1,† entries 1 and 2), K selectride provided the desired β-alcohol 33 exclusively in a 94% isolated yield compared to the observed 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity with NaBH4 (Table S1,† entry 1). Non-ionic boranes favored formation of the desired α-alcohol 34 with the borane 2-picoline complex in the presence of HCl providing the best diastereoselectivity (Table S1,† entries 3–11).11 Solvent optimization (Table S1,† entries 12–15) revealed that reactions in ether provided the desired α-alcohol in 75% isolated yield with 1:10 diastereoselectivity (Table S1,† entry 15). Putative mechanistic rationale for diastereoselectivity in this reaction is based on sterics (favoring hydride attack from the si-face to give (R)-alcohol 33) or partial spiroketal oxygen–borane coordination (favoring hydride attack from the re-face to give (S)-alcohol 34).
:1 diastereoselectivity with NaBH4 (Table S1,† entry 1). Non-ionic boranes favored formation of the desired α-alcohol 34 with the borane 2-picoline complex in the presence of HCl providing the best diastereoselectivity (Table S1,† entries 3–11).11 Solvent optimization (Table S1,† entries 12–15) revealed that reactions in ether provided the desired α-alcohol in 75% isolated yield with 1:10 diastereoselectivity (Table S1,† entry 15). Putative mechanistic rationale for diastereoselectivity in this reaction is based on sterics (favoring hydride attack from the si-face to give (R)-alcohol 33) or partial spiroketal oxygen–borane coordination (favoring hydride attack from the re-face to give (S)-alcohol 34).
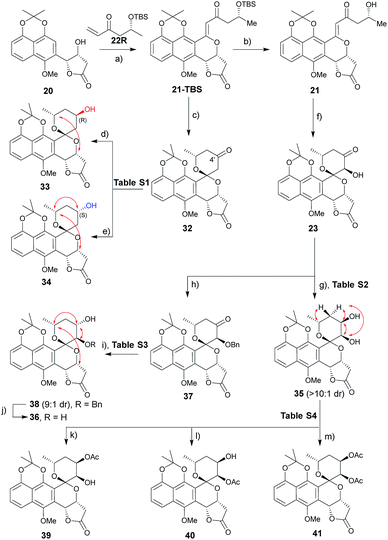 |
| | Scheme 2 Synthesis of griseusin A/C-type analogues. Key NOE crosspeaks are highlighted (red arrows). Reagents and conditions: (a) Pd(OAc)2 (20 mol%), Li2CO3, Ag2CO3 (4 eq.), DCE, 80 °C, 16 h, 40%; (b) HF·pyridine (10 eq.), TEA (2 eq.), dioxane, rt, 16 h, 85%; (c) HF·pyridine (10 eq.), CH3CN, rt, 16 h, 91%; (d) K-selectride, THF, −78 °C, 2 h, 94%; (e) BH3·2-picoline, HCl, Et2O, 16 h, 75%; (f) DMDO, TfOH, DCM, −78–0 °C, >10:1 dr, 80%; (g) LiBH4 (2 eq.), THF, −78 °C, 0.5 h, 85%, β:α > 10:1; (h) KOtBu (2 eq.), BnBr (2 eq.), THF, 0 °C to rt, 16 h, 77%; (i) BH3·Me3N (2 eq.), TFA (3.0 eq.), ether, rt, 36 h, 74%, α:β = 9:1; (j) Pd/C, H2, MeOH, rt, 4 h, 95%; (k) Ac2O (10 eq.), dicyclohexymethyl amine (10 eq.), DCM, rt, 40 h, 70%; (l) Ac2O (5 eq.), pyridine (5 eq.), DCM, rt, 16 h, 88%; (m) Ac2O (10 eq.), DABCO (10 eq.), DCM, rt, 16 h, 93%. | |
A similar optimization strategy was pursued for reduction of the 23 C4′-carbonyl to set the stage for the synthesis of C3′/4′-diols (Table S2†). Electrophilic reducing agents, such as sodium borohydride and lithium borohydride, provided the desired C4′ cis-diol 35 in high yield and si-facial selectivity (Scheme 2). In contrast, this study failed to identify suitable reductive conditions to furnish the corresponding desired trans-diol 36, potentially due to C3′–OH infringement on the putative spiroketal oxygen–borane coordination required for stereoselective hydride delivery. As an alternative, benzyl-protected intermediate 37 led to the re-face C4′-carbonyl stereospecific reduction reminiscent to that for (S)-alcohol 34 (Table S3†). The corresponding 23 protection/reduction/deprotection protocol afforded the desired trans-diol 36 in a 54% cumulative yield for three steps (Scheme 2).
With diol 35 in hand, we next evaluated regioselective O-acetylation (Table S4†). Initial attempts to obtain the desired products 39–41via mild transesterification12 were hampered by poor diol reactivity (Table S4,† entries 1 and 2). Use of acetic anhydride and base optimization led to the desired products in variable yields (Table S4,† entries 3–8). Specifically, use of a sterically-hindered base favored the desired C4′-acetylation (Table S4,† entry 6) while relatively weak base gave exclusive C3′-acetylation (Table S4,† entry 7). Reduction of the amount of base and acetic anhydride further improved yield (Table S4,† entry 8) while the use of a nucleophilic strong base favored peracetylated product 41 (Table S4,† entry 9).
