
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction and interconversion of anion-coordination-based (‘aniono’) grids and double helicates modulated by counter-cations†
Xiaoxia
Fan‡
a,
Dan
Zhang‡
a,
Shiyu
Jiang
a,
Heng
Wang
b,
Lin-Ting
Lin
c,
Bo
Zheng
 a,
Wen-Hua
Xu
a,
Yanxia
Zhao
a,
Benjamin P.
Hay
d,
Yi-Tsu
Chan
c,
Xiao-Juan
Yang
*a,
Xiaopeng
Li
b and
Biao
Wu
*a
a,
Wen-Hua
Xu
a,
Yanxia
Zhao
a,
Benjamin P.
Hay
d,
Yi-Tsu
Chan
c,
Xiao-Juan
Yang
*a,
Xiaopeng
Li
b and
Biao
Wu
*a
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, China. E-mail: yangxiaojuan@nwu.edu.cn; wubiao@nwu.edu.cn
bDepartment of Chemistry, University of South Florida, Tampa, FL 33620, USA
cDepartment of Chemistry, National Taiwan University, Taipei 10617, Taiwan
dSupramolecular Design Institute, Oak Ridge, TN 37830, USA
First published on 22nd May 2019
Abstract
Supramolecular assembly of well-defined discrete architectures has been of great interest due to the tunable properties of these structures in functional materials and bio-mimicking. While metal-coordination-driven assembly has been extensively studied, anion-coordination-driven assembly (ACDA) is just emerging for constructing complex supramolecular structures. Herein two A2nL2n (A = anion, L = ligand; n = 1 or 2) ‘aniono’-supramolecular assemblies, i.e. double helicates and the first anion grid, have been constructed based on the coordination between phosphate (PO43−) anion and a bis–tris(urea) ligand. Moreover, the aniono-grid and double helicate motifs can be readily interconverted under ambient conditions by simply changing the counter-cation. These results redefine the power and scope of ACDA, which may represent a new approach in the assembly of well-defined architectures in parallel with the metal coordination-driven assembly of metallo-supramolecules.
Introduction
Assembly of supramolecular architectures plays an active role in the fields of functional materials and bio-mimicking.1 A broad array of discrete structures has been constructed based on metal-coordination-driven assembly, such as helicates, polygons, polyhedra, cages, and interlocked metallo-supramolecules.2 Meanwhile, such architectures can be accessed by (dynamic) covalent bonding3 or hydrogen bonding.4 During the assembling process, interconversion between two topologies in some systems, e.g., double helicates and mononuclear complexes,5 tetrahedral cages and triple helicates,6 and many others,7 has also been studied in terms of the dynamic functions of different structures.8 Among the various entities, helical and grid complexes are highly interesting due to their specific topologies and properties.9 As mimics of the helical motifs in DNA and proteins, synthetic helicates have fascinated supramolecular chemists with applications in molecular recognition, catalysis, molecular machines, optics, etc.10 With specific arrangement of metal ions within a square or rectangular grid, metallo-grids have been explored in magnetic and electrochemical materials as well as in supramolecular host–guest chemistry.11Compared to the flourishing metal-coordination-driven assembly, anion-coordination-driven assembly (ACDA) is less developed, yet it provides an efficient approach to construct well-defined structures.12 In particular, the coordination between phosphate ions (PO43−) and oligourea ligands has proven to be surprisingly stable.13 As such, we were able to construct a series of anion-based architectures with phosphate ions as the coordination centers and oligo-bis(urea) as the ligands, such as the first A2L3 triple helicate and both face-directed A4L4- and edge-directed A4L6-type (A denotes anion) tetrahedral cages.14 More recently, we further demonstrated the interconversion between the A2L3 triple helicate and A4L6 cage.15 These results imply that the ‘aniono’-supramolecular assemblies might become a new class of superstructures in parallel with the ‘metallo’-systems. In addition, held together by multiple hydrogen bonds, these metal-free ‘aniono’-systems are more bio-compatible, and their assembly–disassembly and isomerization may be readily modulated under mild conditions. However, compared to the well-established metal coordination, there are far fewer known anion complexes that exhibit the stability, predictable geometry, and symmetry needed to form such assemblies.16 These features render ACDA very attractive but also highly challenging.
In the pursuit of more complex anion-based systems, we move forward from the bis–bis(urea)17 to the bis–tris(urea) unit.18 The tris(urea) subunit exhibits stronger coordination affinity than the bis(urea) and forms stable AL2 phosphate complexes with binding constants even comparable to those of some metal complexes.18a In addition, the coordination geometry of these complexes is analogous to the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal complexes of terpyridine (tpy) (Scheme 1a), a versatile subunit for metal-based supramolecular assembly.19 This analogy suggests that the 1:2 phosphate–tris(urea) complex is a potentially viable motif for anion-directed assembly with more predictable structures. Moreover, symmetry-constrained, de novo structure-based design methods20 suggested that the p-xylylene-linked bis–tris(urea) L exhibited a conformation that complemented an A2L2 helical assembly (Fig. S1†). In this work, ligand L readily forms the double helicate and first anion-directed grid complex. Herein we report the crystal structures and solution assembly of these assemblies (Scheme 1b). The double helicate motif features a large, partially open cavity that is ideal for guest encapsulation and exchange, much like the grooves of DNA. Moreover, the grid and double helicate are easily interconverted by changing the counter-cation. Note that the switch between the M2L2 double helicates and 2 × 2 (M4L4) grid complexes had not been reported until recently when Lehn et al. described the first example of such an interconversion between a binuclear CuI double helicate and a tetranuclear CuII grid.21 Hence, the current work also represents the first ‘anion version’ of the double helicate/grid interconversion and broadens the spectrum of ACDA in supramolecular transformation.
