
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-distributed Pt-nanoparticles within confined coordination interspaces of self-sensitized porphyrin metal–organic frameworks: synergistic effect boosting highly efficient photocatalytic hydrogen evolution reaction†
Shuai
Li
,
Hong-Min
Mei
,
Shi-Lin
Yao
,
Zhi-Yao
Chen
,
Yu-Lin
Lu
 ,
Li
Zhang
* and
Cheng-Yong
Su
*
,
Li
Zhang
* and
Cheng-Yong
Su
*
MOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhli99@mail.sysu.edu.cn; cesscy@mail.sysu.edu.cn
First published on 1st October 2019
Abstract
Effective conversion of solar energy into chemical energy by visible light represents a potential strategy for sustainable development. Among which, photocatalytic hydrogen evolution reaction (HER) with a relatively small activation energy (1.23 eV, around 1000 nm light irradiation) is especially attractive. In this work, well-distributed platinum nanoparticles (Pt-NPs) with a width of about 3 nm have been successfully immobilized into the confined coordination interspaces of 3.7 nm diameter, which are facilitated by early transition metal Hf(IV)-based clusters of a self-sensitized palladium porphyrin metal–organic framework. Under visible light irradiation, the resultant Pt@Pd-PCN-222(Hf) (which is also denoted as Pt@Pd-PMOF-2(Hf)) displays superb photocatalytic activity, achieving an unprecedented maximum H2 evolution rate of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 674 μmol g−1 h−1 with a turn-over number (TON) of 4131.2 in 32 h and the highest turn-over frequency (TOF) of 482.5 h−1 based on Pt-NPs. This photocatalyst can be recycled and reused for three successive runs without significant loss of catalytic activity. This effective strategy takes advantage of the synergetic effect between Pd-porphyrin photosensitizers and Pt-NP co-catalysts confined within nanoscale coordination interspaces incorporating hydrophilic Hf(IV)-oxo clusters.
674 μmol g−1 h−1 with a turn-over number (TON) of 4131.2 in 32 h and the highest turn-over frequency (TOF) of 482.5 h−1 based on Pt-NPs. This photocatalyst can be recycled and reused for three successive runs without significant loss of catalytic activity. This effective strategy takes advantage of the synergetic effect between Pd-porphyrin photosensitizers and Pt-NP co-catalysts confined within nanoscale coordination interspaces incorporating hydrophilic Hf(IV)-oxo clusters.
Introduction
Hydrogen evolution reaction (HER) through photocatalytic water splitting is of significant importance to convert and store solar energy in the chemical bonds of H2, which is an intermediate energy carrier.1,2 Although a range of photocatalysts, including molecular catalysts and inorganic semiconductors, have been developed for efficient HER,3 it is yet challenging to realize recyclability and improve visible light conversion. Solar energy contains around 5% ultraviolet light, 42–43% visible light (400–700 nm) and 52–55% infrared light (>700 nm).4 Metal–organic frameworks (MOFs) have emerged as a promising class of HER photocatalysts, featuring a unique capability to merge versatile photosensitizers and co-catalysts, which are able to expand the light absorption band and improve electron–hole separation, respectively, into a single MOF as composite materials.5–8It is convenient to engineer the coordination nanospaces of MOFs purposely for a specific functionality.9–12 Excellent photosensitizers such as porphyrins could be introduced into the frameworks, giving rise to self-sensitized MOFs.13–23 Porphyrins are well-known redox-active photosensitizers, as most of them have the ability to absorb visible light corresponding to the Soret and Q-bands, and display a remarkably long lifetime of the triplet excited-state. For example, the triplet state lifetimes of Rh-, Pd- and Pt-porphyrins at room temperature are typically in the order of tens to hundreds of microseconds (μs).24–26 Furthermore, with the desire to improve the electron–hole separation ability, co-catalysts such as metal nanoparticles (MNPs) could be encapsulated into the coordination interspaces of MOFs. As a consequence, a Schottky barrier can form at the interface of MOFs and MNPs, and the photogenerated electrons might transfer from the LUMO of MOFs to MNPs, which could enhance electron–hole separation and then the photocatalytic performance.18,27–37 Excellent catalytic performance can be achieved if MNPs were uniformly located inside the MOFs due to the synergistic catalysis between self-sensitized MOFs and incorporated MNPs. Nevertheless, the highest H2 evolution rates of the sole MOFs or MNPs@MOFs have not exceeded 10000 μmol g−1 h−1 yet.
For effective water splitting, photocatalysts should have high stability under aqueous conditions. MOFs that are constructed from highly charged metal cations (e.g. Cr3+, Zr4+ and Hf4+) and carboxylates are reported to have high stability in water and even acidic or basic aqueous solutions, displaying high potential for photocatalytic water splitting.38–42 Early transition metals are known to be oxophilic, and thus coordination interspaces comprising early transition metal-oxo clusters and carboxylates could endow the corresponding MOFs with a hydrophilic character,43–45 boosting the photocatalytic water splitting reaction. As a result, we believe that IVA group metal Zr4+/Hf4+-based MOFs could be effective photocatalysts towards water splitting, taking advantage of the H2O concentration effect in confined coordination interspaces as shown in Fig. 1.
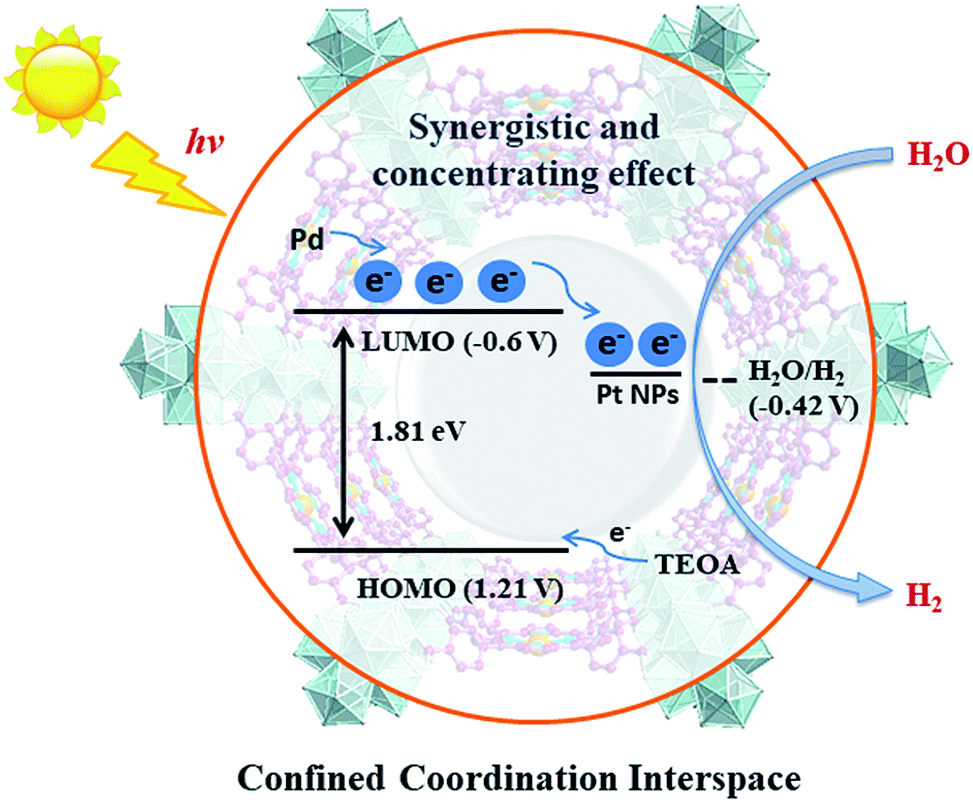 | ||
| Fig. 1 Schematic illustration of the structure and photocatalytic behavior of the MOF composite Pt@Pd-PCN-222(Hf). | ||
In this paper, we report the preparation of a Pd-porphyrin metal–organic framework of Pd-PCN-222(Hf) (which is also denoted as Pd-PMOF-2(Hf)) through the self-assembly of HfCl4 and PdTCPP (TCPP = tetrakis(4-carboxyphenyl)porphyrin). This MOF possessed a csq topology as in PCN-222,40 MOF-545 (ref. 41) or MMPF-6,42 and contained hexagonal and triangular channels with diameters of 3.7 and 1.3 nm, respectively. Pt-nanorods with consistent orientation and a width of about 3 nm were successfully immobilized into Pd-PCN-222(Hf) owing to controlled growth in the confined coordination interspaces of Pd-PCN-222(Hf). Efficient photocatalytic HER activities were achieved in the coordination nanospaces of the resultant MOF composite Pt@Pd-PCN-222(Hf), which displayed the following features: (1) incorporating self-sensitized Pd-porphyrin with visible light absorption and a long triplet lifetime, (2) encapsulating uniformly oriented and distributed Pt-NPs as co-catalysts for effective electron–hole separation, (3) connecting hydrophilic Hf(IV)-oxo clusters with carboxylate linkers for high chemical stability and the H2O concentration effect, and consequently, (4) achieving a synergetic effect based on the Pd-porphyrin photosensitizer, Pt-NP co-catalyst and microenvironmental hydrophilicity. Under visible light irradiation, a maximum H2 evolution rate of 22674 μmol g−1 h−1 was accomplished, which was much higher than those of so far reported MOF-based HER photocatalysts (Table S1†).