2.3 Synthesis of key griseusin B precursors
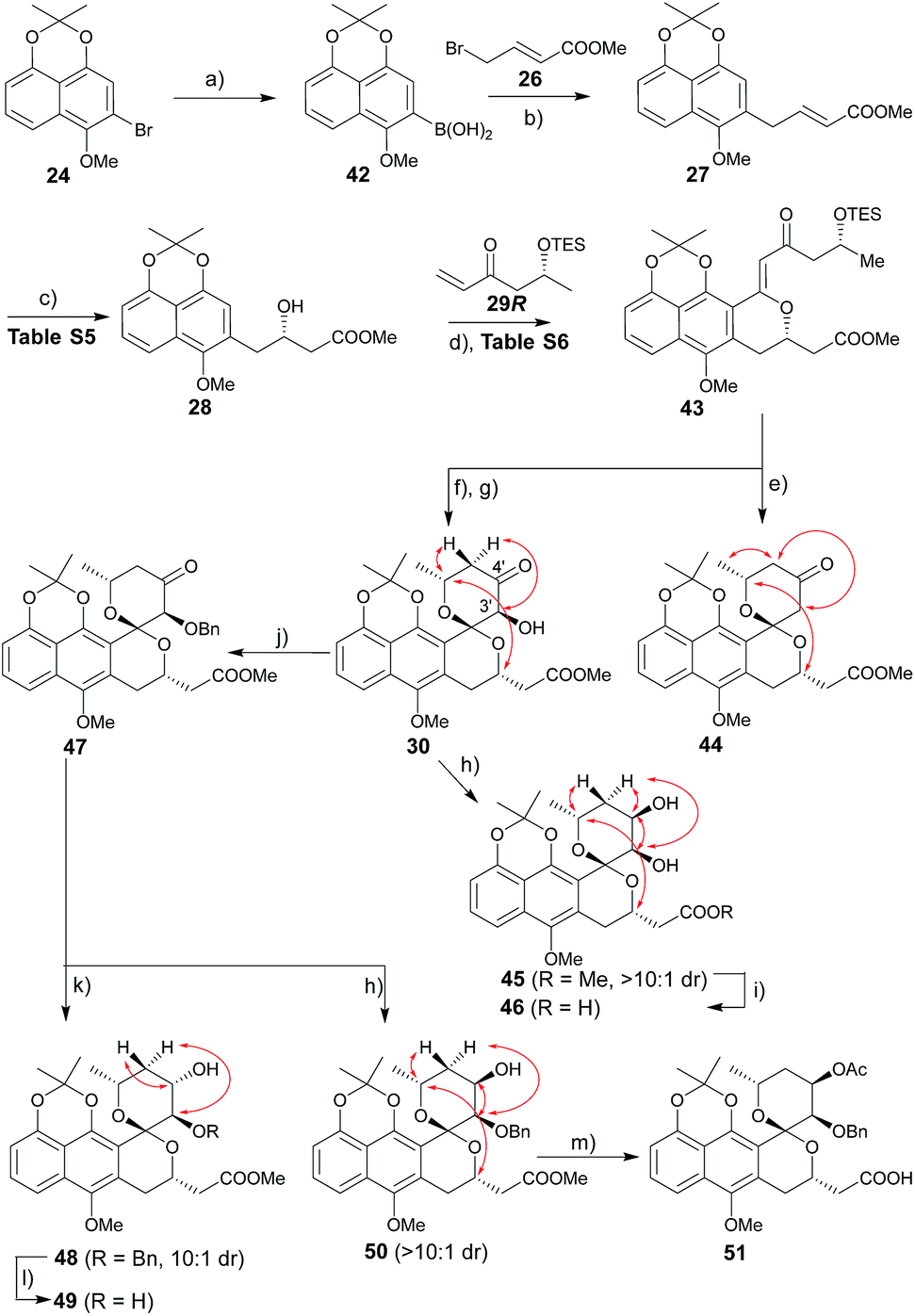
The route to griseusin B analogues began with the synthesis of 42 following standard methodology for lithium–bromine exchange-mediated boronic acid generation (Scheme 3). Palladium-catalyzed Suzuki coupling13 of 42 with bromobutenoate (26) gave 27. With α,β-unsaturated ester 27 in hand, we investigated a range of β-borylation conditions for enantioselective β-conjugate addition (Table S5†). This small study revealed chiral diphosphine ligand10 as advantageous over bisoxazoline and N-heterocyclic carbene catalysts,14 enabling quantitative yield with 90% enantioselectivity on gram scale.
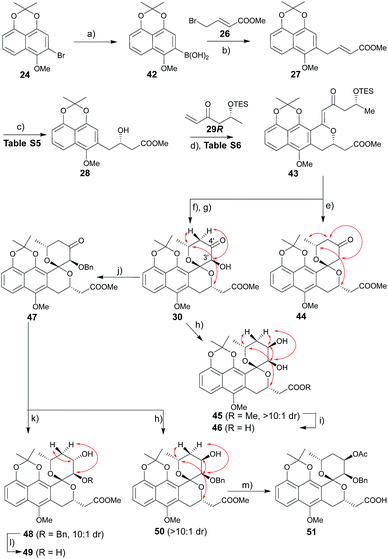 |
| | Scheme 3 Synthesis of griseusin B-type analogues. Key NOE crosspeaks are highlighted (red arrows). Reagents and conditions: (a) nBuLi, B(OiPr)3, THF, −78 °C, 2 h, 80%; (b) 26, Pd(OAc)2, KF, dioxane, rt, 2 h, 74%; (c) 1.5 mol% CuCl, 3 mol% S-Rp-Josiphos, 2.5 mol% NaOtBu, Bpin2, THF, 0 °C to rt, 16 h; then NaBO3, H2O, rt, 2 h, 91%, 90% ee; (d) 29, 40 mol% Pd(OAc)2, Ag2CO3 (2 eq.), Li2CO3, CHCl3, 80 °C, 16 h, 63%; (e) HF·pyridine, CH3CN, rt, 12 h, 81%; (f) HF·pyridine, TEA, dioxane, rt, 1 h, 88%; (g) DMDO, 50 mol% TfOH, DCM, −78–(−20) °C, 2 h, 93%; (h) LiBH4 (2 eq.), THF, −78 °C, 0.5 h, 86%, β:α > 10:1; (i) 1 N LiOH, MeOH, rt, 1 h, 95%; (j) BnBr, Ag2O, NMP, rt, 36 h, 76%; (k) BH3·Me3N (2 eq.), TFA (3.0 eq.), ether, rt, 36 h, 77%, α:β = 10:1; (l) Pd/C, H2, MeOH, rt, 4 h, 93%; (m) 1 N LiOH, MeOH, rt, 1 h; Ac2O, DABCO, DCM, rt, 40 h; 65% for two steps. | |
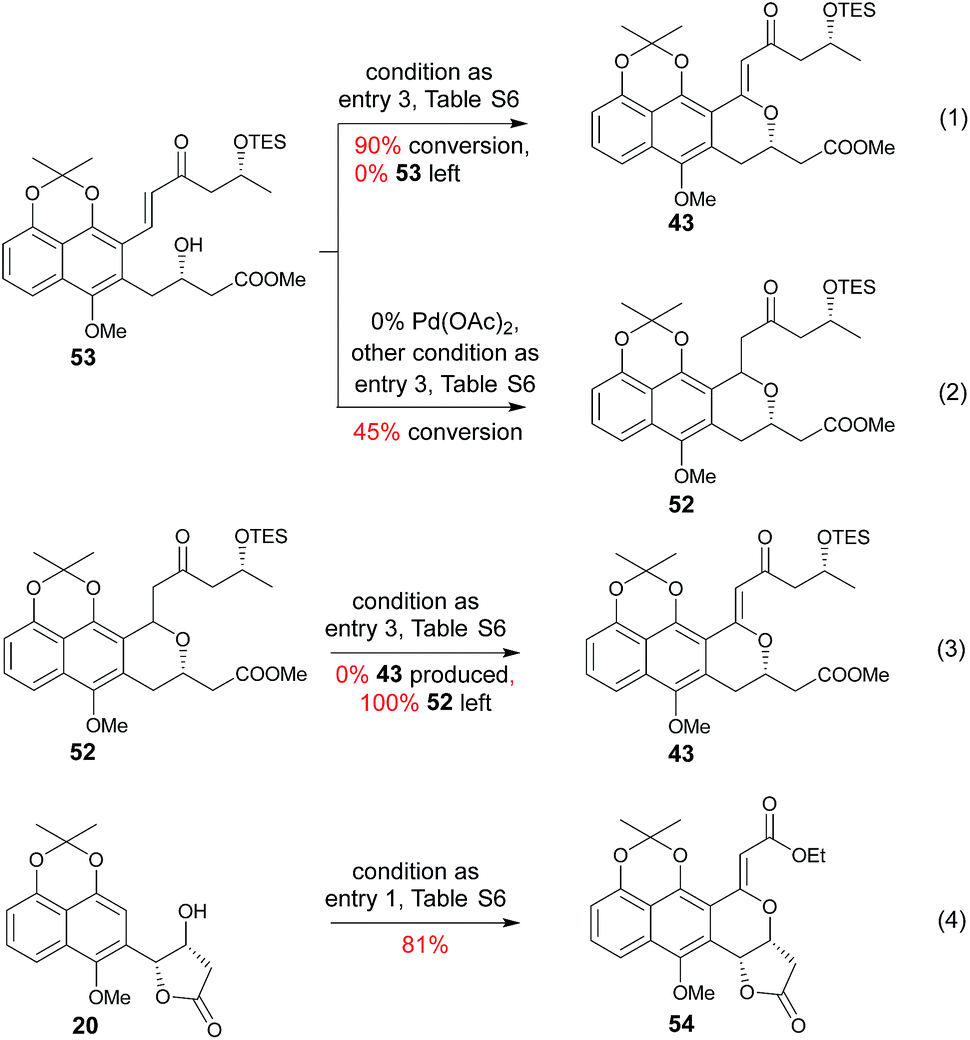
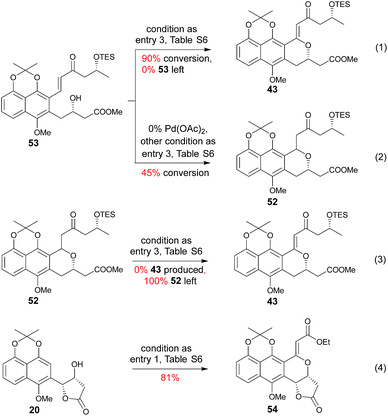
Subsequent installation of 29R (to access the desired 1-methylene isochroman 43, Scheme 4) began with our C–H activation C1 olefination conditions for 20.3m While these initial conditions (Table S6,† entry 1) were sub-optimal in this case, subsequent modification of reaction solvent (Table S6,† entries 2–4) and catalyst/oxidant loading (Table S6,† entries 4–8) gave the desired 1-methylene isochroman 43 in 63% on gram scale (Table S6,† entry 9). The lower observed abundance of side product 52 in these C1 olefination studies implicated the flexible secondary alcohol of 28 (compared to the corresponding rigid secondary alcohol of 20) as advantageous. Access to 53 also facilitated further mechanistic investigation of this C–H olefination event (Scheme 4). Specifically, intermediate 53 results from initial hydroxyl-assisted C–H insertion of Pd(II) followed by β-elimination and subsequently enters the second round of Pd-catalyzed intra-molecular oxidative cyclization (eqn (1)). In contrast, side product 52 cannot undergo β-elimination (eqn (3)) and therefore irreversibly consumes 53 as a major competing reaction (eqn (2)). The bifurcation between the desired product (e.g., 1-methylene isochroman 43) and undesired shunt products (e.g., 52) is presumably dictated