:1 metal complexes of terpyridine (tpy) (Scheme 1a), a versatile subunit for metal-based supramolecular assembly.19 This analogy suggests that the 1:2 phosphate–tris(urea) complex is a potentially viable motif for anion-directed assembly with more predictable structures. Moreover, symmetry-constrained, de novo structure-based design methods20 suggested that the p-xylylene-linked bis–tris(urea) L exhibited a conformation that complemented an A2L2 helical assembly (Fig. S1†). In this work, ligand L readily forms the double helicate and first anion-directed grid complex. Herein we report the crystal structures and solution assembly of these assemblies (Scheme 1b). The double helicate motif features a large, partially open cavity that is ideal for guest encapsulation and exchange, much like the grooves of DNA. Moreover, the grid and double helicate are easily interconverted by changing the counter-cation. Note that the switch between the M2L2 double helicates and 2 × 2 (M4L4) grid complexes had not been reported until recently when Lehn et al. described the first example of such an interconversion between a binuclear CuI double helicate and a tetranuclear CuII grid.21 Hence, the current work also represents the first ‘anion version’ of the double helicate/grid interconversion and broadens the spectrum of ACDA in supramolecular transformation.
 | ||
| Scheme 1 (a) Design of bis–bis(urea) and bis–tris(urea) ligands by mimicking oligopyridine–metal coordination; (b) triple helicate assembled by bis–bis(urea) ligands and TMA3(PO4);14a A2L2 double helicate (complexes 1a–1d) and A4L4 grid (complex 2) (A = anion, L = ligand) assembled by the bis–tris(urea) ligand L and PO43− in the presence of different tetraalkylammonium cations. | ||
Results and discussion
Synthesis, structures, and solution studies of the double helicate 1a and grid complex 2
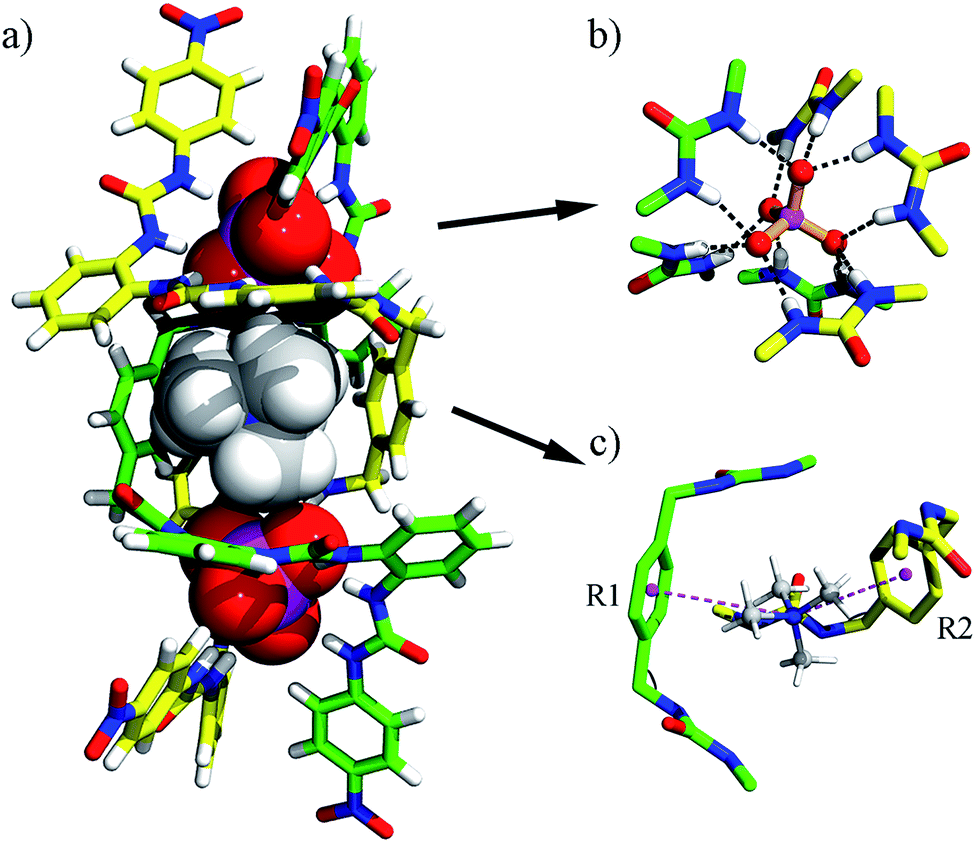 | ||
| Fig. 1 (a) Crystal structure of the double helicate [(TMA)⊂(PO4)2L2]5– (1a); (b) hydrogen bonds between a PO43− ion and six urea units; (c) a TMA+ ion is encapsulated in the central cavity through cation–π interactions (purple-dashed lines; R1 and R2 denote the centroids of the aryl rings that are involved in cation–π interactions). Only an M enantiomer is shown; other counter cations and solvents are omitted for clarity. | ||
In the assembly of anion-centered double helicates, the first one was constructed with polyguanidinium ligands and sulfate ions in solution.22 Later, several other double helicates based on halide anions were reported, e.g., the fluoride-directed helicate with neutral amide ligands23 and chloride-directed double helix with pyrrole-based oligomers.24 Very recently, Flood et al. reported the interconversion of single (foldamers) and double helicates glued by anions.25 However, complex 1a is the first double helicate with PO43− anions.
The assembly of L and PO43− ions and the encapsulation of TMA+ in solution was investigated by 1D and 2D NMR and ESI-MS. Compared to the free ligand, all the urea NH groups in complex 1a show significant downfield shifts (Δδ = 2.89–3.91 ppm, NMR of ligand L was measured in DMSO-d6 for solubility reasons; Fig. 2b), corresponding to the coordination of urea to phosphate. A characteristic upfield shift of the H12 proton on the terminal nitrophenyl group (Δδ = −0.54 ppm; proton numbering, see Scheme 1a) is observed because of the strong shielding effects imposed in the helical structure. Interestingly, the two protons from one methylene of the spacer (H2) appear at different resonances due to their non-equivalent environments owing to the lack of symmetry in the double helicate (Fig. 1). This was also proved by 2D COSY which reveals the correlation between the two split peaks, indicating that they are from the same carbon (Fig. S25†).