Experimental
Synthesis of nanoscale Pd-PCN-222(Hf)
Pd(II) 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin (PdTCPP) was synthesized according to the literature.46 The nanoscale Pd-PCN-222(Hf) was prepared by the reaction of PdTCPP (5 mg, 5.6 μmol) and HfCl4 (10 mg, 31.2 μmol) in DMF (3 mL) in the presence of CF3COOH (150 μL) with heating at 120 °C in an oil bath. After 8 min, red needle-like crystals of Pd-PCN-222(Hf) (8.4 mg, 95% yield) were obtained, washed with DMF and ethanol several times, and dried under vacuum at 120 °C overnight. Anal. calcd for C48H32PdN4O16Hf3·4DMF·5H2O (C60H70PdN8O25Hf3): C, 37.05; H, 3.63; and N, 5.76%. Found: C, 37.67; H, 3.44; and N, 5.25%. FT-IR (KBr) ν 3440 (br), 3133 (br), 1722 (s), 1657 (s), 1390 (s), 1273 (s), 1202 (s), 1012 (s), 798(m), and 711(m) cm−1.Synthesis of Pt@Pd-PCN-222(Hf)
A mixture of pristine Pd-PCN-222(Hf) (20 mg) and K2PtCl4 solution (20 mg mL−1; 100 μL) in DMF (4 mL) was sonicated for 15 min, and then heated at 80 °C in a drying oven. After 4 h, the product was separated as red needle-like crystals via centrifugation at 12000 rpm for 3 min, washed with DMF and ethanol several times, and dried under vacuum at 120 °C overnight. FT-IR (KBr) ν 3440 (br), 3133 (br), 1722 (s), 1657 (s), 1390 (s), 1273 (s), 1202 (s), 1012 (s), 798(m), and 711(m) cm−1.
Photophysical examination
Photoluminescence spectra were recorded on an Edinburgh FLS980 Photoluminescence Spectrometer. Before the fluorescence emission test, the equivalent solid-state Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) samples were finely ground to avoid error. The photoluminescence lifetime experiments were performed by the time-correlated single photo counting (TCSPC) method by using a 406.5 nm picosecond pulsed diode laser, or by using the same spectrometer but with a μF900 Xe lamp.For ultrafast transient absorption (TA) measurements, the samples were dispersed in CH3CN in a 0.7 mL sealed quartz cuvette, and then measurements were carried out using a femtosecond pump-probe setup consisting of Helios (Ultrafast Systems LLC) spectrometers and a regeneratively amplified Ti:sapphire laser source (Coherent Legend, 800 nm, 150 fs, 5 mJ per pulse, and 1 kHz repetition rate) in the transmission mode. A pump light of 400 nm was generated by the frequency doubling portion of the 800 nm output (75%) pulse in a BaB2O4 (BBO) crystal, and the power intensity can be adjusted by a range of neutral-density filters, which were fixed at 125 μJ cm−2 in this experiment. The delay time window between the pump and probe pulse from 0 to 7.7 ns was controlled by a motorized delay time stage.
Electrochemical measurements
Electrochemical measurements were carried out using a CHI 660E electrochemical workstation (Chenhua Instrument, Shanghai, China). Na2SO4 aqueous solution (0.2 mol L−1) was used as the electrolyte medium. The electrochemical cell used was equipped with three electrodes, including a Pt mesh counter electrode, an Ag/AgCl reference electrode (3 mol L−1 KCl) and a fluorine-doped tin oxide (FTO) glass (1 × 1 cm2) functionalized with test materials. The test materials were prepared by the following method: an activated sample (2 mg) was added to a solution of 5 wt% Nafion (10 μL) and ethanol (1 mL), and the resultant suspension was dispersed thoroughly via ultrasonication.The photocurrent response was measured using constant voltage tracking (CVT). A 300 W Xe lamp (λ ≥ 420 nm) was used as the light source, and a shutter was used to modulate the light and dark conditions during the tests. Photoresponsive signals of the samples were measured under chopped light with a bias potential of +0.13 V. The electrochemical impedance spectroscopy (EIS) measurement was performed in the frequency range from 10−2 to 105 Hz with a bias potential of 2.0 V.
Photocatalytic hydrogen evolution reaction
In a typical experiment, Pt@Pd-PCN-222(Hf) (5 mg) was suspended in a mixture of acetonitrile (10 mL), triethanolamine (2.5 mL) and deionized water (250 μL). The reaction mixture in a 40 mL glass vial was sonicated for 10 min, purged with nitrogen for 20 min with gentle stirring to eliminate the air, and then irradiated at room temperature with a 300 W Xe lamp equipped with a UV cut-off filter (λ ≥ 420 nm). After the reaction was complete, the gaseous products were analyzed by gas chromatography (GC), which was performed using GC9790, Fuli Analytical Instrument Co., Ltd.The catalyst was recycled and reused for three successive runs. After each run, the catalyst was separated via centrifugation at 12000 rpm for 3 min and reused for the next run, which was charged with a fresh solvent, a sacrificial agent and water.
