
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic protodeboronation of pinacol boronic esters: formal anti-Markovnikov hydromethylation of alkenes†
Florian
Clausen
,
Marvin
Kischkewitz
,
Klaus
Bergander
and
Armido
Studer
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstraβe 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 22nd May 2019
Abstract
Pinacol boronic esters are highly valuable building blocks in organic synthesis. In contrast to the many protocols available on the functionalizing deboronation of alkyl boronic esters, protodeboronation is not well developed. Herein we report catalytic protodeboronation of 1°, 2° and 3° alkyl boronic esters utilizing a radical approach. Paired with a Matteson–CH2–homologation, our protocol allows for formal anti-Markovnikov alkene hydromethylation, a valuable but unknown transformation. The hydromethylation sequence was applied to methoxy protected (−)-Δ8-THC and cholesterol. The protodeboronation was further used in the formal total synthesis of δ-(R)-coniceine and indolizidine 209B.
Organoboron compounds are highly valuable building blocks in organic synthesis.1,2 The most important application to be named is the Suzuki–Miyaura-coupling.3–5 Moreover, the boron moiety can be converted into a broad range of functional groups.2,6 These transformations include oxidations,7 aminations,8–10 halogenations,11,12 and C–C-bond-formations such as alkenylations,13,14 alkynylations15 and arylations.16 To access the B-building blocks, various borylation approaches have been developed over the years17–19 including the prominent asymmetric hydroboration reaction reported by H. C. Brown in 1961.20
However, considering synthetic applications, drawbacks associated with organoboranes are their limited air and moisture stability.21 Along these lines, the introduction of the more stable boronic ester moiety has significantly expanded the scope of boron chemistry. In particular the pinacol boronic esters, which are usually bench stable,22 easy to purify and often even commercially available have played a prominent role. These features are attractive for chemical transformations, where the valuable boron moiety remains in the product (homologations,23–25 conjunctive cross couplings26 or radical-polar crossover reactions27–32). However, the increased stability also rises new challenges, considering the removal of the boron moiety at the end of a sequence if required. While boranes undergo efficient protodeboronation with propionic acid by protonolysis33–35 or hydrogenation at elevated temperatures,36,37 boronic esters usually do not.21,22 We further noted that the protodeboronation of unactivated alkyl and especially primary alkyl boronic esters is underexplored. Reduction of 3° alkyl boronic esters was recently investigated by Aggarwal and coworkers; however, their method is only applicable to the protodeboronation of activated benzylic substrates.38 In 2005, Renaud and coworkers described an efficient sequence for the formal hydrogenation of unactivated alkenes to alkanes using a hydroboration-deboronation strategy (Scheme 1a).39 The protodeboronation of in situ generated catechol boronic esters proceeds via a radical chain reaction, but this reaction is limited to the more expensive catechol boronic esters and works well for 2° alkyl B-esters.40