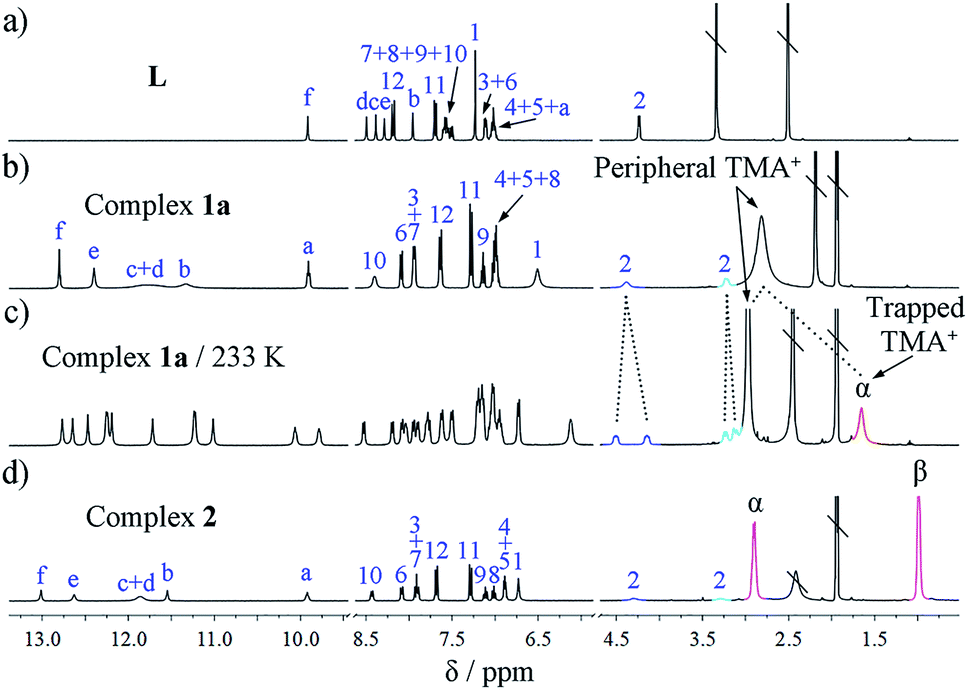 | ||
| Fig. 2 1H NMR spectra (400 MHz) of (a) ligand L in DMSO-d6, 296 K; (b) double helicate 1a in CD3CN, 296 K; (c) 1a in CD3CN, 233 K; and (d) grid 2 in CD3CN, 296 K. Signals of the two hydrogens of the methylene linker are shown in blue and turquoise, while that of trapped TMA+ ion is shown in red and protons of TEA+ in deep pink, respectively; \ represents solvent residue. | ||
Notably, in the room-temperature NMR spectrum, there is only one peak for the two types of cations, i.e., encapsulated and peripheral TMA+ ions. This peak appears at an ‘averaged’ position of 2.80 ppm, only slightly upfield-shifted (Δδ = −0.28 ppm) relative to free TMA+ (3.08 ppm) in (TMA)3PO4, possibly due to the fast exchange between peripheral and trapped TMA+ guests at the NMR time scale. However, when the temperature is lowered to 233 K the peaks of ligands L split into two sets, indicating that the molecular motion is ‘frozen’ and the guest is ‘fastened’ (Fig. 2c). Meanwhile, the two different types of TMA+ cations become distinguishable, with the ‘missing’ signal of the trapped TMA+ (Hα) now showing a much more upfield shift at δ 1.64 ppm (Δδ = −1.44 ppm compared to the free state; see Fig. S9–S11† for variable-temperature 1H NMR). In contrast, the protons of the five peripheral TMA+ ions (2.97 ppm) only shifted slightly upfield (Δδ = −0.11 ppm), revealing the lack of shielding on these ions, with an appropriate ratio (5:1) of the integral for the two types of TMA+ ions.
2D NMR spectra provide further evidence for the TMA+ cation encapsulation by the clear 1H–1H NOE (nuclear Overhauser effect) correlations between Hα of TMA+ and the phenylene protons (H1) of the linker of L (see Scheme 1 for proton numbering; Fig. S26†). The formation of a single host–guest species was confirmed by DOSY (diffusion-ordered nuclear magnetic resonance spectroscopy) (Fig. S27†). The HR ESI-MS spectrum of complex 1a also supports the formation of the A2L2 complex. Specifically, intense peaks at m/z 804.6570 (x = 3) and 1244.0363 (x = 4) corresponding to various A2L2(TMA)n species, such as [(PO4)2L2(TMA)x](6−x)−, are seen in the spectrum (Fig. 4a).
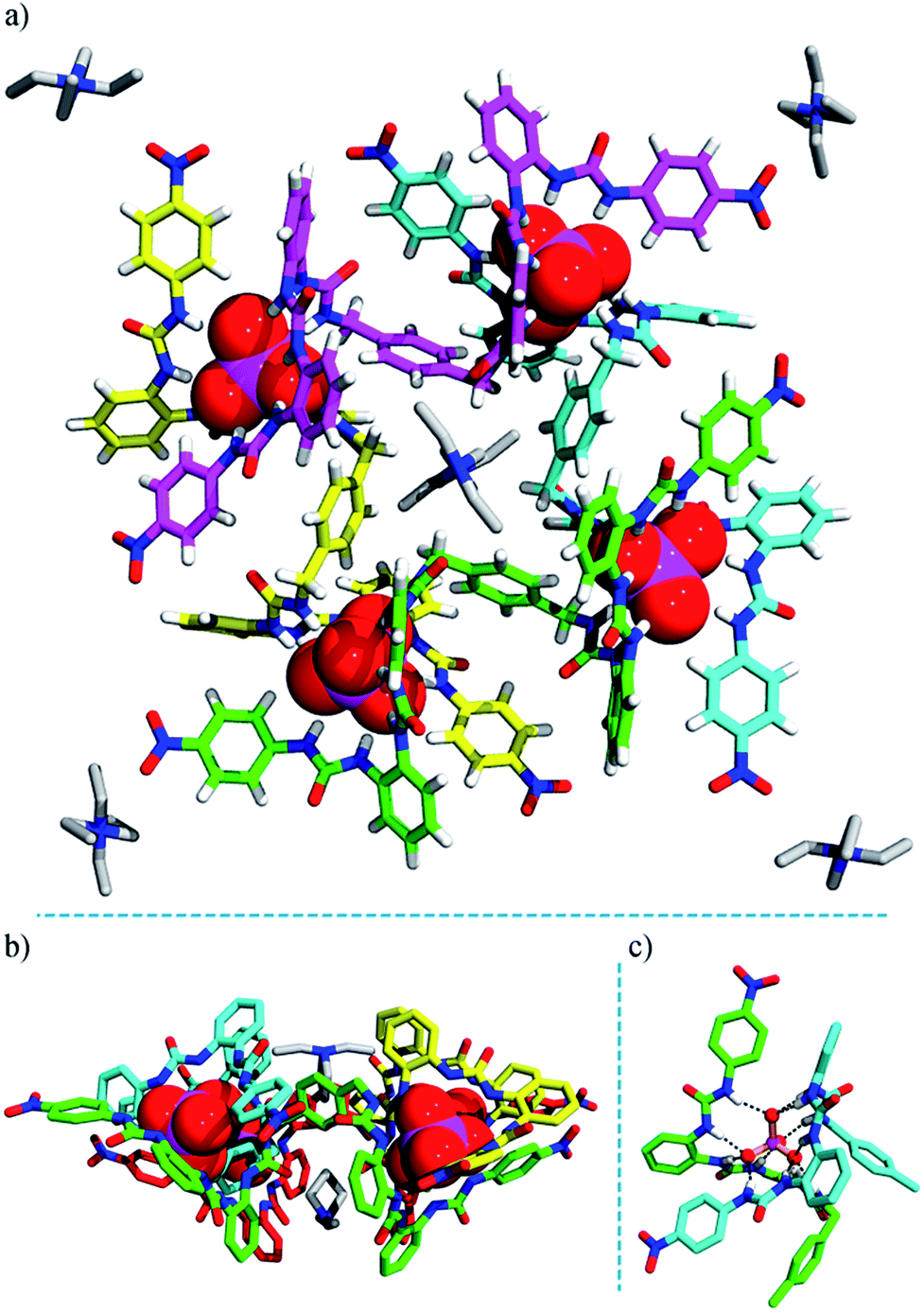 | ||
| Fig. 3 Crystal structure of the heterochiral (ΔΛΔΛ) grid complex 2 (TEA)10[(TEA)2⊂(PO4)4L4]. (a) Top view; (b) side view; (c) hydrogen bonds formed between a PO43− ion and six urea units. Some TEA+ cations are omitted for clarity (see Fig. S18† for the locations and interactions of these counter ions). | ||