by a propensity for palladium re-coordination versus nucleophilic attack of the participant hydroxyl (which can be influenced via nucleophile orientation and/or nucleophilicity as noted in Table S6†). For example, the higher yield of methylene isochroman 54 is attributed to lower nucleophilicity of the 20-derived α,β-unsaturated ester (Scheme 4). Cumulatively, these studies provide further support of Yu's15 and our3m previously proposed step-wise mechanism for this oxidative cyclization event.
 |
| | Scheme 4 Reactions relevant to C–H olefination mechanistic considerations. | |
Guided by our prior success with griseusin A-type spiropyran ring formation, unsaturated ketone 43 was converted to C3′-deshydroxy substrate 44 and C3′-(S)-hydroxy substrate 30, respectively (Scheme 3). The C4′-carbonyl of intermediate 30 was subsequently stereoselectively reduced to obtain 45 followed by hydrolysis with LiOH to give intermediate 46. In parallel, 30 C3′-O-benzyl protection under a mild conditions gave intermediate 47. Subsequent diastereoselective 47 C4′-ketone reduction with either borane or borohydride gave 4′-(S)-alcohol 48 or 4′-(R)-alcohol 50, respectively. Final standard protecting group manipulations gave key subclass B intermediates 49 and 51.
2.4 Completion of total syntheses
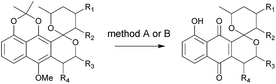
The synthesis of ent-precursors mirrored that conducted for the preparation of griseusin A/C and B subclasses. For example, ent-45 employed R-Sp-Josiphos as a chiral catalyst in the enantioselective β-borylation of 27 and α,β-unsaturated ketone handle 29S in the C–H olefination step, while AD-mix-α and ketone 22S were applied to construct ent-griseusin C and D scaffolds. With all key griseusin and ent-griseusin precursors in hand, final deprotection was accomplished with silver oxide under acidic conditions as summarized in Table 1. Following previously reported oxidative strategies,16 hydrogen peroxide and DABCO furnished the desired 11 C-4a/C-10a epoxide with 8:1 diastereoselectivity (Scheme 5 and Table S7†). In summary, starting from 1,5-dihydroxynaphthalene the divergent strategy put forth enabled access to twelve naturally-occurring griseusins and eight new griseusin analogues in 10–18 steps with total yields ranging from 3–8%.
Table 1 Total synthesis of natural-occurring griseusins (1–10) and their analogs (13–19)a
|
|
| Entry |
Reactant (methodb) |
Product (yieldc) |
Entry |
Reactant (methodb) |
Product (yieldc) |
|
See ESI for experimental details.
Method A: 5 eq. AgO, 10 eq. 6 N HNO3 at 0 °C for 10 min; method B: 2.2 eq. AgO, 6 eq. 3 N HNO3 at −10 °C for 30 min.
Isolated yields.
Deprotection of 51 benzyl ether (hydrogenolysis) was conducted before the deprotection step and reported yield included this conversion.
Ester hydrolysis of 49 and ent-45 were conducted before the deprotection step and the reported yield included this conversion.
See ref. 3m for structural detail.