Results and discussion
Synthesis, structure and stability
Pd-PCN-222(Hf) with up to 95% yield was prepared by the reaction of PdTCPP and HfCl4 in the presence of CF3CO2H as a modulating reagent after being heated at 120 °C in an oil bath for 8 min (Fig. 2). The bulk purity was confirmed by powder X-ray diffraction (PXRD) patterns (Fig. 3a), which fit well with the simulated patterns of PCN-222(Hf) constructed with metal free porphyrin ligand TCPP. The coordination framework of Pd-PCN-222(Hf) comprises Hf6(OH)8 clusters linked by Pd-porphyrin ligands, giving rise to hexagonal mesochannels with a diameter of 3.7 nm as well as trigonal microchannels with a diameter of 1.3 nm.40 The square planar geometry of Pd(II)-porphyrin was clearly shown by X-ray single crystallography of PdTMCPP (TMCPP = 5,10,15,20-tetrakis(4-methoxycarbonylphenyl)porphyrin) (Fig. S1†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) measurements disclosed that the as-prepared Pd-PCN-222(Hf) samples are of nanorod shape with about 300 nm width and 500 nm length (Fig. 4a and b).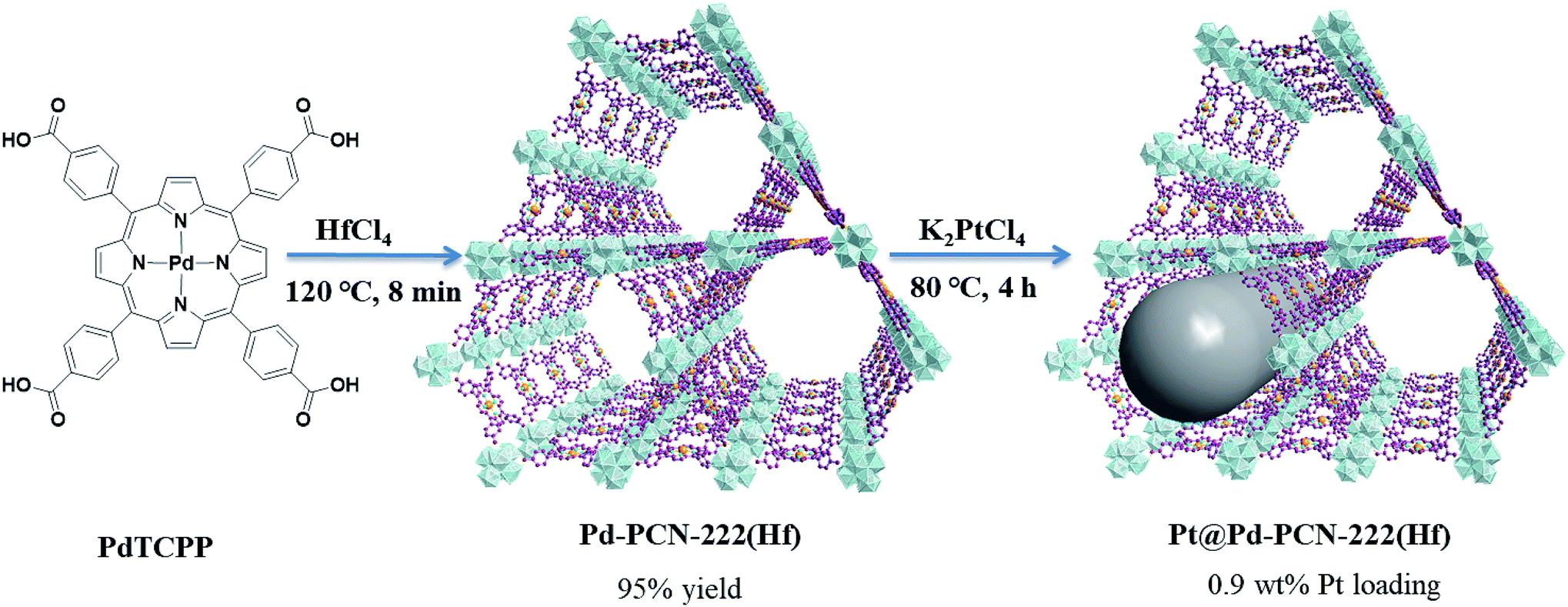 | ||
| Fig. 2 The syntheses of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf). | ||
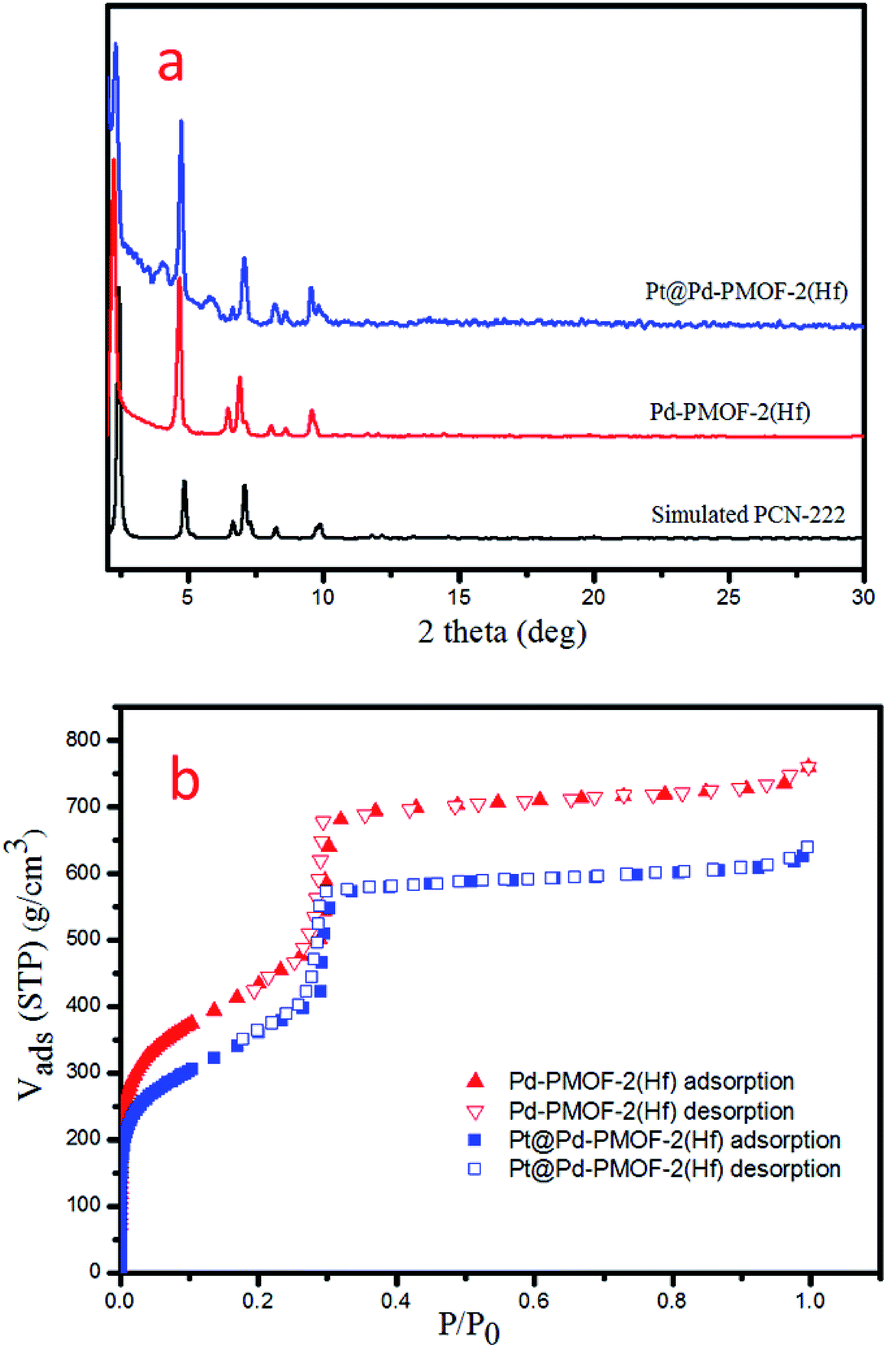 | ||
| Fig. 3 PXRD patterns (a) and N2 sorption isotherms at 77 K (b) of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf). | ||
 | ||
| Fig. 4 SEM (a) and TEM (b) images of Pd-PCN-222(Hf), and median-magnification TEM (c), high-magnification TEM (insert c) and aberration-corrected HAADF-STEM (d) images of Pt@Pd-PCN-222(Hf). | ||
The MOF composite Pt@Pd-PCN-222(Hf) was prepared by the in situ formation of Pt NPs in the coordination interspaces of the hexagonal mesochannels (3.7 nm) in Pd-PCN-222(Hf), which was accomplished by the solvothermal reaction of Pd-PCN-222(Hf) and K2PtCl4 in DMF at 80 °C for 4 h (Fig. 2). The presence of both Pd and Pt was confirmed by the X-ray photoelectron spectroscopy (XPS) examination (Fig. S2†). The metallic Pd(II) and Pt(0) species were clearly identified by the binding energies at 338.09 and 343.34 eV assignable to the 3d5/2 and 3d3/2 levels of Pd(II), respectively, as well as the binding energies at 71.14 and 74.47 eV related to the 4f7/2 and 4f5/2 levels of Pt(0), respectively (Fig. S2b and c†).47,48 The weight loading of Pt-NPs was determined to be 0.92% using an inductively coupled plasma optical emission spectrometer (ICP-OES). The PXRD patterns of Pt@Pd-PCN-222(Hf) closely matched with those of Pd-PCN-222(Hf), indicating that the framework and crystallinity of Pd-PCN-222(Hf) remain well after introduction of Pt-NPs (Fig. 3a).