The negative charges of the anionic grid are balanced by twelve TEA+ counter-cations, which reside in four different environments. Notably, inside the grid, two TEA+ cations (type I) are readily accommodated, with one sitting over the other, an arrangement adopted possibly to facilitate cation⋯π interactions with the ‘aromatic box’ around them (Fig. 3b and S18a†). The two type-II TEA+ ions (distributed to four half-occupancy positions) are located at each vertex of the grid, forming hydrogen bonds with the terminal nitro groups (Fig. 3a and S18b†). Notably, each ligand strand as the ‘edge’ of grid 2 is folded to a helical conformation spanning ca. 1.5 turns. Within each ligand helix, a ‘half cavity’ is formed to encapsulate one of the third-type TEA+ cations (type III), which is surrounded by four aryl rings of the ‘host’ ligand and complemented by a terminal nitrophenyl plane from a neighboring ligand (Fig. S18a†). Finally, the remaining four TEA+ ions (type-IV) each cap a phosphate corner of the grid, forming intra- and inter-molecular interactions with ligand aromatic rings, thus linking two grids (Fig. S19†). All these cations, either internal or ‘peripheral’, may provide important templating or additional stabilizing effects for the grid structure, which also imply the possible guest inclusion ability of the voids in the grid complex 2.
In the 1H NMR spectrum of grid 2, the urea NH groups also show large downfield shifts (Δδ = 2.93–4.14 ppm; Fig. 2d). The signal of the aryl H12 proton (Δδ = −0.49 ppm) undergoes an obvious upfield shift. The two protons from one CH2 group of the spacer also split into two doublets due to their different chemical environments. Notably, although the crystal structure of 2 shows four different locations of TEA+ cations, they appear as only one set of signals in the NMR spectrum. Both of the CH2 (Hα) and CH3 (Hβ) signals of TEA+ shift slightly upfield compared to that of the free (TEA)3PO4 (Δδα = −0.28 ppm and Δδβ = −0.20 ppm) (Fig. 2d), and there is no significant change when the temperature is lowered (down to 233 K). This might reflect the facts that (a) there is a fast exchange among the protons at the four locations; and (b) these cations indeed form similar interactions with ligand L as revealed by the crystal structure. On the other hand, the HR ESI-MS spectrum confirms the composition of the A4L4 complex with the stoichiometry [(TEA)9(PO4)4L4]3− (obsd 1851.9443 and cald 1851.8676, Fig. 4b). It is evident that complexes 1a and 2 display different NMR and ESI-MS signals, which can be used to distinguish the configuration of the double helicate or grid (see also the red dotted box in Fig. 5a).