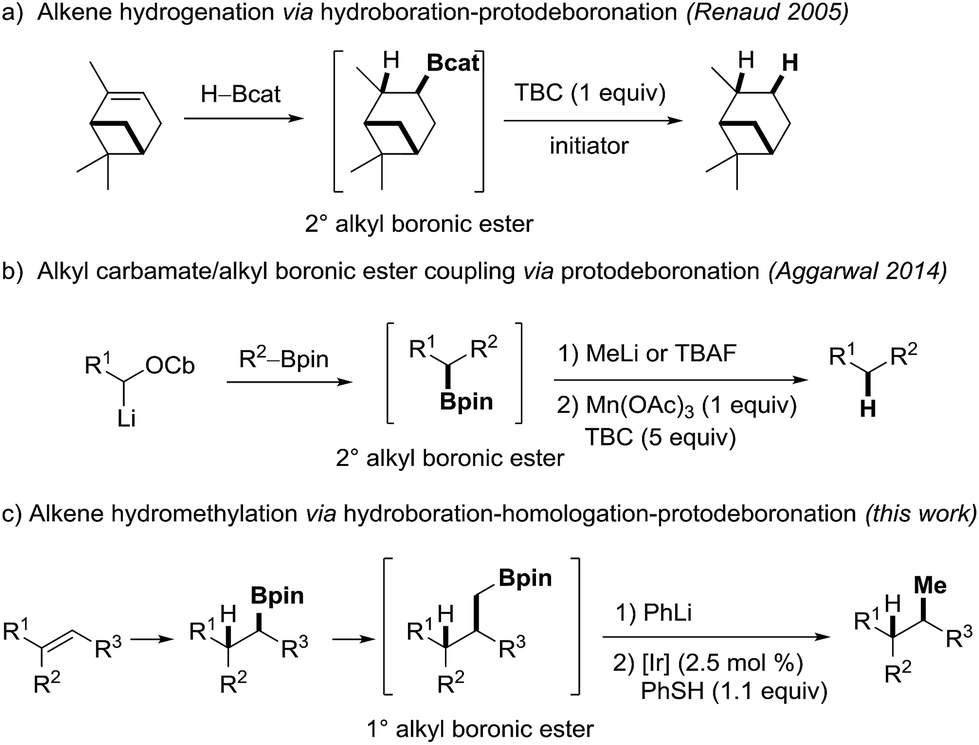 | ||
| Scheme 1 Protodeboronation and its application in synthesis. | ||
An elegant C–C-coupling/protodeboronation strategy was introduced in 2014 by Aggarwal and coworkers (Scheme 1b).41 In this sequence, the protodeboronation of 2° alkyl pinacol boronic esters was achieved by oxidation of their corresponding methyl boron ate complexes with a stoichiometric amount of manganese(III)acetate. The resulting secondary alkyl radicals are then reduced with 4-tert-butylcatechol (TBC). In contrast to the Renaud process, the latter protodeboronation works with pinacol boronic esters but does not proceed on primary B-esters (see below) and a large amount of reductant is required.
During a metabolite study, we targeted an anti-Markovnikov-type hydromethylation of a trisubstituted alkene and noted that different alkene hydromethylation approaches have been published over the years.42 However, all protocols developed so far provide the Markovnikov product. We therefore envisioned a hydroboration/CH2-homologation/protodeboronation strategy to achieve anti-Markovnikov alkene hydromethylation (Scheme 1c). In this sequence, the regioselectivity is controlled in the initial hydroboration step, where suitable methods are available.43,44 The methylene unit can readily be introduced via established Matteson-homologation using CH2Br2 as the methylene source. The real challenge turned out to be the final protodeboronation of the intermediate 1° alkyl boronic ester. The conditions reported by Aggarwal41 were ineffective for our systems and also other methods failed. We therefore initiated a project along those lines and aimed at a catalytic protodeboronation using boron ate complexes as substrates. Single electron oxidation by a redox catalyst should generate the corresponding alkyl radicals.45–47 Reduction of these alkyl radicals to give the targeted products should be achieved by thiols, since they are known to be efficient H-donors and the resulting thiyl radicals can be reduced by the photoredox catalyst thereby regenerating the initial oxidation state of the catalyst.48–51 Hence, an external stoichiometric oxidant is not required increasing the economy of the protodeboronation.
We commenced our investigations by testing different boron ate complexes derived from boronic ester 1a. In each case, the ate complex was generated in Et2O. After removal of the ether, the residual ate complex was reacted with 2.5 mol% of the Ir-catalyst PC1 and thiophenol (1.1 equiv.) in MeOH/acetone at room temperature under blue light irradiation. B-ate complexes derived from TBAF,47 DMAP,47 PPh3 (ref. 47) and 3-quinolidinol47 (1.1 equivalent each) did not afford the desired product 2a. However, with PhLi, the target 2a was obtained in 79% yield (Scheme 2). Phenyl boronic ester 3 was formed as the stoichiometric byproduct. Notably, in a proof of principle experiment we demonstrated that the cascade also works efficiently by using catalytic amounts of the thiol (10 mol%) in combination with diphenyl phosphate (1.1 equiv.) as the external proton source (95%, see ESI†). However, by using 50 mol% thiophenol in MeOH/acetone, the yield dropped significantly, showing the necessity of the phosphate in the thiol catalytic procedure. Since thiophenol is cheap, we decided to use the stoichiometric thiol protocol for the scope studies.