Ester hydrolysis of 44 was conducted prior to deprotection step and the reported yield included this conversion.
|
| 1 |
39 (A) |
1 (81%) |
10 |
ent-46 (A) |
10 (79%) |
| 2 |
35 (A) |
2 (85%) |
11 |
32 (A) |
13 (73%) |
| 3 |
23 (A) |
3 (63%) |
12 |
1,3′-epi-23f, (A) |
14 (63%) |
| 4 |
36 (A) |
4 (62%) |
13 |
40 (A) |
15 (75%) |
| 5 |
ent-35 (A) |
5 (77%) |
14 |
33 (A) |
16 (78%) |
| 6 |
51 (B) |
6 (54%d) |
15 |
34 (A) |
17 (75%) |
| 7 |
46 (B) |
7 (68%) |
16 |
25 (A) |
18 (63%) |
| 8 |
49 (B) |
8 (56%e) |
17 |
44 (B) |
19 (52%g) |
| 9 |
ent-45 (B) |
9 (55%e) |
|
|
|
 |
| | Scheme 5 Synthesis and structural determination of 4a,10a-epoxy-epi-deacetylgriseusin B (11). Key NOE crosspeaks are highlighted (red arrows). Reagents and conditions: (a) H2O2, DABCO, DCM, 0 °C to rt, 2 h, 88%; (b) H2, Pd/C, MeOH, 2 h, 42%. | |
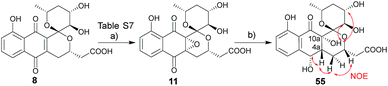
2.5 Configuration assignment of griseusin B epoxide (11)
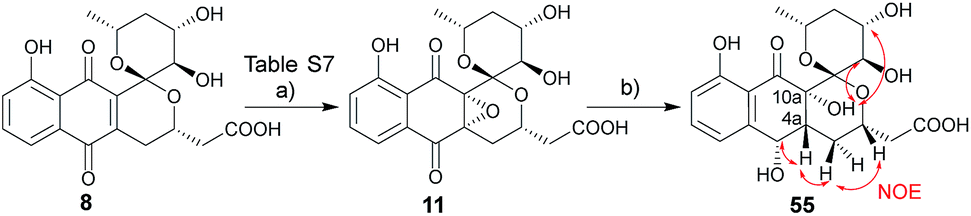
While the spectroscopic data for synthetic 11 and previously-isolated 11 were consistent (Table S8†), the reported epoxide 11 isolated from Nocardiopsis sp. YIM80133 lacked assignment of absolute stereochemistry.6g To address this, NMR studies of tetrol 55, produced via hydrogenation of 11, enabled assignment of the epoxide absolute stereochemistry as C-4aR, 10aS (Scheme 5). As biosynthetic transformations are commonly conserved within a given natural product classes, this definitive stereochemical assignment may also be relevant to other naturally-occurring C-4a/C-10a-epoxy PNQs.1a,c
2.6 Structural revision of griseusin D (12)
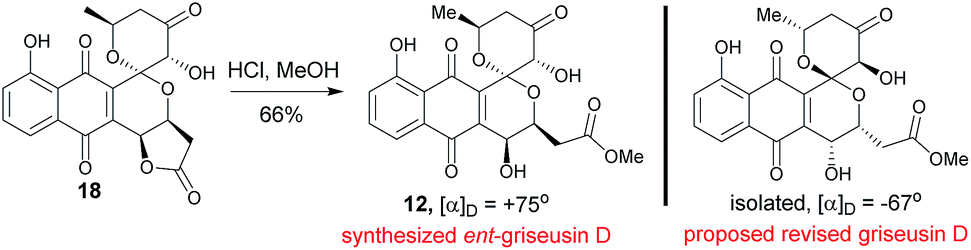
Methanolysis of 18 afforded E-ring lactone 12 in good yield (Scheme 6). While the spectroscopic data for compound 12 was in strong agreement with the reported griseusin D (Table S9†),6f the determined optical rotation of 12 was diametrically opposed to that reported for griseusin D. Thus, we postulate 12 to be ent-griseusin D and the structure of reported griseusin D to be that illustrated in Scheme 6. Consistent with this, most griseusins previously reported from the griseusin D-producing strain (Nocardiopsis sp. YIM80133) also displayed negative optical rotations.6g
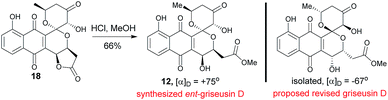 |
| | Scheme 6 Synthesis of ent-griseusin D (12) and proposed griseusin D structural revision. | |
2.7 Griseusin comparative cancer cell line cytotoxicity
All compounds were tested against four cancer cell lines (non-small cell lung A549, prostate PC3 and colorectal HCT116 and DLD-1) (Table 2). While most griseusins were found to be cytotoxic across this panel, A549 was generally (2- to 10-fold) less susceptible to PNQs. An intact D-lactone ring (e.g., as found in griseusins A and C, compounds 1–4) was generally advantageous as compared to D-ring open comparators (e.g., griseusins B and D, compounds 6–8, 12). Surprisingly, active enantiomeric comparators (e.g., 1vs.5 or 7vs.9) displayed similar potencies. E-ring C3′-substitution generally had a negative impact on potency (e.g., 2vs.16, 3vs.13, or 4vs.17), while bioactivity was less impacted by C4′-alterations (ketone, S-hydroxyl, R-hydroxyl). Destruction of the naphthoquinone core also abolished activity (e.g., compounds 11 and 55).
Table 2 Cancer cell cytotoxicity and inhibition of axolotl tail regenerationa
| Cmpd |
A549b |
PC3b |
HCT116b |
DLD-1b |
Axolotl tailc |
|
See ESI for experimental details.
IC50 ± SD values (μM) from experiments performed in triplicate. The highest concentrations tested were 20 μM (A549 and PC3) and 2 μM (HCT116 and DLD-1), respectively.
Results from single dose (10 μM) experiments performed in triplicate where ‘+’ indicates complete inhibition, ‘−’ reflects no effect and ‘toxic’ denotes lethality.
|
|
1
|
1.0 ± 0.2 |
0.1 ± 0.08 |
0.2 ± 0.08 |
0.1 ± 0.02 |
+ |
|
2
|
3.4 ± 1.2 |
0.9 ± 0.3 |
0.5 ± 0.1 |
0.1 ± 0.08 |
+ |
|
3
|
1.8 ± 0.03 |
0.1 ± 0.1 |
0.1 ± 0.02 |
0.1 ± 0.004 |
+ |
|
4
|
10.3 ± 7.9 |
1.9 ± 0.3 |
>2 |
>2 |
− |
|
5
|
6.9 ± 0.1 |
0.6 ± 0.02 |
0.2 ± 0.01 |
0.2 ± 0.06 |
N.D. |
|
6
|
4.1 ± 1.0 |
2.1 ± 0.9 |
1.2 ± 0.9 |
0.6 ± 0.3 |
+ |
|
7
|
11.6 ± 9.0 |
0.9 ± 0.5 |
>2 |
0.3 ± 0.1 |
− |
|
8
|
>20 |
2.9 ± 0.6 |
1.2 ± 0.6 |
>2 |
− |
|
9
|
15.1 ± 0.2 |
0.9 ± 0.08 |
0.4 ± 0.05 |
0.3 ± 0.01 |
− |
|
10
|
5.0 ± 0.1 |
0.7 ± 0.01 |
0.8 ± 0.5 |
0.5 ± 0.4 |
Toxic |
|
11
|
>20 |
>20 |
>2 |
>2 |
− |
|
12
|
3.2 ± 0.6 |
1.0 ± 0.3 |
>2 |
0.1 + 0.02 |
+ |
|
13
|
0.7 ± 0.3 |
0.1 ± 0.02 |
0.1 ± 0.06 |
0.1 ± 0.03 |
Toxic |
|
14
|
2.1 ± 0.1 |
1.1 ± 0.3 |
0.7 ± 0.01 |
0.9 ± 0.4 |
− |
|
15
|
1.2 ± 0.3 |
0.2 ± 0.06 |
0.2 ± 0.02 |
0.1 ± 0.01 |
+ |
|
16
|
1.0 ± 0.2 |
0.2 ± 0.1 |
0.1 ± 0.04 |
0.1 ± 0.01 |
Toxic |
|
17
|
1.2 ± 0.2 |
0.2 ± 0.04 |
0.1 ± 0.01 |
0.04 ± 0.01 |
+ |
|
18
|
2.7 ± 0.5 |
0.6 ± 0.3 |
0.2 ± 0.03 |
0.1 ± 0.03 |
+ |
|
19
|
1.2 ± 0.2 |
0.3 ± 0.05 |
0.1 ± 0.05 |
0.1 ± 0.06 |
Toxic |
|
55
|
>20 |
>20 |
>2 |
>2 |
N.D. |
2.8 Griseusins inhibit peroxiredoxin 1 (Prx1) and glutaredoxin 3 (Grx3) and thereby influence 4E-BP1 phosphorylation
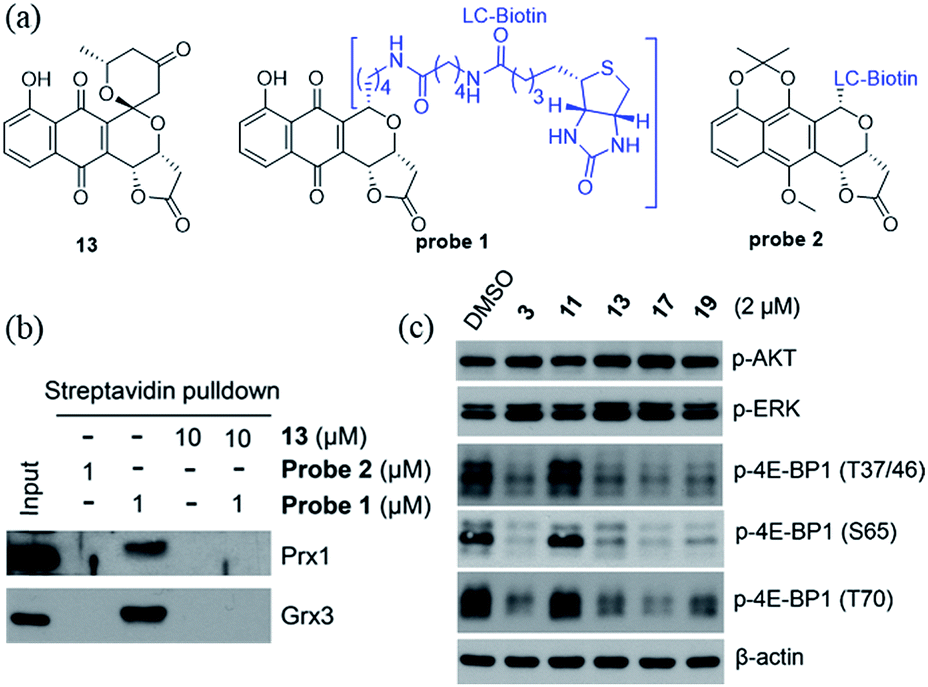
We recently identified the PNQ FB as a potent inhibitor of peroxiredoxin 1 (Prx1) and glutaredoxin 3 (Grx3) and determined FB's antitumor effect to be mediated by inhibition of phosphorylation of 4E-BP1.5 While 4E-BP1 is a cap-dependent translation repressor of oncogenic mRNA translation and tumorigenesis, phosphorylation of 4E-BP1 by mTORC1 relieves its inhibitory control leading to tumor progression.17 To assess whether griseusin-type PNQs can also bind Prx1 and/or Grx3, we conducted a simple competition assay with model griseusin 13 using the FB-based biotinylated probe set from our previously-reported target identification study (Fig. 2a).5 In this study (Fig. 2b), 13 could completely block the binding of the active FB-based probe 1 to Prx1 or Grx3 in HCT116 crude extracts. Consistent with this, 13 also directly inhibited Prx1 and Prx2 catalytic activity with apparent IC50s of 2.3 μM and 7.3 μM, respectively (Fig. S1†). Similar to the inhibitory effect of FB on 4E-BP1 phosphorylation, the representative cytotoxic griseusins tested (e.g., 3, 13, 17, and 19) inhibited 4E-BP1 phosphorylation while non-cytotoxic analogues (e.g., 11) had no effect on 4E-BP1p (Fig. 2c). Collectively, these findings suggest that griseusin-type and FB-type PNQs exhibit a similar mechanism of action.
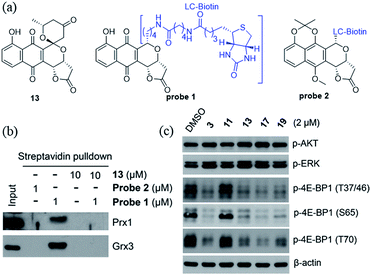 |
| | Fig. 2 (a) Structures of compound 13 and probes 1 and 2. Probe 1 is active (both cytotoxic and as an inhibitor of 4E-BP1 phosphorylation) while the control probe 2 is an inactive comparator. (b) Compound 13 competes with probe 1 binding to Prx1 and Grx3. (c) Cytotoxic griseusin-type PNQs effectively inhibit phosphorylation of 4E-BP1 but not AKT and ERK kinases. | |
2.9 Impact of griseusins on axolotl tail regeneration
Recent studies in a highly regenerative salamander model (the Mexican axolotl, Ambystoma mexicanum) revealed that ROS rapidly increased in response to axolotl tail amputation and was required for tail regeneration.18 These studies also demonstrated pharmacological inhibition of ROS producing enzymes with diphenyleneiodonium chloride (DPI) and VAS2870 reduced ROS and led to inhibition of cellular proliferation and tail outgrowth inhibition. To investigate the impact of the Prx1/Grx3-inhibiting griseusins within this context, representative griseusin analogues were evaluated in the same axolotl embryo tail regeneration (ETR) assay.7 Tail-amputated axolotl embryos were incubated in microtiter plates in the absence (vehicle control, DMSO) or presence of 10 μM agent (1–4, 6–19) and imaged on day 1 (pre-treatment) and day 7. The initial single dose screen revealed a moderate correlation between cancer cell line cytotoxicity and inhibition of tail regeneration (Table 2 and Fig. S2†) where divergence may result, in part, from yet to be determined factors that contribute to differences in uptake and/or in vivo exposure. Importantly, these single dose studies, in conjunction with the subsequent established dose response of both inhibition of tail regeneration and lethality with representative 13 (Fig. S3†), are consistent with a mechanistic relationship between Prx1/Grx3, ROS and tail regeneration and provide preliminary evidence for in vivo application of griseusin-based probes.
3 Conclusions
This comprehensive study highlights the first concise and divergent synthetic approach to prepare griseusin A–E analogs. The enabling chemistry put forth facilitated a broad comparative cancer cell line cytotoxicity SAR analysis and in-depth mechanistic studies that established griseusins as inhibitors of Prx1/Grx3 and 4E-BP1 phosphorylation. Importantly, Prx1 and Grx3 overexpression is a hallmark of a variety of cancers and is associated with redox adaptation that promotes tumor progression and resistance to many anticancer agents and radiation.19 Consistent with this, knockdown of Prx1 and Grx3 expression in cancer cells leads to an increase in ROS levels, resulting in inhibition of proliferation, survival, invasion, metastasis, and sensitivity to chemotherapy and radiation.19d,20 These studies suggest that similar subtle shifts in the balance of [ROS] within the axolotl tail regeneration model may also contribute to the observed anti-proliferative effects of griseusins in vivo. Cumulatively, the facile access to the griseusins described herein is expected to enable new molecular probe development to advance the study the role of Prx1 and Grx3 in biology and may also serve as a starting point for early anticancer lead development. Given the established mechanistic relationship between griseusins and FB (an effective anticoccidial and antimalarial agent),1a,4 the current study may also prompt similar functional evaluation of griseusins.
Ethical statement
While the use of pre-feeding stage axolotls does not require a University of Kentucky Institutional Animal Care and Use Committee (IACUC)-approved protocol, embryos used in this study were treated according to the same ethical standards that apply to feeding axolotls.
Conflicts of interest
The authors declare the following competing financial interest: J. S. T. is a co-founder of Centrose (Madison, WI, USA).
Acknowledgements
This work was supported by National Institutes of Health grants R01 CA203257 (QBS, JST), R24 OD21479 (SRV, JST), R01 CA175105 (QBS), T32 DA016176 (YZ), the University of Kentucky College of Pharmacy, the National Center for Advancing Translational Sciences (UL1TR000117 and UL1TR001998), and the Start-up funding of Jiangsu Specially-Appointed Professor and National Natural Science Foundation of China (No. 21877062, YZ). We also thank Prof. Tyler D. McQuade (Florida State University) generously providing McQuade I NHC copper catalyst.
Notes and references
-
(a) G. A. Ellestad, M. P. Kunstmann, H. A. Whaley and E. L. Patterson, J. Am. Chem. Soc., 1968, 90, 1325–1332 CrossRef CAS PubMed
 ;
(b) S. Omura, K. Tsuzuki, Y. Iwai, M. Kishi, S. Watanabe and H. Shimizu, J. Antibiot., 1985, 38, 1447–1448 CrossRef CAS PubMed ;
(c) T. Okabe, K. Nomoto and N. Tanaka, J. Antibiot., 1986, 39, 1–5 CrossRef CAS PubMed ;
(d) T. W. Yu, M. J. Bibb, W. P. Revill and D. A. Hopwood, J. Bacteriol., 1994, 176, 2627–2634 CrossRef CAS PubMed ;
(e) R. J. Cox, T. S. Hitchman, K. J. Byrom, I. S. Findlow, J. A. Tanner, J. Crosby and T. J. Simpson, FEBS Lett., 1997, 405, 267–272 CrossRef CAS PubMed ;
(f) R. J. Zawada and C. Khosla, J. Biol. Chem., 1997, 272, 16184–16188 CrossRef CAS PubMed ;
(g) J. Crosby, K. J. Byrom, T. S. Hitchman, R. J. Cox, M. P. Crump, I. S. Findlow, M. J. Bibb and T. J. Simpson, FEBS Lett., 1998, 433, 132–138 CrossRef CAS PubMed ;
(h) R. E. Armer, C. J. Dutton, B. R. Fenner, S. D. Greenwood, K. T. Hall and A. J. Rudge, Bioorg. Med. Chem. Lett., 1998, 8, 139–142 CrossRef CAS PubMed ;
(i) R. J. Zawada and C. Khosla, Chem. Biol., 1999, 6, 607–615 CrossRef CAS PubMed ;
(j) T. Taguchi, S. Okamoto, K. Hasegawa and K. Ichinose, ChemBioChem, 2011, 12, 2767–2773 CrossRef CAS PubMed ;
(k) X. Wang, K. A. Shaaban, S. I. Elshahawi, L. V. Ponomareva, M. Sunkara, Y. Zhang, G. C. Copley, J. C. Hower, A. J. Morris, M. K. Kharel and J. S. Thorson, J. Nat. Prod., 2013, 76, 1441–1447 CrossRef CAS PubMed ;
(l) X. Wang, S. I. Elshahawi, K. A. Shaaban, L. Fang, L. V. Ponomareva, Y. Zhang, G. C. Copley, J. C. Hower, C. G. Zhan, M. K. Kharel and J. S. Thorson, Org. Lett., 2014, 16, 456–459 CrossRef CAS PubMed .
;
(b) S. Omura, K. Tsuzuki, Y. Iwai, M. Kishi, S. Watanabe and H. Shimizu, J. Antibiot., 1985, 38, 1447–1448 CrossRef CAS PubMed ;
(c) T. Okabe, K. Nomoto and N. Tanaka, J. Antibiot., 1986, 39, 1–5 CrossRef CAS PubMed ;
(d) T. W. Yu, M. J. Bibb, W. P. Revill and D. A. Hopwood, J. Bacteriol., 1994, 176, 2627–2634 CrossRef CAS PubMed ;
(e) R. J. Cox, T. S. Hitchman, K. J. Byrom, I. S. Findlow, J. A. Tanner, J. Crosby and T. J. Simpson, FEBS Lett., 1997, 405, 267–272 CrossRef CAS PubMed ;
(f) R. J. Zawada and C. Khosla, J. Biol. Chem., 1997, 272, 16184–16188 CrossRef CAS PubMed ;
(g) J. Crosby, K. J. Byrom, T. S. Hitchman, R. J. Cox, M. P. Crump, I. S. Findlow, M. J. Bibb and T. J. Simpson, FEBS Lett., 1998, 433, 132–138 CrossRef CAS PubMed ;
(h) R. E. Armer, C. J. Dutton, B. R. Fenner, S. D. Greenwood, K. T. Hall and A. J. Rudge, Bioorg. Med. Chem. Lett., 1998, 8, 139–142 CrossRef CAS PubMed ;
(i) R. J. Zawada and C. Khosla, Chem. Biol., 1999, 6, 607–615 CrossRef CAS PubMed ;
(j) T. Taguchi, S. Okamoto, K. Hasegawa and K. Ichinose, ChemBioChem, 2011, 12, 2767–2773 CrossRef CAS PubMed ;
(k) X. Wang, K. A. Shaaban, S. I. Elshahawi, L. V. Ponomareva, M. Sunkara, Y. Zhang, G. C. Copley, J. C. Hower, A. J. Morris, M. K. Kharel and J. S. Thorson, J. Nat. Prod., 2013, 76, 1441–1447 CrossRef CAS PubMed ;
(l) X. Wang, S. I. Elshahawi, K. A. Shaaban, L. Fang, L. V. Ponomareva, Y. Zhang, G. C. Copley, J. C. Hower, C. G. Zhan, M. K. Kharel and J. S. Thorson, Org. Lett., 2014, 16, 456–459 CrossRef CAS PubMed .
-
(a) M. A. Brimble, L. J. Duncalf and M. R. Nairn, Nat. Prod. Rep., 1999, 16, 267–281 RSC ;
(b) M. A. Brimble, Pure Appl. Chem., 2000, 72, 1635–1639 CAS ;
(c) J. Sperry, P. Bachu and M. A. Brimble, Nat. Prod. Rep., 2008, 25, 376–400 RSC ;
(d) J. Jacobs, S. Claessens, K. Huygen, K. A. Tehrani and N. De Kimpe, Pure Appl. Chem., 2011, 83, 1651–1674 CAS ;
(e) B. J. Naysmith, P. A. Hume, J. Sperry and M. A. Brimble, Nat. Prod. Rep., 2017, 34, 25–61 RSC ;
(f) Y. Zhang, X. Wang, M. Sunkara, Q. Ye, L. V. Ponomereva, Q. B. She, A. J. Morris and J. S. Thorson, Org. Lett., 2013, 15, 5566–5569 CrossRef CAS PubMed .
-
(a) T. Kometani, Y. Takeuchi and E. Yoshii, J. Org. Chem., 1982, 47, 4725–4730 CrossRef CAS ;
(b) T. Kometani, Y. Takeuchi and E. Yoshii, J. Org. Chem., 1983, 48, 2311–2314 CrossRef CAS ;
(c) N. Tsuji, T. Kamigauchi, H. Nakai and M. Shiro, Tetrahedron Lett., 1983, 24, 389–390 CrossRef CAS ;
(d) M. A. Brimble and M. R. Nairn, J. Chem. Soc., Perkin Trans. 1, 1990, 1, 169–171 RSC ;
(e) M. A. Brimble and M. R. Nairn, J. Chem. Soc., Perkin Trans. 1, 1992, 5, 579–583 RSC ;
(f) M. A. Brimble and M. R. Nairn, Molecules, 1996, 1, 3–14 CrossRef CAS ;
(g) M. A. Brimble, M. R. Nairn and J. Park, Org. Lett., 1999, 1, 1459–1462 CrossRef CAS ;
(h) M. A. Brimble, M. R. Nairn and J. Park, J. Chem. Soc., Perkin Trans. 1, 2000, 5, 697–709 RSC ;
(i) K. A. Parker, T. L. Mindt and Y. H. Koh, Org. Lett., 2006, 8, 1759–1762 CrossRef CAS PubMed ;
(j) B. J. Naysmith and M. A. Brimble, Org. Lett., 2013, 15, 2006–2009 CrossRef CAS PubMed ;
(k) B. J. Naysmith, D. Furkert and M. A. Brimble, Tetrahedron, 2014, 70, 1199–1206 CrossRef CAS ;
(l) X. Jiang, M. Wang, S. Song, Y. Xu, Z. Miao and A. Zhang, RSC Adv., 2015, 5, 27502–27508 RSC ;
(m) Y. Zhang, Q. Ye, X. Wang, Q. B. She and J. S. Thorson, Angew. Chem., Int. Ed. Engl., 2015, 54, 11219–11222 CrossRef CAS PubMed .
- J. T. Fitzgerald, P. P. Henrich, C. O'Brien, M. Krause, E. H. Ekland, C. Mattheis, J. M. Sa, D. Fidock and C. Khosla, J. Antibiot., 2011, 64, 799–801 CrossRef CAS PubMed .
- Q. Ye, Y. Zhang, Y. Cao, X. Wang, Y. Guo, J. Chen, J. Horn, L. V. Ponomareva, L. Chaiswing, K. A. Shaaban, Q. Wei, B. D. Anderson, D. K. St Clair, H. Zhu, M. Leggas, J. S. Thorson and Q. B. She, Cell Chem. Biol., 2019, 26, 366–377 CrossRef CAS PubMed .
-
(a) N. Tsuji, M. Kobayashi, Y. Wakisaka, Y. Kawamura and M. Mayama, J. Antibiot., 1976, 29, 7–9 CrossRef CAS PubMed ;
(b) N. Tsuji, M. Kobayashi, Y. Terui and K. Tori, Tetrahedron, 1976, 32, 2207–2210 CrossRef CAS ;
(c) M. Maruyama, C. Nishida, Y. Takahashi, H. Naganawa, M. Hamada and T. Takeuchi, J. Antibiot., 1994, 47, 952–954 CrossRef CAS PubMed ;
(d) M. Igarashi, W. Chen, T. Tsuchida, M. Umekita, T. Sawa, H. Naganawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 1502–1505 CrossRef CAS PubMed ;
(e) X. Li, Y. Zheng, I. Sattler and W. Lin, Arch. Pharmacal Res., 2006, 29, 942–945 CrossRef CAS ;
(f) Y. Q. Li, M. G. Li, W. Li, J. Y. Zhao, Z. G. Ding, X. L. Cui and M. L. Wen, J. Antibiot., 2007, 60, 757–761 CrossRef CAS PubMed ;
(g) J. He, E. Roemer, C. Lange, X. Huang, A. Maier, G. Kelter, Y. Jiang, L. H. Xu, K. D. Menzel, S. Grabley, H. H. Fiebig, C. L. Jiang and I. Sattler, J. Med. Chem., 2007, 50, 5168–5175 CrossRef CAS PubMed ;
(h) M. S. Abdelfattah, T. Kazufumi and M. Ishibashi, J. Antibiot., 2011, 64, 729–734 CrossRef CAS PubMed ;
(i) Z. G. Ding, J. Y. Zhao, M. G. Li, R. Huang, Q. M. Li, X. L. Cui, H. J. Zhu and M. L. Wen, J. Nat. Prod., 2012, 75, 1994–1998 CrossRef CAS PubMed .
-
(a) X. Wang, Y. Zhang, L. V. Ponomareva, Q. Qiu, R. Woodcock, S. I. Elshahawi, X. Chen, Z. Zhou, B. E. Hatcher, J. C. Hower, C. G. Zhan, S. Parkin, M. K. Kharel, S. R. Voss, K. A. Shaaban and J. S. Thorson, Angew. Chem., Int. Ed. Engl., 2017, 56, 2994–2998 CrossRef CAS PubMed ;
(b) L. V. Ponomareva, A. Athippozhy, J. S. Thorson and S. R. Voss, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2015, 178, 128–135 CAS .
- Conditions required to remove other potential naphthyl moiety protecting groups (e.g., trimethyl or triacetyl) were anticipated to contribute to C1 epimerization and/or scaffold decomposition.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS .
- J. E. Lee and J. Yun, Angew. Chem., Int. Ed. Engl., 2008, 47, 145–147 CrossRef CAS PubMed .
- S. Sato, T. Sakamoto, E. Miyazawa and Y. Kikugawa, Tetrahedron, 2004, 60, 7899–7906 CrossRef CAS .
-
(a) J. W. Bosco and A. K. Saikia, Chem. Commun., 2004, 1116–1117, 10.1039/b401218f ;
(b) R. Singh, R. M. Kissling, M. A. Letellier and S. P. Nolan, J. Org. Chem., 2004, 69, 209–212 CrossRef CAS PubMed .
- L. Chiummiento, M. Funicello, P. Lupattelli and F. Tramutola, Org. Lett., 2012, 14, 3928–3931 CrossRef CAS PubMed .
-
(a) T. Shiomi, T. Adachi, K. Toribatake, L. Zhou and H. Nishiyama, Chem. Commun., 2009, 5987–5989, 10.1039/b915759j ;
(b) J. K. Park, H. H. Lackey, M. D. Rexford, K. Kovnir, M. Shatruk and D. T. McQuade, Org. Lett., 2010, 12, 5008–5011 CrossRef CAS PubMed .
- Y. Lu, D. H. Wang, K. M. Engle and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916–5921 CrossRef CAS PubMed .
-
(a) D. A. Henderson, P. N. Collier, G. Pave, P. Rzepa, A. J. White, J. N. Burrows and A. G. Barrett, J. Org. Chem., 2006, 71, 2434–2444 CrossRef CAS PubMed ;
(b) K. Tatsuta, Y. Suzuki, T. Toriumi, Y. Furuya and S. Hosokawa, Tetrahedron Lett., 2007, 48, 8018–8021 CrossRef CAS .
-
(a) A. C. Hsieh, Y. Liu, M. P. Edlind, N. T. Ingolia, M. R. Janes, A. Sher, E. Y. Shi, C. R. Stumpf, C. Christensen, M. J. Bonham, S. Wang, P. Ren, M. Martin, K. Jessen, M. E. Feldman, J. S. Weissman, K. M. Shokat, C. Rommel and D. Ruggero, Nature, 2012, 485, 55–61 CrossRef CAS PubMed ;
(b) Q. Ye, W. Cai, Y. Zheng, B. M. Evers and Q. B. She, Oncogene, 2014, 33, 1828–1839 CrossRef CAS PubMed ;
(c) J. Wang, Q. Ye, Y. Cao, Y. Guo, X. Huang, W. Mi, S. Liu, C. Wang, H. S. Yang, B. P. Zhou, B. M. Evers and Q. B. She, Nat. Commun., 2017, 8, 2207 CrossRef PubMed .
- N. W. Al Haj Baddar, A. Chithrala and S. R. Voss, Dev. Dyn., 2019, 248, 189–196 CrossRef CAS PubMed .
-
(a) M. K. Cha and I. H. Kim, Cancer Epidemiol., 2009, 33, 281–287 CrossRef CAS PubMed ;
(b) K. Iwao-Koizumi, R. Matoba, N. Ueno, S. J. Kim, A. Ando, Y. Miyoshi, E. Maeda, S. Noguchi and K. Kato, J. Clin. Oncol., 2005, 23, 422–431 CrossRef CAS PubMed ;
(c) M. H. Park, M. Jo, Y. R. Kim, C. K. Lee and J. T. Hong, Pharmacol. Ther., 2016, 163, 1–23 CrossRef CAS PubMed ;
(d) Y. Qu, J. Wang, P. S. Ray, H. Guo, J. Huang, M. Shin-Sim, B. A. Bukoye, B. Liu, A. V. Lee, X. Lin, P. Huang, J. W. Martens, A. E. Giuliano, N. Zhang, N. H. Cheng and X. Cui, J. Clin. Invest., 2011, 121, 212–225 CrossRef CAS PubMed ;
(e) D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed ;
(f) C. M. Woolston, S. J. Storr, I. O. Ellis, D. A. Morgan and S. G. Martin, Radiother. Oncol., 2011, 100, 308–313 CrossRef CAS PubMed .
-
(a) M. Bajor, A. O. Zych, A. Graczyk-Jarzynka, A. Muchowicz, M. Firczuk, L. Trzeciak, P. Gaj, A. Domagala, M. Siernicka, A. Zagozdzon, P. Siedlecki, M. Kniotek, P. C. O'Leary, J. Golab and R. Zagozdzon, Br. J. Cancer, 2018, 119, 873–884 CrossRef CAS PubMed ;
(b) M. F. Chen, P. C. Keng, H. Shau, C. T. Wu, Y. C. Hu, S. K. Liao and W. C. Chen, Int. J. Radiat. Oncol., Biol., Phys., 2006, 64, 581–591 CrossRef CAS PubMed ;
(c) H. Jiang, L. Wu, M. Mishra, H. A. Chawsheen and Q. Wei, Am. J. Cancer Res., 2014, 4, 445–460 Search PubMed ;
(d) G. Poschmann, M. Grzendowski, A. Stefanski, E. Bruns, H. E. Meyer and K. Stuhler, Biochim. Biophys. Acta, 2015, 1854, 624–631 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02289a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Qing
Ye‡
d,
Larissa V.
Ponomareva
bc,
Yanan
Cao
d,
Yang
Liu
bc,
Zheng
Cui
c,
Steven G.
Van Lanen
c,
S. Randal
Voss
e,
Qing-Bai
She
abc,
Qing
Ye‡
d,
Larissa V.
Ponomareva
bc,
Yanan
Cao
d,
Yang
Liu
bc,
Zheng
Cui
c,
Steven G.
Van Lanen
c,
S. Randal
Voss
e,
Qing-Bai
She