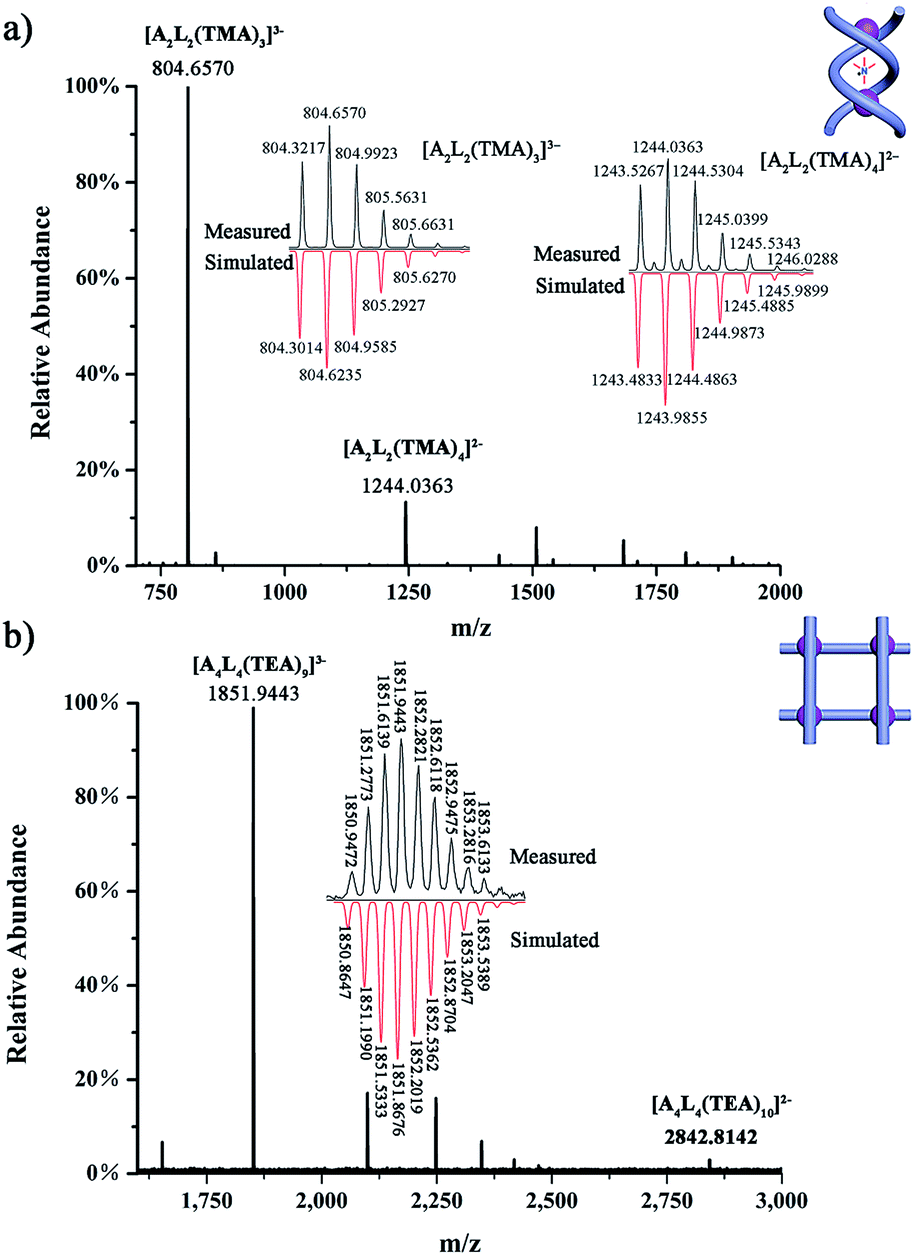 | ||
| Fig. 4 HR ESI-MS spectra of (a) complex (TMA)5[(TMA)⊂(PO4)2L2] (1a); and (b) complex (TEA)10[(TEA)2⊂(PO4)4L4] (2). | ||
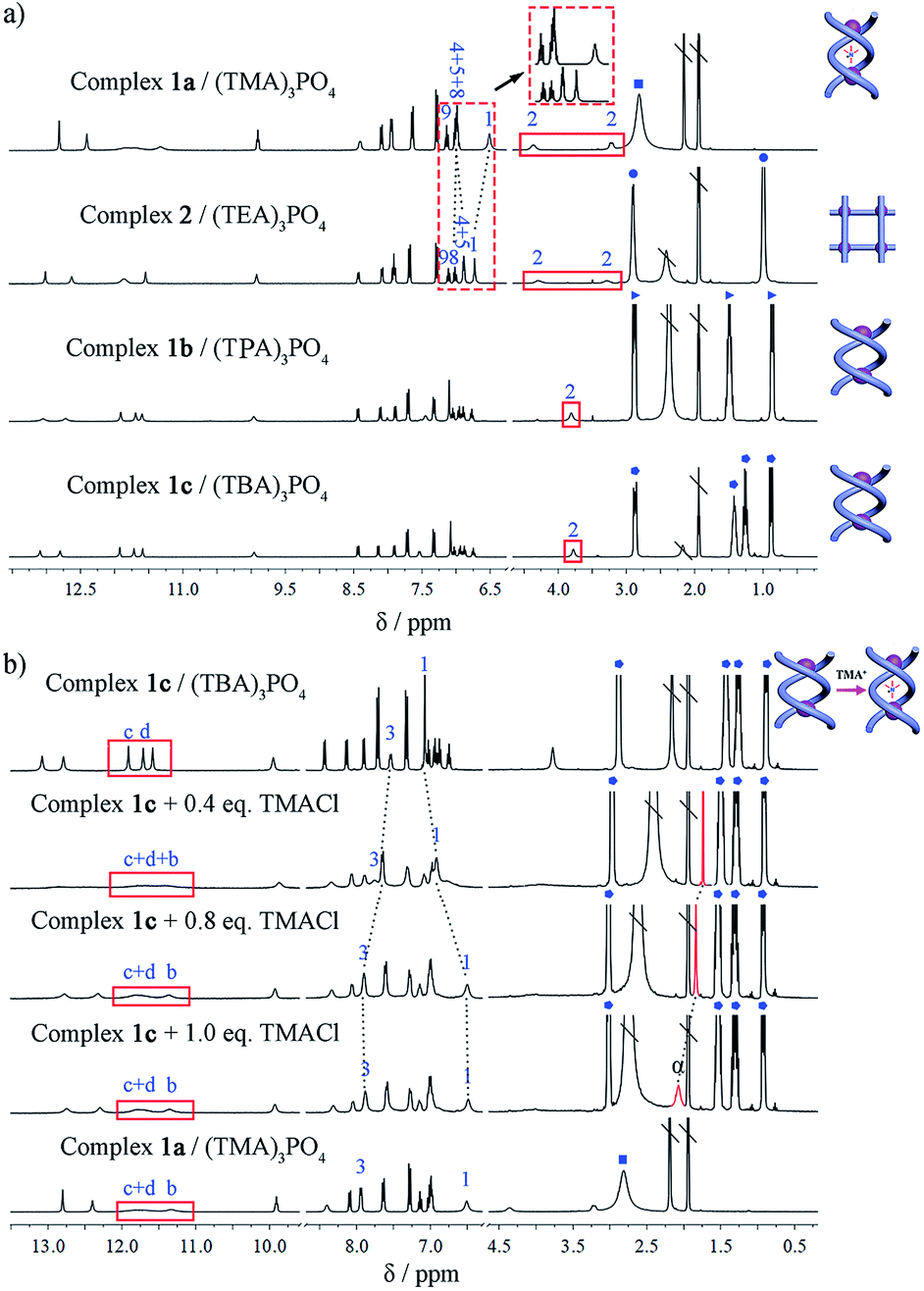 | ||
Fig. 5
1H NMR spectra demonstrating the assembly of different phosphate salts with ligand L (400 MHz, CD3CN, 296 K). (a) L with 1 equiv. of (TMA)3PO4, (TEA)3PO4, (TPA)3PO4 or (TBA)3PO4. (b) Stacking spectra of complexes 1a and 1c, and titration of 1c with (TMA)Cl ( denotes signals for TMA+, denotes signals for TMA+,  for TEA+, for TEA+,  for TPA+, and for TPA+, and  for TBA+. The signal of trapped TMA+ is shown in red; \ represents solvent residue). for TBA+. The signal of trapped TMA+ is shown in red; \ represents solvent residue). | ||
Assembly of the double helicates 1b and 1c
Considering the different outcomes in the two cases where only the counter cation varies, it appears that the counter cation (as the guest) plays a critical role in determining the supramolecular assembling process. To further elucidate the effect of the cation, we studied the assembly of L with one equivalent of (TPA)3PO4 or (TBA)3PO4 which contains the larger tetraalkylammonium homologues (TPA = tetrapropylammonium; TBA = tetrabutylammonium). In both cases, crystalline products (complexes 1b and 1c) were yielded. Although single crystals suitable for X-ray diffraction could not be obtained despite many attempts, the configuration of these complexes can be identified by a combination of 1H NMR, HR ESI-MS, and theoretical calculations. The two products show 1H NMR features that are similar to each other (Fig. 5a) but different from those of either complex 1a or 2, and the methylene H2 protons of the linker do not show any split, implying no significant guest interaction with the host. Their ESI-MS spectra are also similar, revealing a 2:2 anion-to-ligand ratio (Fig. S21 and S22†). Thus, we speculate that both complexes may have the double-stranded configuration, either double helicate or mesocate, with an ‘empty’ central cavity (solvent-filled) because either TPA or TBA is too large to fit. DFT calculations yield representative geometries for the helicate (Fig. S28†) and mesocate (Fig. S29†) assemblies in the absence of a guest. In the former case, optimization of the observed helicate geometry suggests that although the cavity becomes slightly smaller, the double helicate structure persists after the guest is removed.