 | ||
Scheme 2 Substrate scope of the photoredox catalyzed protodeboronation. Conditions: 1a–n (0.2 mmol), PhLi (0.22 mmol), Et2O (2 mL), thiophenol (0.22 mmol), Ir-catalyst PC1 (2.5 mol%) in MeOH/acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2 mL). Isolated yields, unless otherwise noted. aYield determined by GC. [Ir] = Ir(dFCF3ppy)2(dtbbpy)PF6 (PC1). :1, 2 mL). Isolated yields, unless otherwise noted. aYield determined by GC. [Ir] = Ir(dFCF3ppy)2(dtbbpy)PF6 (PC1). | ||
Under optimized conditions, various primary alkyl boronic esters 1b–n were tested (Scheme 2). Reactions proceeded smoothly and the products 2b–n were isolated in moderate to excellent yields (52–93%). Electron-rich arenes are well tolerated, as documented by the successful preparation of 2b–c, 2g and 2k. Furthermore, substrates bearing a triphenyl silane (1i) and a carbazole moiety (1j) performed well and the desired products 2i–j were isolated in 72% and 88% yield, respectively. As expected, the protodeboronation of enantioenriched boronic ester 1k, readily prepared via stereoselective hydroboration,52 occurs with high stereospecificity (96% es). The reason for the slight drop in ee might be reversible H-abstraction at the benzylic position by the thiyl radical. In any case, this sequence represents a new strategy for the asymmetric synthesis of aryl dialkyl methanes of type 2k.
The method was also applied to the protodeboronation of the 2° alkyl boronic esters 1l and 1m to yield the Boc-protected pyrrolidine 2l and sulfone 2m in 85 and 93% yield. Furthermore, the 3° alkyl boronic ester 1n bearing a tert-butylester group was smoothly protodeboronated (73%). A gram scale experiment (preparation of 2h) proved the robustness of the procedure (61%).
In order to verify that the protodeboronation occurs via a radical pathway, a probe experiment was conducted. The phenyl boron ate complex derived from the cyclopropylmethyl substituted boronic ester 1o reacted under standard conditions to the ring opened alkene 2o (E/Z = 4:1, 84%), clearly supporting the radical nature of the cascade (Scheme 3a).53 A possible mechanism is depicted in Scheme 3b. Photoexcited PC1 first oxidizes the boron ate complex A to generate an alkyl radical R˙ along with Ph-Bpin. C-radical R˙ then abstracts an H-atom from thiophenol to form the product 2 and the phenyl thiyl radical, which is reduced by the Ir(II)-complex to regenerate the starting Ir(III)-complex PC1.
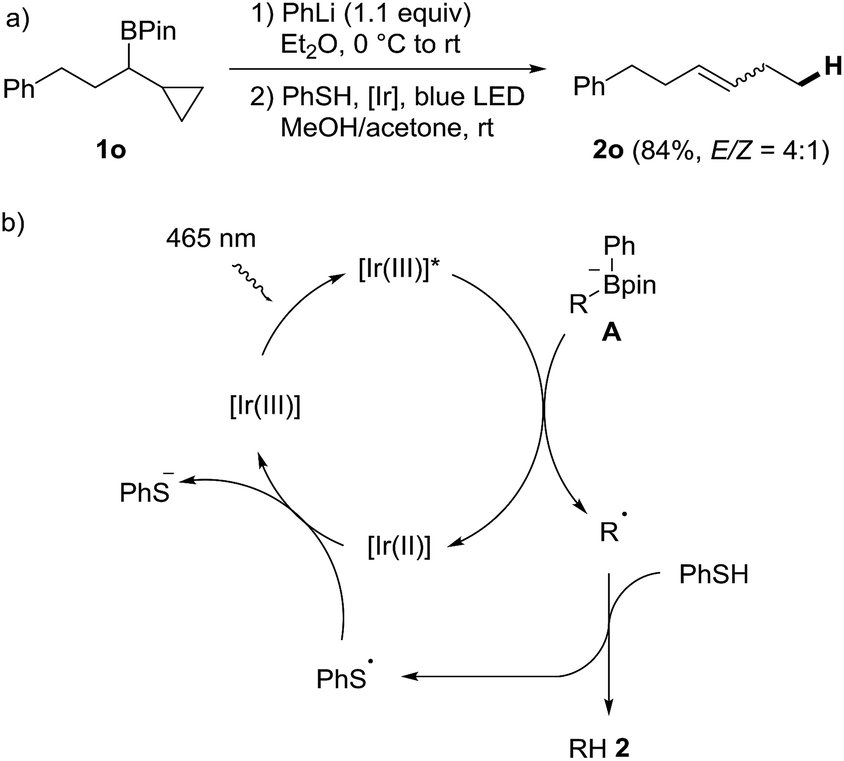 | ||
| Scheme 3 Radical probe experiment and proposed mechanism. | ||
To further document the potential of the protodeboronation, we applied the novel protocol to the formal total synthesis of two indolizidine-core containing secondary metabolites, δ-(R)-coniceine (9) and indolizidine 209B (13) (Scheme 4). Starting with commercial N-Boc-pyrrolidine (4), enantiopure boronic ester 5 was prepared via a (+)-sparteine mediated asymmetric lithiation/borylation.54–56 The ester moiety was introduced via stereospecific radical-induced 1,2-migration27,28 of the corresponding in situ generated vinyl and isopropenyl boron ate complexes with α-iodo acetates as radical precursors to provide 6 and 10 in 52% and 65% yield. In the coniceine route, 6 was directly subjected to the protodeboronation to provide the pyrrolidine 7 in 61% yield. After ester cleavage and Boc-deprotection, the corresponding carboxylic acid was cyclised with EDC and DMAP to yield the bicyclic lactam 8 in 87% yield, completing the formal total synthesis57 of δ-(R)-coniceine (9). A crucial step in the indolizidine 209B synthesis was the build-up of the additional C-8 stereocenter via a stereoselective protodeboronation. To this end, the 3° boronic ester 10 was converted into the rigid indolizidine precursor 11via Boc-deprotection and lactamization (68%). Protodeboronation by using the less nucleophilic (3,5-bis(trifluoromethyl)phenyl)lithium for boron ate complex formation in place of PhLi, to prevent aryl addition to the lactam moiety, afforded the indolizidine 12 in 59% yield and good diastereoselectivity (dr = 5:1). Final transformation into indolizidine 209B (13) can be achieved by Grignard-addition to the lactam followed by stereoselective reductive deoxygenation.58
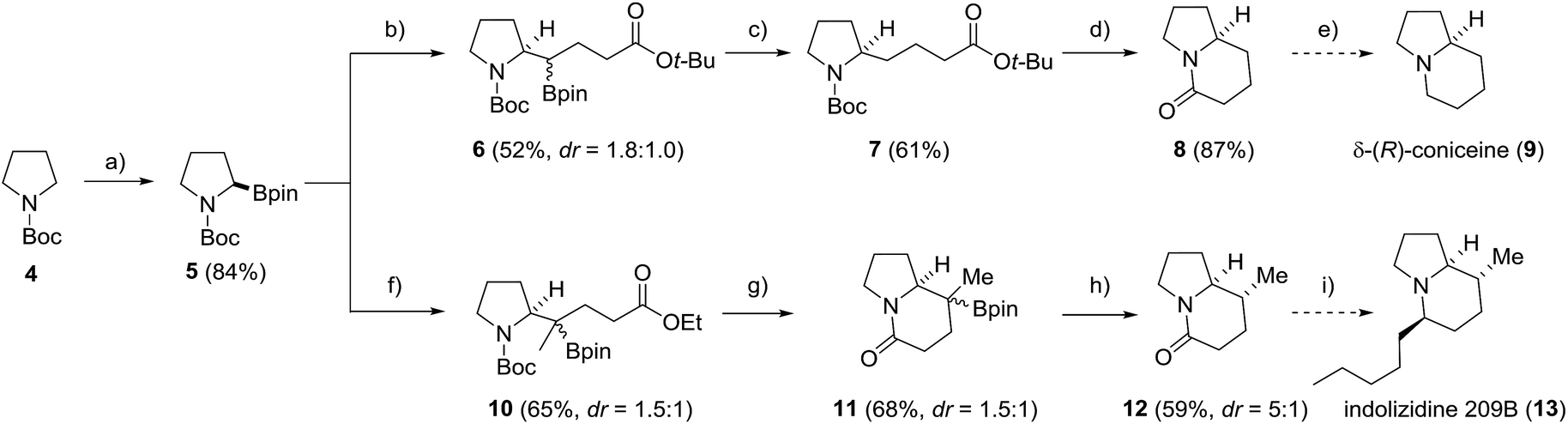 | ||
| Scheme 4 Formal total synthesis of δ-(R)-coniceine (9) and indolizidine 209B (13). Reagents and conditions: (a) s-BuLi (1.2 equiv.), (+)-sparteine (1.2 equiv.) Et2O, −78 °C, 3 h then isopropyl pinacol borate (1.3 equiv.), −78 °C, 2 h; (b) vinyllithium (1.3 equiv.), Et2O, −78 °C to rt, 30 min then tert-butyl 2-iodoacetate (2.0 equiv.), MeCN, hν, 50 °C, 24 h; (c) PhLi (1.1 equiv.), Et2O, 0 °C to rt, 30 min then PhSH (1.1 equiv.), PC1 (2.5%), blue LED, MeOH/acetone, 16 h; (d) HCl in dioxane, rt, 8 h then EDC·HCl (2.0 equiv.), DMAP (20 mol%), Hünig's base (2.0 equiv.), CH2Cl2, rt, over night; (e) see ref. 57; (f) isopropenyllithium (1.3 equiv.), Et2O, −78 °C to rt, 30 min then ethyl 2-iodoacetate (5.0 equiv.), MeCN, hν, 50 °C, 24 h; (g) TFA (10 eq.), CH2Cl2, 0 °C to rt, 2 h then NEt3 (20 equiv.), CH2Cl2, rt, 12 h; (h) (3,5-bis(trifluoromethyl)phenyl)lithium (1.3 equiv.), Et2O, −78 °C, 1 h then PhSH (1.3 equiv.), PC1 (2.5%), blue LED, MeOH/acetone, 16 h; (i) see ref. 58. | ||
Finally, we focused on the formal alkene hydromethylation sequence (Scheme 5). Initial studies were conducted with the boronic esters 14a–d which can be obtained via hydroboration of the corresponding styrene derivatives (see the ESI†). The Matteson-CH2-homologation was carried out with in situ generated CH2BrLi in THF at low temperature. After removal of the solvent, the crude homologated boronic esters 15a–d were directly subjected to the protodeboronation to provide the targeted compounds 16a–d in 65–66% overall yields. The heteroarenes 14e, 14f and a silylated boronic ester were eligible substrates and provided 16e–g in 51–70% yield. When the OTs-protected phenol 14h was subjected to the sequence, the deprotected phenol 16h resulted (48%). The sequence was also applied to the hydromethylation of more complex alkenes such as the fluorine containing system 14d to obtain the hydromethylated compound 16d in 66% yield. The hetero arenes 14e and 14f were tolerated and provided the products 16e and 16f in 51–55% and yields. When the OTs-protected phenol 14h was subjected to the sequence the deprotected compound 16h was isolated in 48% yield. The method was also applied to cyclic olefins such as the cyclohexane derivative 14i to give the product 16i in 61% yield, the methyl protected (−)-Δ8-THC 14j and the cholesterol derivative 14k. Stereo- and regioselective hydroboration using catecholborane under neat conditions16 followed by transesterification with pinacol gave the 2° alkyl boronic esters 14j and 14k in 55% to 65% yield as single diastereoisomers (see ESI†). CH2-homologation and protodeboronation afforded the methylated products 16j and 16k in excellent overall yields (89% and 90%), convincingly documenting the potential of our novel sequence.
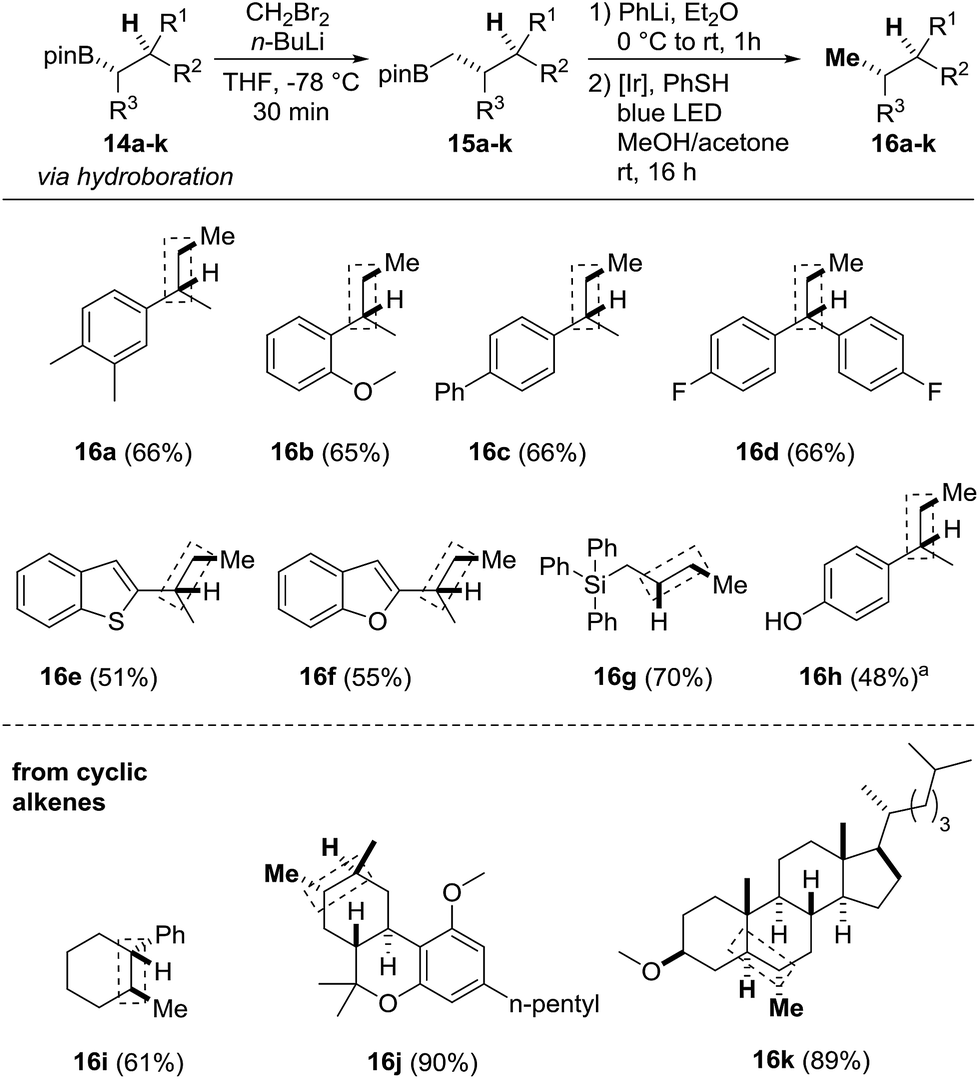 | ||
| Scheme 5 Formal alkene hydromethylation. Homologation: 14a–k (0.05–0.2 mmol, 1.0 equiv.), CH2Br2 (2.0–3.0 equiv.), n-butyllithium (1.5–2.0 equiv.) in THF or Et2O: protodeboronation: 15a–k (0.05–0.2 mmol, 1.0 equiv., used as crude from homologation step), PhLi (1.1 equiv.), Et2O (0.5–2.0 mL), thiophenol (1.1 equiv.), PC1 (2.5 mol%) in MeOH/acetone (1:1, 0.5–2.0 mL). Yield corresponds to the two-step homologation-protodeboronation sequence. For the hydroboration step, we refer to the ESI.†aDerived from O-Ts-protected phenol, O–S cleavage during reaction. | ||
In summary, we have developed the first catalytic protodeboronation of unactivated 1°, 2° and 3° alkyl pinacol boronic esters utilizing photoredox catalysis. We were able to carry out a formal methane addition to various alkenes combining the protodeboronation protocol with a Matteson-CH2-homologation. The sequence was applied to the hydromethylation of secondary metabolite derivatives, a (−)-Δ8-THC derivative and O-methylated cholesterol. Furthermore, the protodeboronation was successfully also used in formal total syntheses of δ-(R)-coniceine (9) and indolizidine 209B (13).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the European Research Council (ERC Advanced Grant agreement no. 692640) for financial support.Notes and references
- Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, 2015 Search PubMed.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS.
- H. K. Scott and V. K. Aggarwal, Chem.–Eur. J., 2011, 17, 13124–13132 CrossRef CAS.
- H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1956, 78, 5694–5695 CrossRef CAS.
- H. C. Brown, M. M. Midland and A. B. Levy, J. Am. Chem. Soc., 1973, 95, 2394–2396 CrossRef CAS.
- H. C. Brown, A. M. Salunkhe and A. B. Argade, Organometallics, 1992, 11, 3094–3097 CrossRef CAS.
- S. N. Mlynarski, A. S. Karns and J. P. Morken, J. Am. Chem. Soc., 2012, 134, 16449–16451 CrossRef CAS PubMed.
- H. C. Brown and C. F. Lane, J. Chem. Soc., Chem. Commun., 1971, 521–522 RSC.
- H. C. Brown, N. R. De Lue, G. W. Kabalka and H. C. Hedgecock, J. Am. Chem. Soc., 1976, 98, 1290–1291 CrossRef CAS.
- R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 49, 3323–3336 CrossRef CAS.
- G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS.
- Y. Wang, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 4270–4274 CrossRef CAS PubMed; Y. Wang, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., 2016, 128, 4342 CrossRef.
- A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed; Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., 2018, 130, 17074 CrossRef.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson and A. K. Chatterjee, et al. , Science, 2017, 356, 7355 CrossRef PubMed.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 486–487 Search PubMed.
- D. S. Matteson, J. Organomet. Chem., 1999, 581, 51–65 CrossRef CAS.
- R. Bernardini, A. Oliva, A. Paganelli, E. Menta, M. Grugni, S. D. Munari and L. Goldoni, Chem. Lett., 2009, 38, 750–751 CrossRef CAS.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033–2036 CrossRef CAS PubMed.
- D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed.
- L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed.
- M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Science, 2017, 355, 936–938 CrossRef CAS PubMed.
- C. Gerleve, M. Kischkewitz and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 2441–2444 CrossRef CAS PubMed; C. Gerleve, M. Kischkewitz and A. Studer, Angew. Chem., 2018, 130, 2466 CrossRef.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed.
- N. D. C. Tappin, M. Gnägi-Lux and P. Renaud, Chem.–Eur. J., 2018, 24, 11498–11502 CrossRef CAS PubMed.
- M. Kischkewitz, Org. Synth., 2018, 95, 205–217 CrossRef CAS.
- M. Kischkewitz, C. Gerleve and A. Studer, Org. Lett., 2018, 20, 3666–3669 CrossRef CAS PubMed.
- H. C. Brown and K. Murray, J. Org. Chem., 1961, 26, 631–632 CrossRef CAS.
- H. C. Brown and K. Murray, J. Am. Chem. Soc., 1959, 81, 4108–4109 CrossRef CAS.
- H. C. Brown and K. J. Murray, Tetrahedron, 1986, 42, 5497–5504 CrossRef CAS.
- R. Köster, Angew. Chem., 1956, 68, 383 Search PubMed.
- F. L. Ramp, E. J. DeWitt and L. E. Trapasso, J. Org. Chem., 1962, 27, 4368–4372 CrossRef CAS.
- S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096–17098 CrossRef CAS PubMed.
- D. Pozzi, E. M. Scanlan and P. Renaud, J. Am. Chem. Soc., 2005, 127, 14204–14205 CrossRef CAS PubMed.
- G. Villa, G. Povie and P. Renaud, J. Am. Chem. Soc., 2011, 133, 5913–5920 CrossRef CAS PubMed.
- R. Rasappan and V. K. Aggarwal, Nat. Chem., 2014, 6, 810–814 CrossRef CAS PubMed.
- H. T. Dao, C. Li, Q. Michaudel, B. D. Maxwell and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 8046–8049 CrossRef CAS PubMed.
- M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef CAS PubMed.
- R. Soundararajan and D. S. Matteson, J. Org. Chem., 1990, 55(8), 2274–2275 CrossRef CAS.
- G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366–5382 RSC.
- C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870 CrossRef CAS PubMed; C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., 2019, 131, 3910 CrossRef.
- F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085 CrossRef CAS PubMed; F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., 2016, 128, 14291 CrossRef.
- Q. Zhu, D. E. Graff and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 741–747 CrossRef CAS PubMed.
- A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Chem.–Eur. J. DOI:10.1002/chem.201901566.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 1993, 115, 939–947 CrossRef.
- J. Chen, T. Xi, X. Ren, B. Cheng, J. Guo and Z. Lu, Org. Chem. Front., 2014, 1, 1306–1309 RSC.
- K. E. Liu, C. C. Johnson, M. Newcomb and S. J. Lippard, J. Am. Chem. Soc., 1993, 115, 939–947 CrossRef CAS.
- A. Varela, L. K. B. Garve, D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 2127–2131 CrossRef CAS PubMed; A. Varela, L. K. B. Garve, D. Leonori and V. K. Aggarwal, Angew. Chem., 2017, 129, 2159 CrossRef.
- D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282–2316 CrossRef CAS.
- S. Wu, S. Lee and P. Beak, J. Am. Chem. Soc., 1996, 118, 715–721 CrossRef CAS.
- S. H. Park, H. J. Kang, S. Ko, S. Park and S. Chang, Tetrahedron: Asymmetry, 2001, 12, 2621–2624 CrossRef CAS.
- H. Wu, M. Yu, Y. Zhang and G. Zhao, Chin. J. Chem., 2009, 27, 183–188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02067e |
| This journal is © The Royal Society of Chemistry 2019 |