The N2 uptakes of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) at 77 K were 760 and 640 cm3·g−1 (STP), respectively, and the Brunauer–Emmett–Teller (BET) surface areas were calculated to be 1511 and 1263 m2 g−1, respectively. This showed a slight decrease upon the loading of Pt-NPs into the pores of Pd-PCN-222(Hf) (Fig. 3b). The activated samples of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) exhibited remarkable water adsorption capacities with uptakes of 881 and 1035 cm3 g−1, respectively, at 298 K and P/P0 = 0.85 (Fig. S3†). The water uptake ability of Pt@Pd-PCN-222(Hf) was even higher than that of the primitive Pd-PCN-222(Hf). Significant hysteresis loops between the adsorption–desorption curves were observed, indicative of physical interactions between the adsorbed water molecules and the hydrophilic Hf(IV)-oxo clusters.43–45 Although the inner coordination interspaces contain hydrophilic Hf(IV)-oxo clusters, the overall porous Pd-PCN-222(Hf) displayed some kind of water resistance at a relatively low pressure, and water uptake abruptly occurred at P/P0 ≈ 0.6. The hysteresis behaviors also suggested that desorption of water molecules was lagging. This implied that the porous Pd-PCN-222(Hf) was able to prevent catalytic sites inside the coordination interspaces from immediate exposure to enormous water molecules, which might cause deterioration of catalysts and hamper the HER process. In comparison, the adsorbed water molecules were transiently concentrated inside the coordination interspaces for photocatalysis rather than freely releasing from Pt@Pd-PCN-222(Hf).
The presence of Pt-NPs in Pt@Pd-PCN-222(Hf) was clearly observable in TEM images, as marked with the red circles in Fig. 4c and the inset, and more discernible as bright spots in the high-angle annular dark-field scanning TEM (HAADF-STEM) tomogram (Fig. 4d). The Pt-NPs with a width of around 3 nm and a length range of 7–15 nm (Fig. S4†) displayed consistent orientation, suggesting a confined growth of Pt-NPs in the coordination interspaces of the hexagonal mesochannels (3.7 nm) in Pd-PCN-222(Hf). The internal Pt-NP location was further demonstrated by the electron tomographic reconstruction of Pt@Pd-PCN-222(Hf) shown in Fig. S5.†
In comparison, the majority of the reported Pt-NPs@MOF materials displayed several issues: (1) the morphology of Pt-NPs was unclear due to the lack of related characterization; (2) the Pt-NPs were deposited on the external surfaces of MOFs; (3) the agglomeration of Pt-NPs occurred; (4) the sizes of Pt-NPs were larger than those of MOF inner cavities, indicative of the distortion/degradation of MOFs during the formation of Pt-NPs; or (5) the Pt-NPs weren't dispersed uniformly in the pores of MOFs.49,50
Both Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) exhibited excellent chemical stability. The fresh samples were soaked in various organic solvents, including methanol, acetonitrile, acetone, dichloromethane, ethyl acetate and water for seven days, and then the PXRD patterns of the recycled samples were checked, which suggested that the crystallinity of the samples was well retained after these treatments (Fig. S6 and S7†). Furthermore, both Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) were stable after being immersed in aqueous solutions of a wide pH range from 1 to 12 for 24 h (Fig. S8 and S9†).
Photocatalytic activities
Prior to the photocatalytic study, the light absorption range and the LUMO potential of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) were examined. Solid UV-vis absorption spectroscopy showed that both Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) possessed strong light absorption extending to around 700 nm with two main peaks, which were the Soret band (∼415 nm) and Q band (∼523 nm) (Fig. S10†). The Mott–Schottky (M–S) plots were measured at frequencies of 500, 1000 and 1500 Hz (Fig. S11–S13†). The positive slopes of the M–S plots were characteristic of n-type semiconductors. The flat band positions determined from the intersections were approximately −0.6 V (vs. NHE) for PdTMCPP, Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf). It was generally accepted that the flat band was equal to the conduction band (LUMO) for an n-type semiconductor, which suggested that the LUMO positions of PdTMCPP, Pd-PCN-222(Hf), and Pt@Pd-PCN-222(Hf) were also approximately −0.6 V (vs. NHE). Based on Tauc plots, the band gap energies (Eg) of PdTMCPP, Pd-PCN-222(Hf), and Pt@Pd-PCN-222(Hf) were calculated to be 1.81, 2.13 and 2.14 eV, respectively. Therefore, the valence bands (HOMO) were accordingly obtained (Fig. S14–S16†). Given the more negative LUMO potentials in comparison to the proton reduction (−0.42 V vs. NHE, pH = 7), it was thermodynamically feasible for the Pd-porphyrin MOF-based materials to behave as HER photocatalysts.Encouraged by high chemical stability, strong visible light absorptivity and appropriate LUMO potentials, the photocatalytic HER for water splitting was carried out. The hydrogen evolution experiments were firstly conducted using triethanolamine (TEOA) as a sacrificial agent in different aqueous-organic solvents, including N,N-dimethylformamide (DMF), dimethylacetamide (DMAC), pyrrole, dimethyl sulfoxide (DMSO), acetone, isopropanol (iPrOH), and acetonitrile (CH3CN), under visible light irradiation (λ ≥ 420 nm) (Table S2,† entries 1–7). Catalytic results disclosed that a H2 evolution rate up to 22674 μmol g−1 h−1 after irradiation for 3 h was achieved in CH3CN, which was around twice that in iPrOH, DMF, acetone and DMAC. For comparison, the H2 evolution rates in DMSO and pyrrole were 2470 and 117 μmol g−1 h−1, respectively, which were rather lower. In addition to TEOA, other sacrificial agents such as methanol (CH3OH), N,N-dimethylaniline (DMA), ascorbic acid and 1,3-dimethyl-2-phenyl-2H-benzimidazole (BIH) were also screened, unveiling that TEOA was the best sacrificial agent in this system (Table S2,† entries 1, 8–11). The optimal amounts of TEOA and water were explored, and the results are shown in entries 1 and 12–15.
Under the optimal reaction conditions, the catalyst durability test of Pt@Pd-PCN-222(Hf) was performed and the H2 evolution rate was monitored every 1 h (Fig. 5). The H2 evolution rate increased from the initial 18920 μmol g−1 h−1 after irradiation for 1 h to a maximum 22674 μmol g−1 h−1 after 3 h, and then gradually decreased to 14510 μmol g−1 h−1 after 8 h. After irradiation for 32 h, the H2 evolution amount arrived at an equilibrium of 194168 μmol g−1, while the corresponding rate dropped to 6068 μmol g−1 h−1 (Table S3†). Based on the loaded Pt-NPs, the initial TON and TOF were calculated to be 1447.3 and 482.5 h−1 for 3 h photoreaction, respectively. After 32 h, the accumulated TON reached 4131.2 while the TOF decreased to 129.1 h−1 without replenishing the sacrificial agent. This suggested that Pt@Pd-PCN-222(Hf) had excellent catalytic durability. For further proof, Pt@Pd-PCN-222(Hf) can be recycled and reused for three successive runs. The recycled Pt@Pd-PCN-222(Hf) did not show a significant loss of catalytic activity (Fig. 6), indicating that the HER photocatalytic ability of Pt@Pd-PCN-222(Hf) was well recovered when reused under fresh solvent, sacrificial agent and water conditions. As shown in the TEM image, no obvious morphology change occurred after the first run (Fig. S17†). PXRD and XPS examination of the recycled samples of Pt@Pd-PCN-222(Hf) verified that its main composition and crystallinity remained intact and the valences of Pd(2+) and Pt(0) did not change after the catalytic reactions (Fig. S18 and S19†). ICP-OES analysis of the filtrate after catalysis indicated that the amount of Pd leaching into the reaction mixture was around 3.5%.
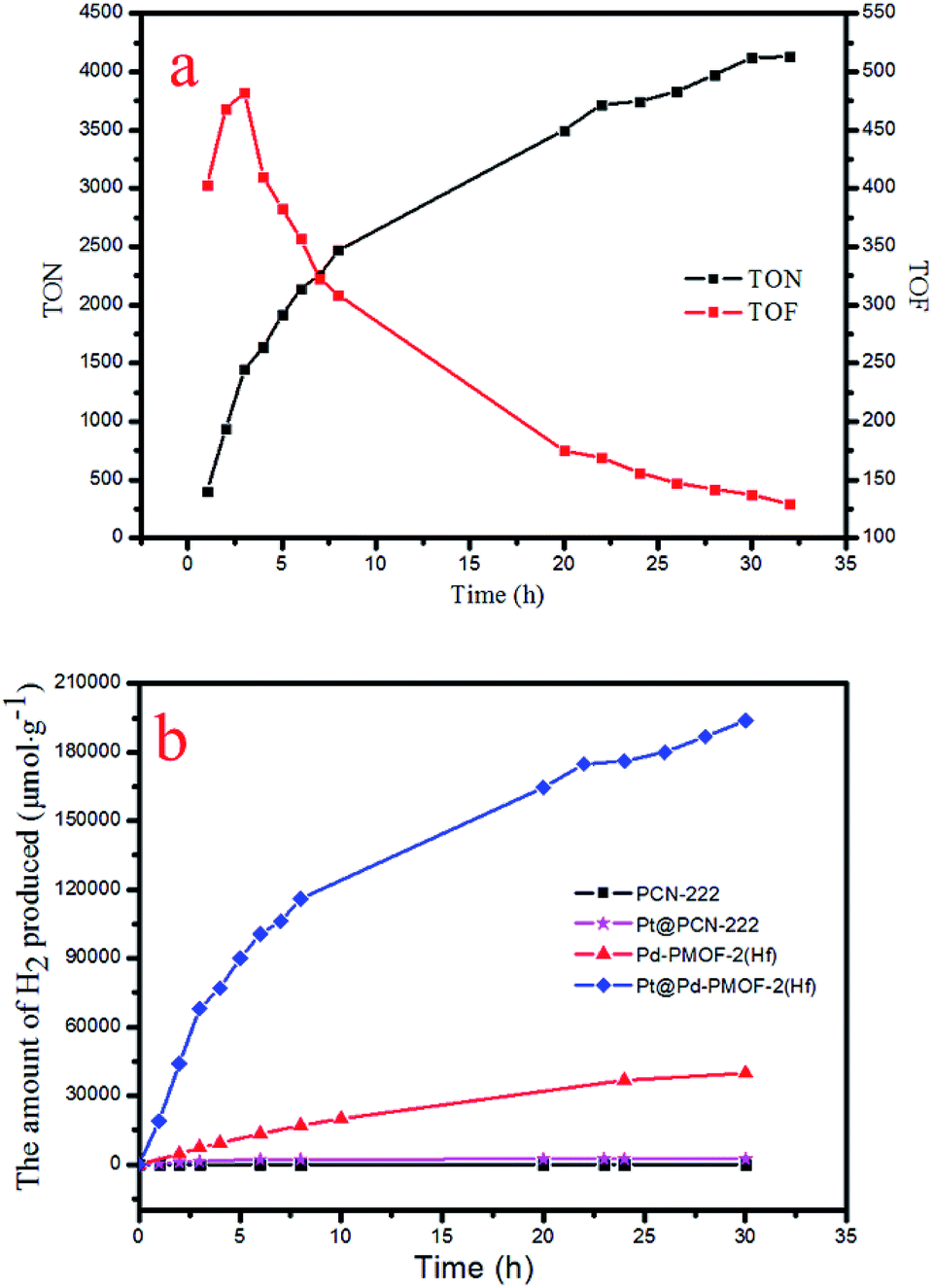 | ||
| Fig. 5 Accumulated TONs (TON = n(H2)/n(Pt)) and TOFs (TOF = d(TON)/dt) based on Pt-NPs in the catalyst durability test of Pt@Pd-PCN-222(Hf) over 32 h (a), and the amount of H2 (μmol g−1) produced by different photocatalysts (b). | ||
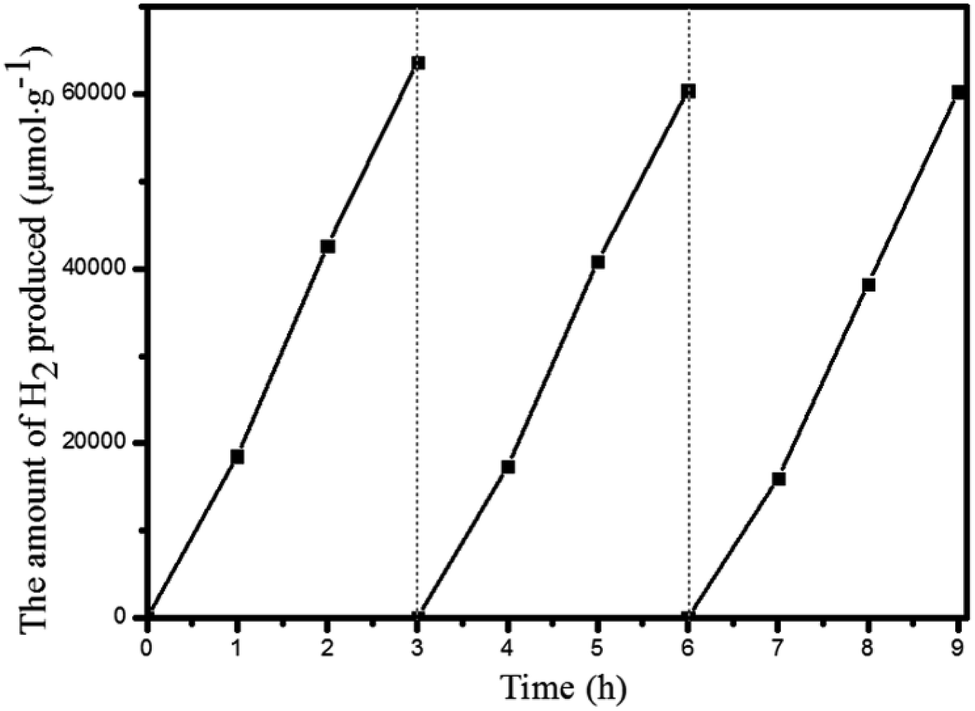 | ||
| Fig. 6 Recycling experiments of Pt@Pd-PCN-222(Hf). | ||
The photocatalytic HER performances of PCN-222(Hf), Pd-PCN-222(Hf) and Pt@PCN-222(Hf) were compared (Fig. 5b and Table S4†). It was reported that, for MOFs constructed from Zr–O clusters and organic linkers with π conjugation, the ligands could behave as light harvesters. After the ligands were photoexcited, the photogenerated electrons transferred to Zr-oxo clusters via linker-to-cluster charge transfer (LCCT). Zr(IV) ions were then reduced to Zr(III), which promoted the subsequent photoreduction reaction.51 Nonetheless, the Hf-based PCN-222(Hf) did not show any activity of photocatalytic HER. After the loading of Pt-NPs as a co-catalyst, a catalytic rate of 441 μmol g−1 h−1 was detected. For comparison, Pd-PCN-222(Hf) that was isostructural with PCN-222(Hf) but had a Pd metal in the porphyrin center exhibited a higher photocatalytic efficiency (2476 μmol g−1 h−1), indicating that Pd-porphyrin itself played a critical role in the photocatalytic reaction. Furthermore, after the loading of Pt-NPs as a co-catalyst, a much higher H2 production rate (22674 μmol g−1 h−1) of Pt@Pd-PCN-222(Hf) was accomplished, which was about 51 and 10 times higher than those of Pt@PCN-222(Hf) and Pd-PCN-222(Hf), respectively.
The catalytic performance of Pt@Pd-PCN-222(Hf) was also compared with those of known Pt@MOF (or Pt/MOF) composites under visible light irradiation (Table S1†). Pt-NPs were deposited in a range of stable MOFs, such as MIL-100(Fe), UiO-66-NH2(Zr), and MIL-125-NH2(Ti), which gave H2 evolution rates of 98, 257 and 367 μmol g−1 h−1, respectively.27–29 Taking advantage of the strong absorption of visible light by porphyrin compounds, porphyrin-based MOFs loaded with Pt-NPs were reported to offer a H2 evolution rate up to 341 μmol g−1 h−1.18,31,32 Dye-sensitization became a relatively mature technology for visible-light harvesting, which was employed to enhance the catalytic efficiency of Pt@UiO-66 (116 μmol g−1 h−1) and Pt/NH2-MIL-101 (477 μmol g−1 h−1).33,34 Ternary MOF composites of Pt@MIL-125/Au and Pt@MIL-101/CdS with additional plasmonic Au-NP and CdS semiconductors, respectively, were also prepared and examined in photocatalytic HER, and the H2 evolution rates increased from a hundred scale to a thousand range up to 7550 μmol g−1 h−1.35,36 In short, Pt@Pd-PCN-222(Hf) represented an exceptionally efficient HER photocatalyst among MOF composites in terms of the H2 evolution rate and catalytic durability.
Reaction mechanism
For a better understanding of the reaction mechanism, the electron–hole separation efficiency was firstly studied via ultrafast transient absorption (TA) spectroscopy.52–55 In the TA measurements, a pump–probe configuration with a femtosecond visible pump and a white-light continuum probe was adopted. The center wavelength of the pump pulses was chosen to be 400 nm, which could effectively excite porphyrin MOFs. The TA of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) both featured a broad excited-state absorption (ESA) around 505 nm accompanied by a ground-state bleaching (GSB) centered around 450 nm, which displayed positive and negative absorbance changes (ΔA), respectively (Fig. 7a and b). The recovery around 505 nm was characterized by two time constants, that is, τ1 = 17500 ps (76.7%) and τ2 = 11.4 ps (23.3%) for Pd-PCN-222(Hf), and τ1 = 2.19 ps (16.6%) and τ2 = 7260 ps (83.4%) for Pt@Pd-PCN-222(Hf) (Fig. 7c and Table S5†). For comparison, Pt@Pd-PCN-222(Hf) exhibited an acceleration of the TA kinetics compared to Pd-PCN-222(Hf). Within the probe-delay limit of our pump/probe spectrometer (∼7.7 ns), the ΔA recovery converged to an asymptote with a non-zero, indicative of an extra extremely long lifetime.
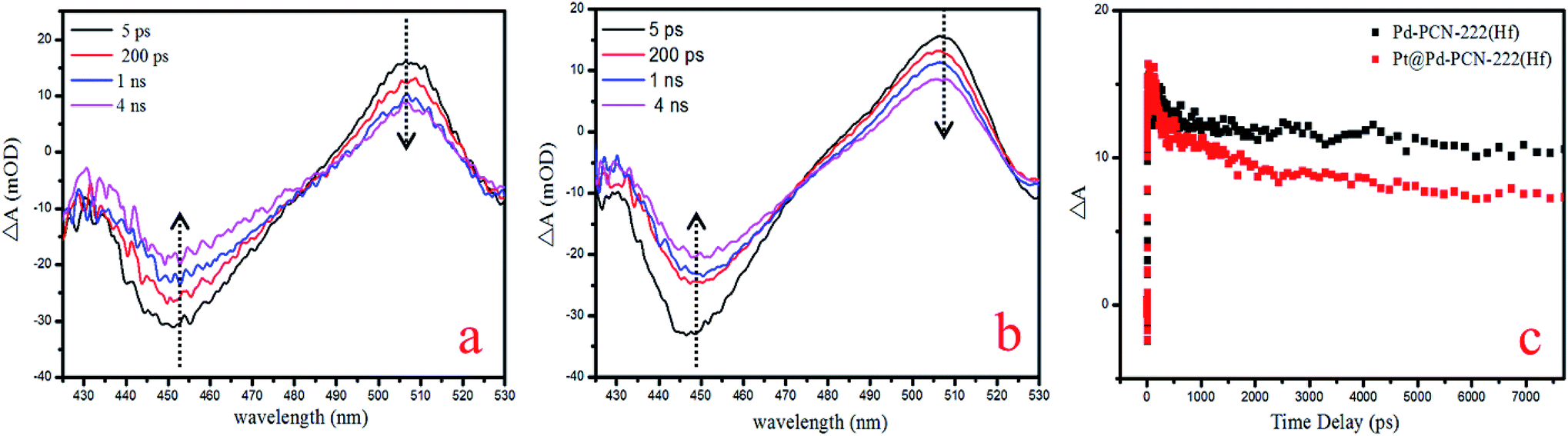 | ||
| Fig. 7 TA spectra of Pd-PCN-222(Hf) (a) and Pt@Pd-PCN-222(Hf) (b), and TA kinetics at the probing wavelength of 505 nm (c). | ||
Next, steady-state photoluminescence (PL) spectroscopy was carried out to characterize the photophysical behaviors of Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) (Fig. S20†). Upon excitation (λex = 422 nm), they both displayed two main PL peaks around 654 and 722 nm. The emission intensity of Pt@Pd-PCN-222(Hf) was remarkably reduced in comparison to that of Pd-PCN-222(Hf), suggesting that the recombination rate of photo-generated electron–hole pairs was decreased after the Pt-NPs were introduced into the coordination interspaces of Pd-PCN-222(Hf), due to the formation of a Schottky barrier at the interface of Pd-PCN-222(Hf) and Pt-NPs. The PL lifetimes of PCN-222, Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) were then examined, which were calculated to be 1.2 ns, 113.4 μs and 91.6 μs, respectively, at 298 K under vacuum (Fig. S21 and Table S6†). Such significantly long-lived triplet states in Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf) were competent for electron transfer, leading to subsequent photoreactions. According to the iterative extended Hückel (IEH) model, the strong phosphorescence of d8 Pd(II) porphyrins should be due to the mixing of empty eg (π*) of porphyrin and the filled dπ of the metal, which was also called π-back bonding, giving rise to a large spin–orbit coupling, rapid intersystem crossing and high triplet yields.56
Finally, the photocurrent response and electrochemical impedance spectroscopy (EIS) measurements were performed to unveil the charge-separation efficiency. Pt@Pd-PCN-222(Hf) possessed the strongest photocurrent response among all samples, indicating that the recombination of photogenerated electrons and holes was largely inhibited and effective charge transfer occurred from Pd-PCN-222(Hf) to Pt-NPs under visible-light irradiation (Fig. 8a). This argument was also supported by EIS results (Fig. 8b), where Pt@Pd-PCN-222(Hf) exhibited the smallest radius, indicative of its fastest interfacial charge transfer.
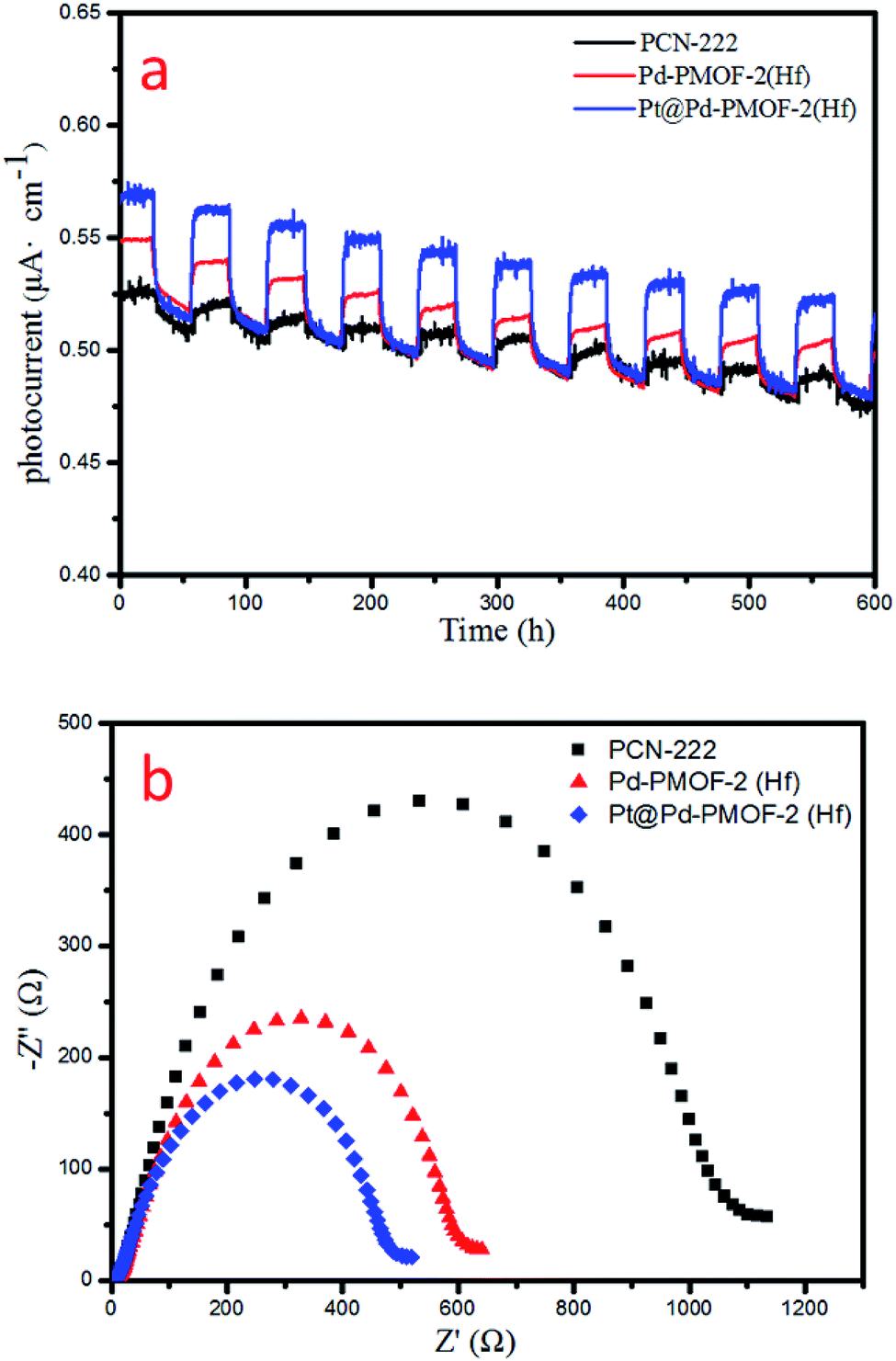 | ||
| Fig. 8 Photocurrent responses (a) and EIS Nyquist plots (b) of PCN-222(Hf), Pd-PCN-222(Hf) and Pt@Pd-PCN-222(Hf). | ||
According to photocatalytic results and physical characterization, a possible mechanism was proposed (Fig. 1 and 9): the Pd-porphyrin was excited by visible light, giving rise to the excited-states. And then, the electrons transferred from the LUMO of the Pd-porphyrin ligand to Pt-NPs, where the H2O molecules were reduced to generate H2. Nevertheless, the LCCT mechanism, in which the photogenerated electrons firstly transferred to the Hf–O clusters, and then flowed to Pt-NPs, could not be ruled out.
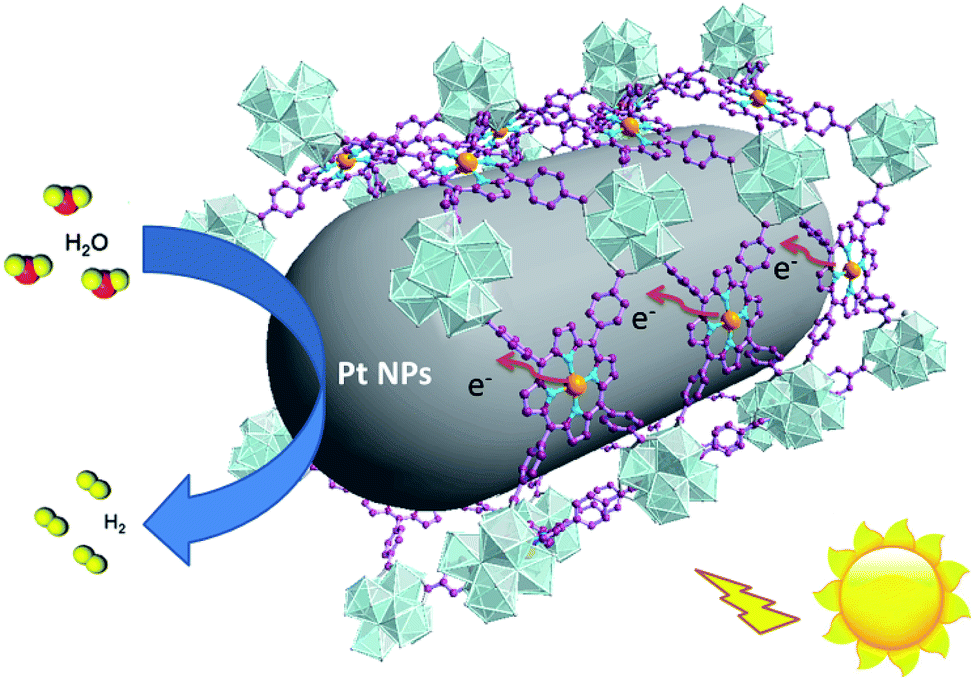 | ||
| Fig. 9 The possible HER reaction mechanism of Pt@Pd-PCN-222(Hf). | ||
The remarkable H2 evolution activity of Pt@Pd-PCN-222(Hf) might be ascribed to three factors. Firstly, the Pd-porphyrin photosensitizers featuring long-lived room temperature phosphorescence were spatially arranged in Pd-PCN-222(Hf), assisting a wide range of visible light absorption without mutual electron coupling quenching. Secondly, the Pt-NPs were well oriented and distributed into the confined coordination interspaces of Pd-PCN-222(Hf) to form a Schottky barrier on the interface with Pd-porphyrin photosensitizers, accelerating the charge transfer and electron–hole separation. Thirdly, the Hf(IV)-based MOF provided hydrophilic Hf(IV)-oxo clusters with H2O accumulation for photocatalytic water splitting. In short, these collaborative effects inside the functional coordination interspaces of Pd-PCN-222(Hf) boosted highly efficient photocatalytic HER.
Conclusion
Photocatalytic water splitting for H2 evolution is vital to energy and environmental science. In this work, we demonstrate a strategy for the design of a powerful HER photocatalyst by taking advantage of the synergistic effect among the Pd-porphyrin photosensitizer with long-lived room temperature phosphorescence, Pt-NPs as co-catalysts that grew in the confined coordination interspaces of Pd-PCN-222(Hf), and the water concentration effect that was enforced by hydrophilic Hf(IV)-oxo clusters. The developed MOF composite Pt@Pd-PCN-222(Hf) can avoid the aggregation of Pt-NPs, assist better electron transfer between Pd-porphyrin and Pt-NPs units, and promote the H2 evolution rate. Further work on the design and catalytic applications of porphyrin-based MOFs is in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Natural Science Foundation of China (21773314, 21720102007, 21821003, and 21890380), Science and Technology Planning Project of Guangzhou (201707010168), and Local Innovative and Research Teams Project of Guangdong Peal River Talents Program (2017BT01C161).Notes and references
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed.
- M. Reza Gholipour, C.-T. Dinh, F. Beland and T.-O. Do, Nanoscale, 2015, 7, 8187–8208 RSC.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 RSC.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed.
- M. Wen, K. Mori, Y. Kuwahara, T. An and H. Yamashita, Appl. Catal., B, 2017, 218, 555–569 CrossRef CAS.
- B. Zhu, R. Zou and Q. Xu, Adv. Energy Mater., 2018, 8, 1801193 CrossRef.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- C.-C. Cao, C.-X. Chen, Z.-W. Wei, Q.-F. Qiu, N.-X. Zhu, Y.-Y. Xiong, J.-J. Jiang, D. Wang and C.-Y. Su, J. Am. Chem. Soc., 2019, 141, 2589–2593 CrossRef CAS PubMed.
- C.-X. Chen, Z.-W. Wei, J.-J. Jiang, S.-P. Zheng, H.-P. Wang, Q.-F. Qiu, C.-C. Cao, D. Fenske and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 6034–6037 CrossRef CAS PubMed.
- Y. Zhang, J. Guo, L. Shi, Y. Zhu, K. Hou, Y. Zheng and Z. Tang, Sci. Adv., 2017, 3, e1701162 CrossRef PubMed.
- Q. Lin, X. Bu, A. Kong, C. Mao, X. Zhao, F. Bu and P. Feng, J. Am. Chem. Soc., 2015, 137, 2235–2238 CrossRef CAS PubMed.
- M. H. Beyzavi, N. A. Vermeulen, A. J. Howarth, S. Tussupbayev, A. B. League, N. M. Schweitzer, J. R. Gallagher, A. E. Platero-Prats, N. Hafezi, A. A. Sarjeant, J. T. Miller, K. W. Chapman, J. F. Stoddart, C. J. Cramer, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13624–13631 CrossRef CAS PubMed.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
- G. Lan, Y.-Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS PubMed.
- X.-S. Wang, L. Meng, Q. Cheng, C. Kim, L. Wojtas, M. Chrzanowski, Y.-S. Chen, X. P. Zhang and S. Ma, J. Am. Chem. Soc., 2011, 133, 16322–16325 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- J. Zheng, M. Wu, F. Jiang, W. Su and M. Hong, Chem. Sci., 2015, 6, 3466–3470 RSC.
- Y. Zhao, N. Kornienko, Z. Liu, C. Zhu, S. Asahina, T.-R. Kuo, W. Bao, C. Xie, A. Hexemer, O. Terasaki, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2199–2202 CrossRef CAS PubMed.
- J. A. Johnson, B. M. Petersen, A. Kormos, E. Echeverria, Y.-S. Chen and J. Zhang, J. Am. Chem. Soc., 2016, 138, 10293–10298 CrossRef CAS PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- Y. Wang, H. Cui, Z.-W. Wei, H.-P. Wang, L. Zhang and C.-Y. Su, Chem. Sci., 2017, 8, 775–780 RSC.
- J. Liu, Y.-Z. Fan, X. Li, Z. Wei, Y.-W. Xu, L. Zhang and C.-Y. Su, Appl. Catal., B, 2018, 231, 173–181 CrossRef CAS.
- O. S. Finikova, A. V. Cheprakov and S. A. Vinogradov, J. Org. Chem., 2005, 70, 9562–9572 CrossRef CAS PubMed.
- L. Zang, H. Zhao, J. Hua, W. Cao, F. Qin, J. Yao, Y. Tian, Y. Zheng and Z. Zhang, J. Mater. Chem. C, 2016, 4, 9581–9587 RSC.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- D. Wang, Y. Song, J. Cai, L. Wu and Z. Li, New J. Chem., 2016, 40, 9170–9175 RSC.
- J.-D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CrossRef CAS.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H.-L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- F. Leng, H. Liu, M. Ding, Q.-P. Lin and H.-L. Jiang, ACS Catal., 2018, 8, 4583–4590 CrossRef CAS.
- J. He, J. Wang, Y. Chen, J. Zhang, D. Duan, Y. Wang and Z. Yan, Chem. Commun., 2014, 50, 7063–7066 RSC.
- M. Wen, K. Mori, T. Kamegawa and H. Yamashita, Chem. Commun., 2014, 50, 11645–11648 RSC.
- J.-D. Xiao, L. Han, J. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1103–1107 CrossRef CAS PubMed.
- J. He, Z. Yan, J. Wang, J. Xie, L. Jiang, Y. Shi, F. Yuan, F. Yu and Y. Sun, Chem. Commun., 2013, 49, 6761–6763 RSC.
- J. Guo, Y. Zhang, L. Shi, Y. Zhu, M. F. Mideksa, K. Hou, W. Zhao, D. Wang, M. Zhao, X. Zhang, J. Lv, J. Zhang, X. Wang and Z. Tang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Ferey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gandara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- Y. Chen, T. Hoang and S. Ma, Inorg. Chem., 2012, 51, 12600–12602 CrossRef CAS PubMed.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- J.-P. Zhang, A.-X. Zhu, R.-B. Lin, X.-L. Qi and X.-M. Chen, Adv. Mater., 2011, 23, 1268–1271 CrossRef CAS PubMed.
- M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 11050–11053 CrossRef CAS PubMed.
- R. Fareghi-Alamdari, M. Golestanzadeh and O. Bagheri, RSC Adv., 2016, 6, 108755–108767 RSC.
- Y.-Z. Chen and H.-L. Jiang, Chem. Mater., 2016, 28, 6698–6704 CrossRef CAS.
- F. Su, C. K. Poh, Z. Tian, G. Xu, G. Koh, Z. Wang, Z. Liu and J. Lin, Energy Fuels, 2010, 24, 3727–3732 CrossRef CAS.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- C. Gomes Silva, I. Luz, F. X. Llabrés i Xamena, A. Corma and H. García, Chem.–Eur. J., 2010, 16, 11133–11138 CrossRef PubMed.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- J.-D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H.-L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- A. Antipas and M. Gouterman, J. Am. Chem. Soc., 1983, 105, 4896–4901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of comparison of H2 evolution rates of different MOF composites, general information, physical characterization, chemical stability study, photocatalytic reactions, mechanistic study. CCDC 1920896. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01866b |
| This journal is © The Royal Society of Chemistry 2019 |