Interconversion between the double helicate and grid motifs
Indeed, the guest-free complexes 1b/c provide an ideal model to examine the templating effect of the cation. For complex 1c, [(TBA)6(PO4)2L2], when (TMA)Cl was titrated in, the NMR spectrum gradually changed to that of the double helicate 1a. The methylene protons split into two peaks, and the encapsulation of TMA+ ions can be monitored. With the addition of less than 1 equiv. of TMA+ ions (per A2L2 unit) to complex 1c, the methyl signal Hα showed a dramatic upfield resonance at 1.74 ppm (Δδ = −1.34 ppm compared to free TMA+), indicating the encapsulation of TMA+. However, this signal shifted back downfield when more than 1 equiv. of TMA+ cations was added (Fig. 5b), which was caused by the fast exchange of the encapsulated and excess peripheral TMA+ cations, as in the case of complex 1a. Finally, with more TMA+ cations (6 equiv.), the titration reached saturation and the spectrum is almost the same as that of 1a.Inspired by the NMR titration results, which indicated that the initially ‘empty’ double helicate (TBA)6[(PO4)2L2] (1c) can encapsulate one TMA+ cation (and thus displace one TBA+ ion), we were able to isolate single crystals of the double helicate complex 1d, (TBA)5[(TMA)⊂(PO4)2L2] (Fig. S16 and Table S3†) from L and a mixture of phosphate salts of TMA+ and TBA+ in a 1:5 ratio. The main structure [(TMA)⊂(PO4)2L2]5− in 1d is essentially the same as that in complex 1a, and the difference lies only in the peripheral counter cations, TBA+ in 1dversus TMA+ in 1a. In addition, HR ESI-MS also shows the signal of [(TBA)3(TMA)(PO4)2L2]2− (m/z obsd 1496.3825 versus calcd 1496.2675, Fig. S20†).
Subsequently, the conversion of double helicate to grid motifs was studied using 1H NMR titration (Fig. 6a) by introducing the TEA+ ions into helicate 1c. With the addition of 1 equiv. of (TEA)Cl to a solution of 1c in CD3CN, the methylene protons H2 of the spacer split to two peaks, a characteristic feature of grid 2. In the aromatic area, the H1 signal shifts upfield due to encapsulation of a TEA+ ion with the conformational change. Meanwhile, both peaks of TEA+ appear in the upfield region (without peripheral TEA+ ions) compared to those of complex 2 but shift downfield gradually with more added TEA+ ions until becoming identical to complex 2. On the other hand, the reverse process, i.e. transformation of the grid structure 2 to double helicate (1a) is also readily realized upon addition of the TMA+ template (Fig. 6b). With the encapsulation of TMA+, the aryl H1 signal of the spacer shifts slightly upfield until reaching the same as complex 1a, and H4 and H5 peaks shift downfield due to the deshielding effect after encapsulation of the TMA+.
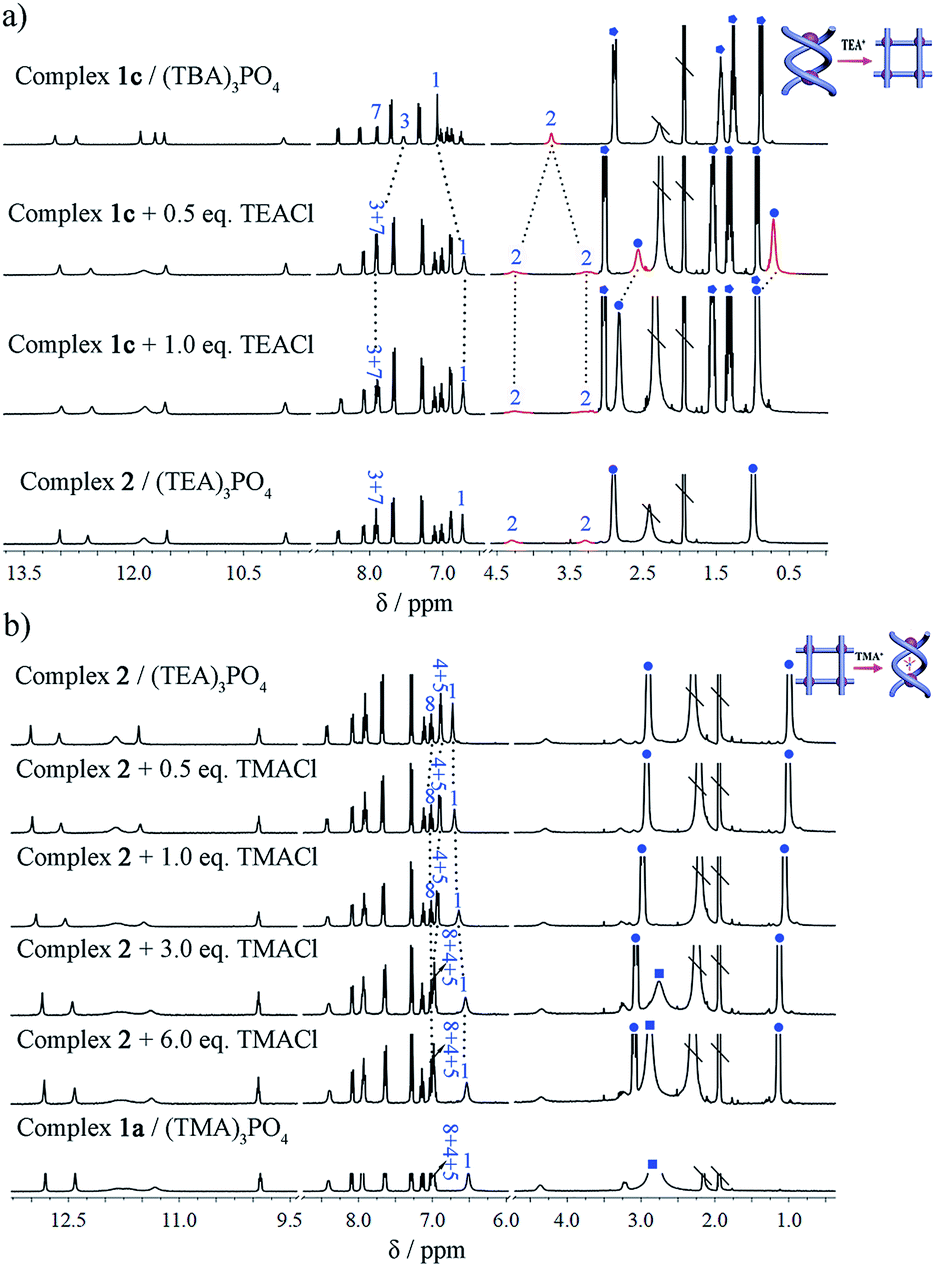 | ||
Fig. 6 Partial 1H NMR titration spectra demonstrating the interconversion of different complexes (400 MHz, CD3CN, 296 K). (a) From ‘empty’ double helicate to grid 2 by titrating 1c with (TEA)Cl. (b) From grid 2 to double helicate 1a by titrating complex 2 with (TMA)Cl ( denotes signals for TMA+, denotes signals for TMA+,  for TEA+, and for TEA+, and  for TBA+. Signals of trapped TEA+ ions are shown in red; \ represents solvent residue). for TBA+. Signals of trapped TEA+ ions are shown in red; \ represents solvent residue). | ||
Thus, we have observed the conversion not only from complexes 1b/c to 1a, namely from ‘empty’ to the TMA+-filled double helicate, but also from 1b/c to grid 2 under the modulation of TEA+ cations. Conversely, due to the strong binding of the helical cage to TMA+, transformation of grid 2 to double helicate 1a is also very feasible. Such template-induced supramolecular transformations have also been reported for metal-coordination driven assembly, in which the counter-anions are responsible for the conformational changes.2c,6c,7d,9a,b
Conclusions
We reported the design and synthesis of a C2-symmetric bis–tris(urea) ligand with a p-xylylene linker, which is assembled with a phosphate anion to form the first anion coordination-based 2 × 2 grid (A4L4) and a double helicate (A2L2) featuring an open cavity similar to DNA grooves. Moreover, facile interconversion between these two motifs can be realized simply by modulation with the counter-cation under ambient conditions, which is also complementary to anion-controlled structural interconversion. This work reveals the power of anion coordination in supramolecular assembly. Such aniono-supramolecular systems could be a new class of architectures for promising applications in many fields. Construction of more complex structures and exploration of their functionality is currently underway.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21772154, 91856102 and 21771144). BPH acknowledges the sponsorship from the 100 Talents Award, Shaan’xi Province.Notes and references
- (a) B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS; (b) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC; (c) W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC; (d) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (e) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed; (f) T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS; (g) S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- (a) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed; (b) R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794–8824 CrossRef CAS; (c) I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 751–756 CrossRef CAS PubMed; (d) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (e) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (f) X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed; (g) Y. Lu, H.-N. Zhang and G.-X. Jin, Acc. Chem. Res., 2018, 51, 2148–2158 CrossRef CAS.
- (a) H. Li, H. Zhang, A. D. Lammer, M. Wang, X. Li, V. M. Lynch and J. L. Sessler, Nat. Chem., 2015, 7, 1003 CrossRef CAS PubMed; (b) X. Wang, Y. Wang, H. Yang, H. Fang, R. Chen, Y. Sun, N. Zheng, K. Tan, X. Lu, Z. Tian and X. Cao, Nat. Commun., 2016, 7, 12469 CrossRef PubMed; (c) H. Qu, Y. Wang, Z. Li, X. Wang, H. Fang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2017, 139, 18142–18145 CrossRef CAS PubMed.
- (a) G. Portella, M. W. Germann, N. V. Hud and M. Orozco, J. Am. Chem. Soc., 2014, 136, 3075–3086 CrossRef CAS PubMed; (b) L. B. Favero, W. Li, L. Spada, L. Evangelisti, G. Visentin and W. Caminati, Chem.–Eur. J., 2015, 21, 15970–15973 CrossRef CAS PubMed.
- (a) Y. Yao, M. W. Perkovic, D. P. Rillema and C. Woods, Inorg. Chem., 1992, 31, 3956–3962 CrossRef CAS; (b) V. Amendola, L. Fabbrizzi, L. Gianelli, C. Maggi, C. Mangano, P. Pallavicini and M. Zema, Inorg. Chem., 2001, 40, 3579–3587 CrossRef CAS PubMed.
- (a) W. Meng, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 1017–1021 CrossRef CAS PubMed; (b) X.-Z. Li, L.-P. Zhou, L.-L. Yan, D.-Q. Yuan, C.-S. Lin and Q.-F. Sun, J. Am. Chem. Soc., 2017, 139, 8237–8244 CrossRef CAS PubMed; (c) T. Zhang, L.-P. Zhou, X.-Q. Guo, L.-X. Cai and Q.-F. Sun, Nat. Commun., 2017, 8, 15898 CrossRef CAS PubMed.
- (a) S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596–13599 CrossRef CAS PubMed; (b) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (c) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC; (d) H. B. T. Jeazet, K. Gloe, T. Doert, O. N. Kataeva, A. Jager, G. Geipel, G. Bernhard, B. Buchner and K. Gloe, Chem. Commun., 2010, 46, 2373–2375 RSC.
- (a) H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977–6991 RSC; (b) S. Otto, Acc. Chem. Res., 2012, 45, 2200–2210 CrossRef CAS; (c) M. Barboiu, A.-M. Stadler and J.-M. Lehn, Angew. Chem., Int. Ed., 2016, 55, 4130–4154 CrossRef CAS.
- (a) B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed., 1996, 35, 1838–1840 CrossRef CAS; (b) B. Hasenknopf, J.-M. Lehn, N. Boumediene, A. Dupont-Gervais, A. Van Dorsselaer, B. Kneisel and D. Fenske, J. Am. Chem. Soc., 1997, 119, 10956–10962 CrossRef CAS; (c) M. Fujita, Chem. Soc. Rev., 1998, 27, 417–425 RSC; (d) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS PubMed; (e) H. Juwarker and K.-S. Jeong, Chem. Soc. Rev., 2010, 39, 3664–3674 RSC; (f) J. G. Hardy, Chem. Soc. Rev., 2013, 42, 7881–7899 RSC.
- (a) J.-m. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938–13941 CrossRef CAS PubMed; (b) Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed; (c) Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS PubMed; (d) M. Albrecht and S. Kotila, Angew. Chem., Int. Ed., 1996, 35, 1208–1210 CrossRef CAS; (e) M. Albrecht, Chem.–Eur. J., 2000, 6, 3485–3489 CrossRef CAS; (f) J. Sahoo, R. Arunachalam, P. S. Subramanian, E. Suresh, A. Valkonen, K. Rissanen and M. Albrecht, Angew. Chem., Int. Ed., 2016, 55, 9625–9629 CrossRef CAS PubMed.
- (a) M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef CAS; (b) M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 3644–3662 CrossRef CAS PubMed; (c) S. Dhers, A. Mondal, D. Aguilà, J. Ramírez, S. Vela, P. Dechambenoit, M. Rouzières, J. R. Nitschke, R. Clérac and J.-M. Lehn, J. Am. Chem. Soc., 2018, 140, 8218–8227 CrossRef CAS PubMed.
- (a) K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed; (b) K. M. Mullen and P. D. Beer, Chem. Soc. Rev., 2009, 38, 1701–1713 RSC; (c) R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC; (d) P. A. Gale, E. N. W. Howe and X. Wu, Chem, 2016, 1, 351–422 CrossRef CAS; (e) Y. Liu, C. Hu, A. Comotti and M. D. Ward, Science, 2011, 333, 436–440 CrossRef CAS; (f) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS; (g) Q. He, P. Tu and J. L. Sessler, Chem, 2018, 4, 46–93 CrossRef CAS.
- (a) B. P. Hay, T. K. Firman and B. A. Moyer, J. Am. Chem. Soc., 2005, 127, 1810–1819 CrossRef CAS; (b) D. Yang, J. Zhao, X.-J. Yang and B. Wu, Org. Chem. Front., 2018, 5, 662–690 RSC; (c) J. Zhao, D. Yang, X.-J. Yang and B. Wu, Coord. Chem. Rev., 2019, 378, 415–444 CrossRef CAS.
- (a) S. Li, C. Jia, B. Wu, Q. Luo, X. Huang, Z. Yang, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 5721–5724 CrossRef CAS PubMed; (b) B. Wu, F. Cui, Y. Lei, S. Li, N. d. S. Amadeu, C. Janiak, Y.-J. Lin, L.-H. Weng, Y.-Y. Wang and X.-J. Yang, Angew. Chem., Int. Ed., 2013, 52, 5096–5100 CrossRef CAS PubMed; (c) W. Zuo, Z. Huang, Y. Zhao, W. Xu, Z. Liu, X.-J. Yang, C. Jia and B. Wu, Chem. Commun., 2018, 54, 7378–7381 RSC.
- X. Bai, C. Jia, Y. Zhao, D. Yang, S.-C. Wang, A. Li, Y.-T. Chan, Y.-Y. Wang, X.-J. Yang and B. Wu, Angew. Chem., Int. Ed., 2018, 57, 1851–1855 CrossRef CAS PubMed.
- C. J. Massena, D. A. Decato and O. B. Berryman, Angew. Chem., Int. Ed., 2018, 57, 16109–16113 CrossRef CAS.
- (a) B. Wu, S. Li, Y. Lei, H. Hu, N. d. S. Amadeu, C. Janiak, J. S. Mathieson, D.-L. Long, L. Cronin and X.-J. Yang, Chem.–Eur. J., 2015, 21, 2588–2593 CrossRef CAS; (b) C. Jia, W. Zuo, D. Yang, Y. Chen, L. Cao, R. Custelcean, J. Hostaš, P. Hobza, R. Glaser, Y.-Y. Wang, X.-J. Yang and B. Wu, Nat. Commun., 2017, 8, 938 CrossRef PubMed.
- (a) C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q.-S. Li and X.-J. Yang, Chem. Commun., 2010, 46, 5376–5378 RSC; (b) S. Li, M. Wei, X. Huang, X.-J. Yang and B. Wu, Chem. Commun., 2012, 48, 3097–3099 RSC.
- (a) J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS; (b) U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892–2926 CrossRef CAS.
- (a) B. P. Hay, Chem. Soc. Rev., 2010, 39, 3700–3708 RSC; (b) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354–1379 RSC.
- A.-M. Stadler, C. Burg, J. Ramírez and J.-M. Lehn, Chem. Commun., 2013, 49, 5733–5735 RSC.
- J. Sánchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277–278 CrossRef.
- S. J. Coles, J. G. Frey, P. A. Gale, M. B. Hursthouse, M. E. Light, K. Navakhun and G. L. Thomas, Chem. Commun., 2003, 568–569 RSC.
- Y. Haketa and H. Maeda, Chem.–Eur. J., 2011, 17, 1485–1492 CrossRef CAS.
- Y. Liu, F. C. Parks, W. Zhao and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 15477–15486 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data and corresponding CIF files. CCDC 1892629–1892631. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02012h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |