
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed acylation-reactions of (hetero)arylzinc pivalates with thiopyridyl ester derivatives†
Ferdinand H.
Lutter‡
 ,
Lucie
Grokenberger‡
,
Maximilian S.
Hofmayer‡
and
Paul
Knochel
*
,
Lucie
Grokenberger‡
,
Maximilian S.
Hofmayer‡
and
Paul
Knochel
*
Ludwig-Maximilians-Universität München, Department Chemie, Butenandtstraße 5-13, Haus F, 81377 München, Germany. E-mail: paul.knochel@cup.uni-muenchen.de
First published on 11th July 2019
Abstract
A cobalt-catalyzed acylation reaction of various primary, secondary and tertiary alkyl, benzyl and (hetero)aryl S-pyridyl thioesters with (hetero)arylzinc pivalates is reported. The thioesters were prepared directly from the corresponding carboxylic acids under mild conditions, thus tolerating sensitive functional groups. Acylations of α-chiral S-pyridyl esters proceeded with very high stereoretention leading to optically enriched α-chiral ketones.
Introduction
The carbonyl group is a central functionality in organic chemistry and the performance of acylation reactions employing organometallic reagents represents a general access to various ketones.1 A major drawback of these reactions is the moderate chemoselectivity or the use of expensive catalysts.1 Acid chlorides are the most common acylation reagents.1,2 However, their preparation requires harsh conditions, thus lowering the functional group tolerance. In contrast, the use of thioesters is a valuable alternative since Fukuyama showed in pioneering work that these acylating reagents react readily with organozinc halides in the presence of a palladium catalyst.3 Additionally, Seki,4 Rovis,5 Fleischer,6 and others7 showed that these reactions can be performed using various transition metal catalysts. Recently, we have shown that organozinc pivalates (RZnOPiv) are an attractive class of zinc organometallics due to their enhanced air- and moisture stability and their excellent compatibility with various transition metal-catalyzed transformations.8 Especially, cobalt-catalyzed reactions have proven to be advantageous.9Herein, we wish to report a new cobalt-catalyzed acylation reaction of various saturated and unsaturated thioesters of type R1C(O)SPy (1) with aryl- and heteroarylzinc pivalates of type R2ZnOPiv (2), leading to a broad range of polyfunctional ketones of type 3. Although thioesters are readily available from the corresponding acid chlorides and thiols,10 the pyridyl thioesters 1 were prepared under exceedingly mild and neutral conditions from the corresponding carboxylic acid of type 4 using Mukaiyama's method (Scheme 1).11
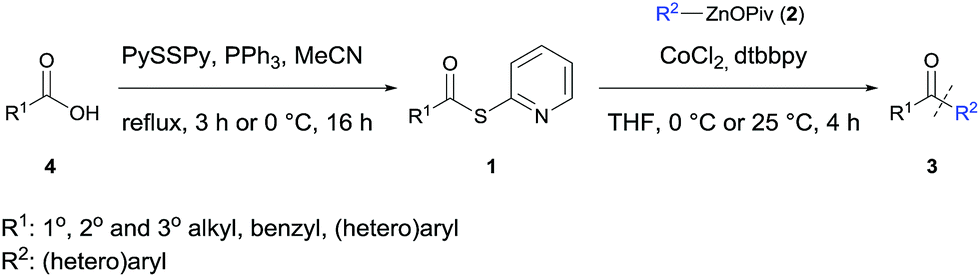 | ||
| Scheme 1 Preparation of thiopyridyl esters of type 1 from carboxylic acids 4 and cobalt-catalyzed acylation with organozinc pivalates 2, affording ketones of type 3. Py = 2-pyridyl. | ||
Results and discussion
In preliminary experiments, S-(pyridin-2-yl)-cyclohexanecarbo-thioate (1a) was treated with 4-(methoxyphenyl)zinc pivalate (2a) under various conditions (Table 1). In the absence of a catalyst, ketone 3a was obtained in only 9% yield (Table 1, entry 1). Although palladium and nickel are well-known metal catalysts for the Fukuyama acylation, the use of cheaper12 and more abundant catalysts is highly desirable. Whereas, MnCl2, CrCl2, FeCl2 or CuCl2 gave unsatisfying results (entries 2–5), CoCl2 proved to be an excellent catalyst for this transformation (entry 6). Its catalytic efficiency could be further improved by the addition of various ligands. After a short screening it became clear that 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbbpy) gave the best results leading to the ketone 3a in 88% isolated yield (entry 11). At this point, we verified that no other metal contaminations are responsible for this catalysis. Thus, using CoCl2 (99.99% purity)13 led to 3a in 86% yield (entry 12). Furthermore, a screening showed that RC(O)SPy thioesters are superior to thioesters of type RC(O)SEt or RC(O)SPh.14|
|
|||
|---|---|---|---|
| Entry | Catalyst | Ligand | Yield of 3aa [%] |
| a Reactions were performed on a 0.5 mmol scale. Determined by GC-analysis. Tetradecane (C14H30) was used as internal standard. b 20% of the ligand was used. c 2,2′-Bipyridine. d 4,4′-Di-tert-butyl-2,2′-dipyridyl. e Isolated yield. f Reaction was performed on a 5 mmol scale. g CoCl2 (99.99% purity) was used. | |||
| 1 | — | — | 9 |
| 2 | MnCl2 | — | Traces |
| 3 | CrCl2 | — | Traces |
| 4 | FeCl2 | — | 50 |
| 5 | CuCl2 | — | 29 |
| 6 | CoCl2 | — | 67 |
| 7 | CoCl2 | PPh3b | 63 |
| 8 | CoCl2 | TMEDA | 64 |
| 9 | CoCl2 | Neocuproine | 49 |
| 10 | CoCl2 | bipyc | 71 |
| 11 | CoCl 2 | dtbbpy | 90 (88) (87) |
| 12 | CoCl2g | dtbbpyd | 86 |
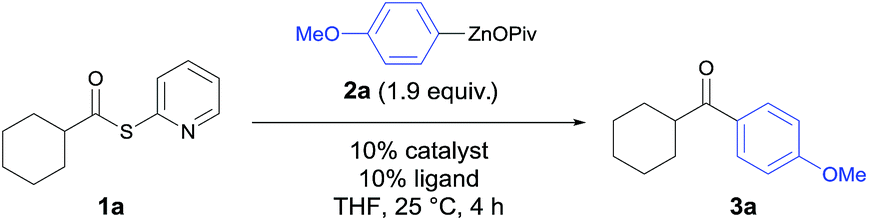
In a typical experiment palmitic acid (4b) was treated with 2,2′-dipyridyl disulfide (1.1 equiv.) and PPh3 (1.5 equiv.) in acetonitrile (0.3 M) under reflux for 3 h. Short purification using flash column chromatography afforded 1b in 98% yield. The required zinc pivalate 2b was prepared by treating 1-bromo-3,4-(methylene-dioxy)benzene (5b) with Mg (1.2 equiv.) and anhydrous LiCl (1.2 equiv.) for 2 h at 0 °C leading to the corresponding arylmagnesium derivative (91% yield).15 Transmetalation with Zn(OPiv)2 (1.0 equiv.) afforded the zinc organometallic 2b in 93% yield.15 The thioester 1b reacted with 3,4-(methylene-dioxy)-1-phenylzinc pivalate (2b) in the presence of 10% CoCl2 and 10% dtbbpy in THF (25 °C, 4 h) furnishing after standard workup and chromatographic purification the ketone 3b in 90% yield (Table 2, entry 1).
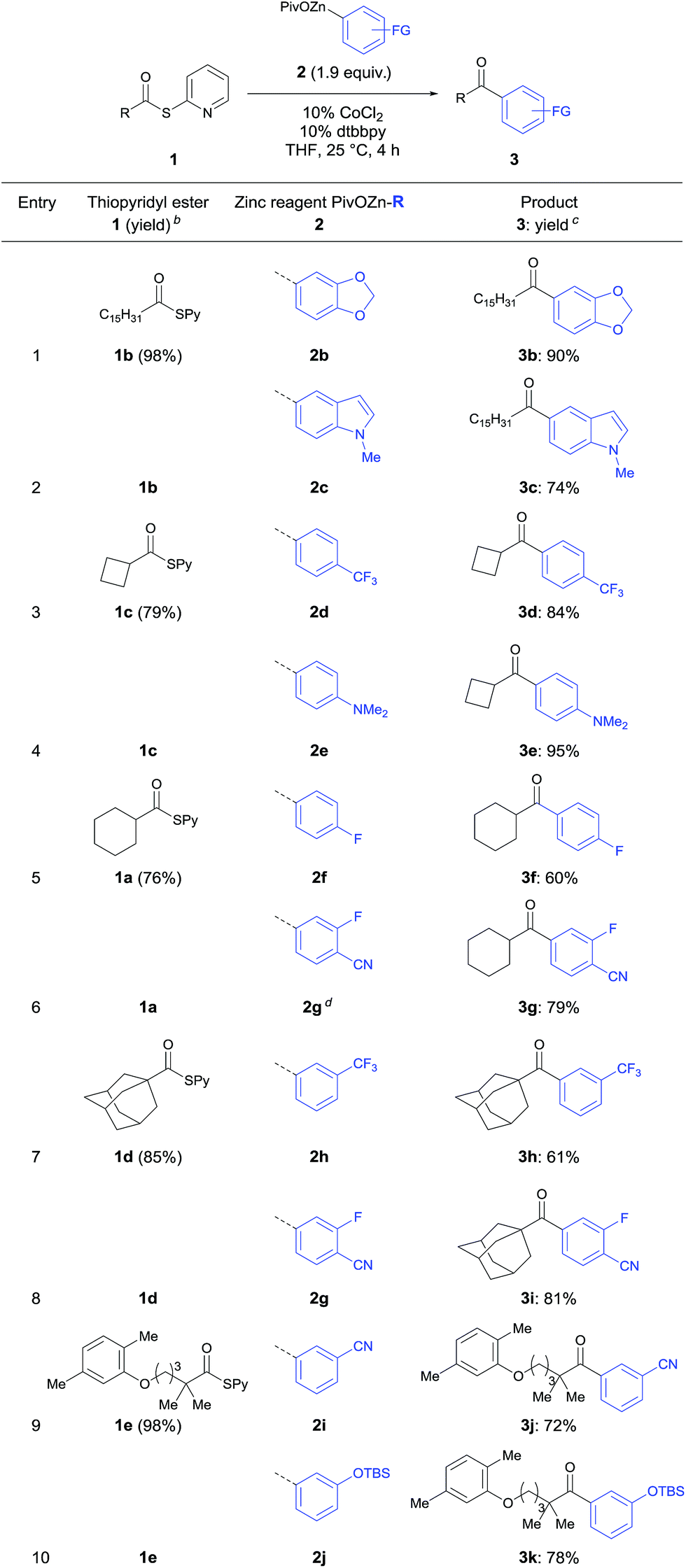
According to this procedure various ketones of type 3 were prepared. Hence, the heterocyclic indolylzinc pivalate (2c) was acylated with palmitic S-pyridyl thioate (1b) furnishing ketone 3c in 74% yield (entry 2). Additionally, secondary thioesters derived from cyclobutane- (4c) and cyclohexanecarboxylic acid (4a) were employed to this acylation procedure leading to the corresponding ketones (3d–3g) in 60–95% yield (entries 3–6). Tertiary S-pyridyl thioesters 1d and 1e derived from 1-adamantanecarboxylic acid (4d) and the lipid regulating drug gemfibrozil16 (4e) reacted smoothly with various functionalized arylzinc pivalates affording acylation products (3h–3k) in 61–81% yield (entries 7–10).
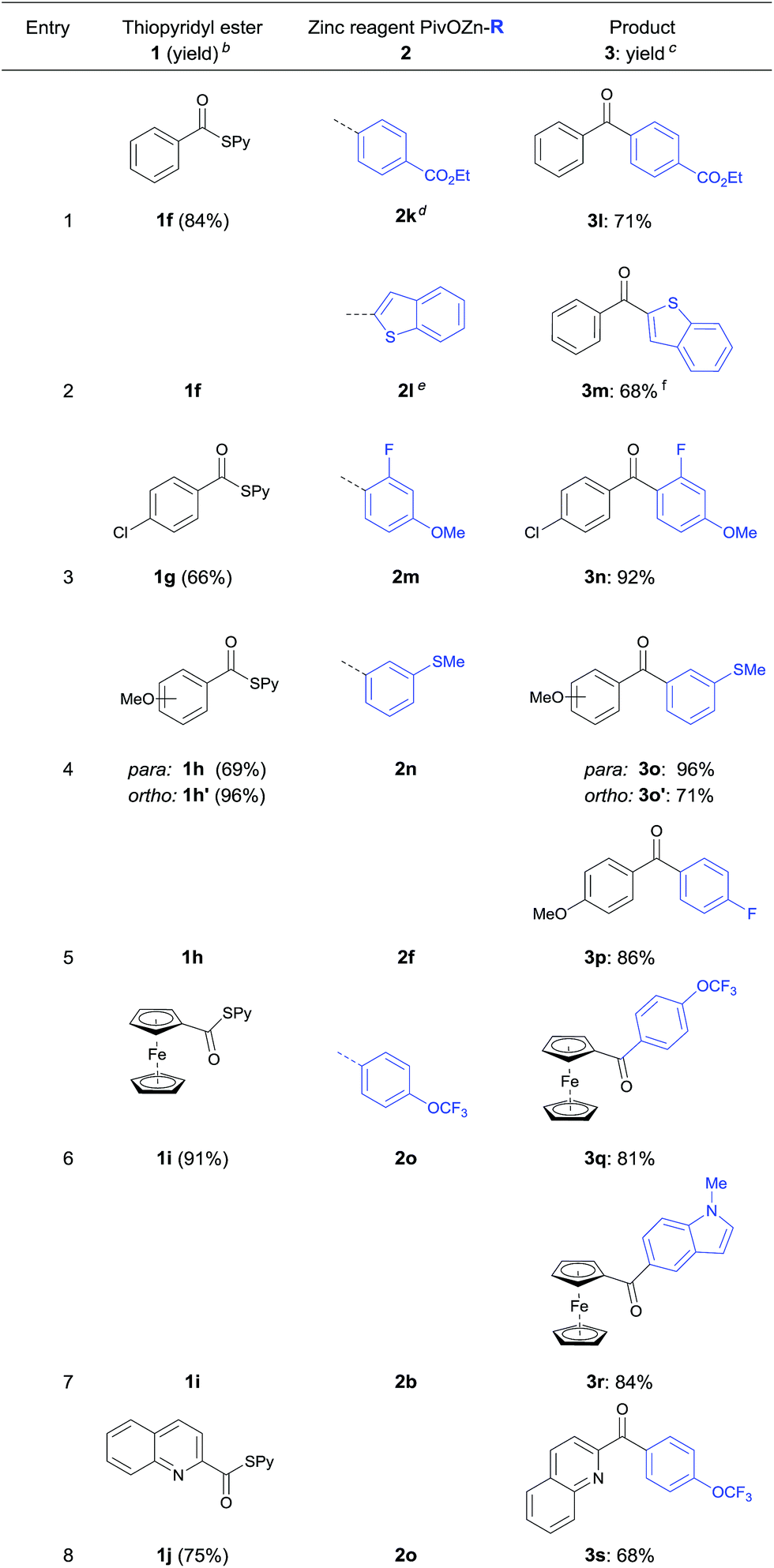
Furthermore, the acylation reaction was extended to aryl- and heteroaryl-S-pyridyl thioesters (Table 3). Thus, (4-(ethoxycarbonyl)-phenyl)-zinc pivalate (2k) prepared via I/Mg-exchange using iPrMgCl·LiCl followed by transmetalation with Zn(OPiv)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8d was readily acylated with S-pyridyl thioester 1f affording the benzophenone 3l in 71% yield (entry 1).
8d was readily acylated with S-pyridyl thioester 1f affording the benzophenone 3l in 71% yield (entry 1).
| a The reactions were performed on a 0.5 mmol scale. b Isolated yield of the S-pyridyl thioester prepared from the corresponding carboxylic acid, PySSPy (1.1 equiv.), PPh3 (1.5 equiv.), MeCN, reflux, 3 h. c Isolated yield. d Prepared using iPrMgCl·LiCl (1.1 equiv.), THF, −40 °C, 2 h. e Prepared using TMPMgCl·LiCl (1.0 equiv.), THF, 0 °C, 3 h. f TMEDA was used instead of dtbbpy. |
|---|
|
Also, 2-benzothiophenylzinc pivalate 2l generated via directed metalation of benzothiophene using TMPMgCl·LiCl and subsequent transmetalation with Zn(OPiv)28d underwent a cobalt catalyzed acylation reaction with 1f leading to the ketone 3m in 68% yield (entry 2). Various substituted aryl thioesters and ferrocenyl derivatives reacted successfully with functionalized (hetero)arylzinc pivalates affording the diaryl ketones 3n–3r in 81–96% yield (entries 3–7). Additionally, 4-trifluoro-methoxyphenylzinc pivalate (2o) was acylated using quinoline thioester 1j furnishing ketone 3s in 68% yield (entry 8).17
The synthesis of α-chiral ketones is of great interest3,5,7c,7f,18 but often challenging under basic conditions due to epimerization. Also, reactions under pH-neutral conditions have been reported by Liebeskind et al. for the synthesis of highly enantiopure peptidyl ketones.19
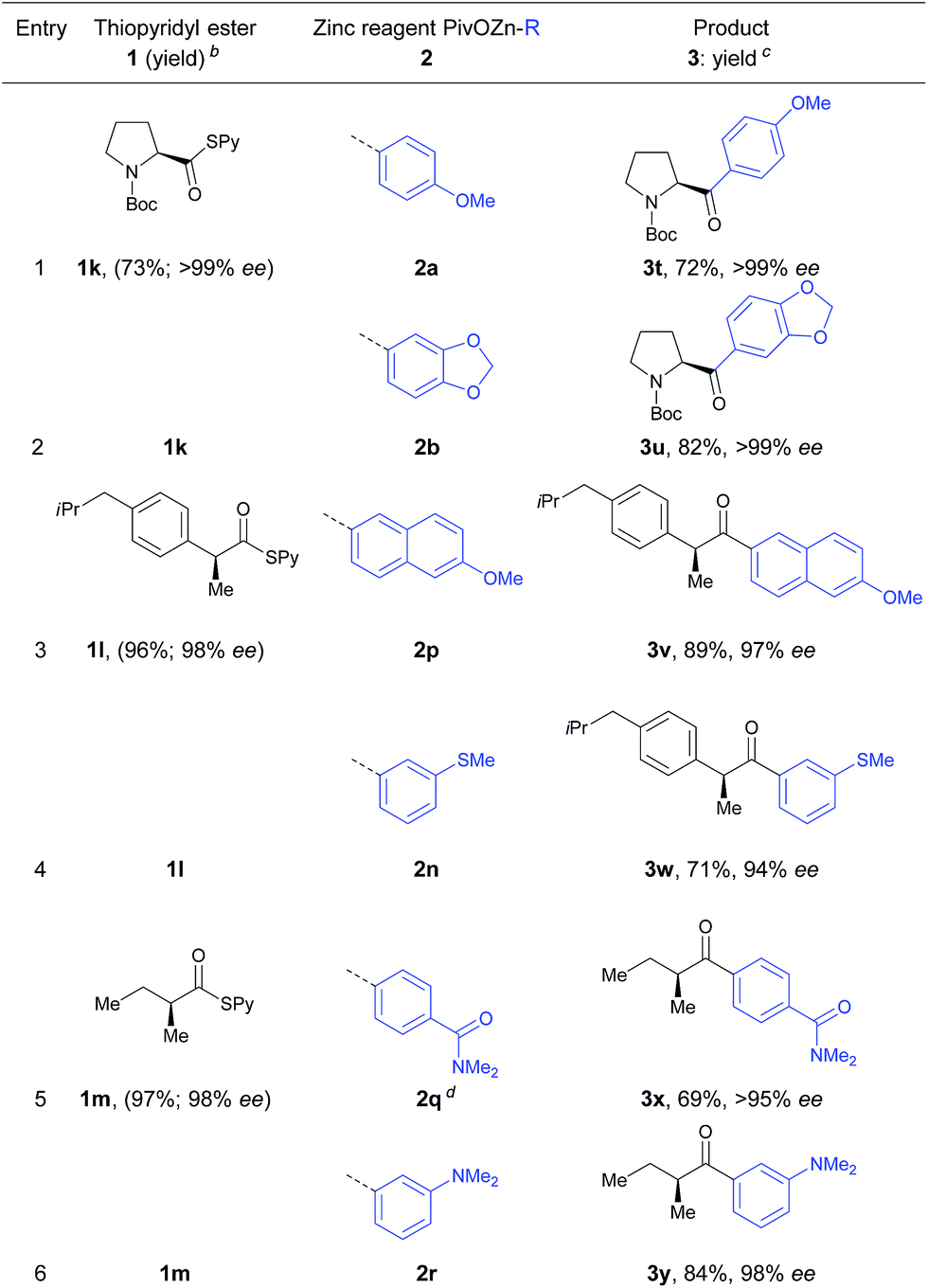
We also tested the applicability of this cobalt-catalyzed acylation to the synthesis of optically enriched α-chiral ketones. Using α-chiral S-pyridyl thioesters at 0 °C afforded several α-chiral ketones with high stereoretention (Table 4). Thus, S-pyridyl thioester 1k prepared from N-Boc protected (S)-proline was treated with arylzinc reagents 2a and 2b leading to the corresponding α-chiral ketones in 72–82% yield and >99% ee (entries 1 and 2).
| a The reactions were performed on a 0.5 mmol scale and at 0 °C instead of 25 °C. b Isolated yield of the S-pyridyl thioester prepared from the corresponding carboxylic acid, PySSPy (1.0 equiv.), PPh3 (1.0 equiv.), MeCN, 0 °C to 25 °C, 16 h. c Isolated yield. d Prepared using iPrMgCl·LiCl (1.1 equiv.), THF, 0 °C, 2 h. |
|---|
|
Furthermore, thioester 1l derived from enantiopure (S)-ibuprofen reacted smoothly with the functionalized arylzinc pivalates 2p and 2n in 71–89% yield and 94–97% ee (entries 3 and 4).
Also, arylzinc pivlalates 2q and 2r bearing an amide or dimethylamino functionality were acylated using optically pure S-(pyridin-2-yl)-(S)-2-methylbutanethioate (1m) furnishing the α-chiral ketones 3x and 3y in 69–84% yield and 95–98% ee (entries 5 and 6).
To gain insights into the reaction mechanism, radical trapping experiments were carried out. Thus, to a standard acylation setup of the developed protocol using S-pyridyl thioester 1a and organozinc pivalate 2a, various amounts of the radical trapping agent 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) were added. With 10% of the trapping reagent a decrease of the yield by 19% was observed for the acylation product 3a. However, using 1.5 equiv. of TEMPO the product formation is almost completely suppressed. This may indicate the involvement of radical intermediates within this acylation reaction.14
The utility of this acylation was demonstrated in the synthesis of the antilipidemic drug fenofibrate20 (3z, Scheme 2). Alkylation of 4-iodophenol (6) with isopropyl 2-bromo-2-methyl-propanoate (7) affords the corresponding iodo-aryl ether 8 in 70% yield. 8 was treated with Mg, LiCl and Zn(OPiv)2 generating the arylzinc pivalate 2s in 72% yield.8a Using the new cobalt-catalyzed acylation procedure, fenofibrate (3z) was obtained in 65% yield.
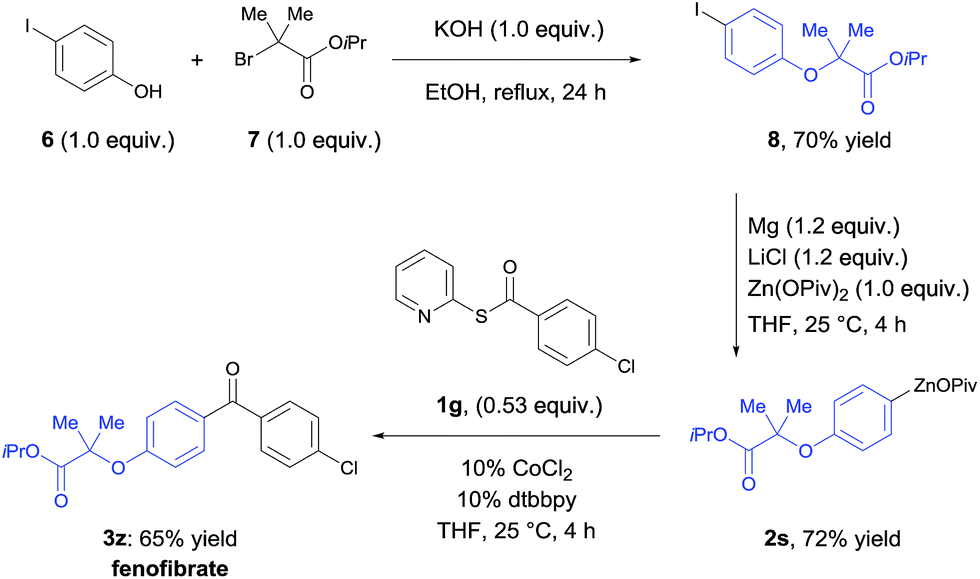 | ||
| Scheme 2 Synthesis of fenofibrate (3z) using the Co-catalyzed acylation. | ||
Conclusion
In conclusion, we have reported a cobalt-catalyzed acylation procedure of a variety of functionalized (hetero)arylzinc pivalates utilizing primary, secondary and tertiary alkyl, benzyl and (hetero)aryl S-pyridyl thioesters as mild acylating agents. These thioesters were readily prepared under neutral conditions from the corresponding carboxylic acid thus allowing their synthesis in the presence of various sensitives functional groups. Several α-chiral ketones were prepared with high stereoretention (94% to >99% ee). Further investigations are currently underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DFG for financial support. We also thank Albemarle (Hoechst, Germany) and BASF SE (Ludwigshafen, Germany) for the generous gift of chemicals. We also thank Vanessa Braun, Constantin Nuber, and Arne Stolpmann for the preparation of starting materials.Notes and references
- (a) R. K. Dieter, Tetrahedron, 1999, 55, 4177–4236 CrossRef CAS; (b) D. A. Shirley, Organic Reactions, Wiley-VCH, Weinheim, 2011 Search PubMed.
- (a) P. Knochel and R. D. Singer, Chem. Rev., 1993, 93, 2117–2188 CrossRef CAS; (b) C. K. Reddy and P. Knochel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1700–1701 CrossRef CAS; (c) H. Fillon, C. Gosmini and J. Périchon, Tetrahedron, 2003, 59, 8199–8202 CrossRef CAS; (d) Handbook of Functionalized Organometallics, ed. P. Knochel, Wiley-VCH, Weinheim, 2005 Search PubMed; (e) S.-H. Kim and R. D. Rieke, Tetrahedron Lett., 2011, 52, 1523–1526 CrossRef CAS.
- H. Tokuyama, S. Yokoshima, T. Yamashita and T. Fukuyama, Tetrahedron Lett., 1998, 39, 3189–3192 CrossRef CAS.
- T. Shimizu and M. Seki, Tetrahedron Lett., 2002, 43, 1039–1042 CrossRef CAS.
- Y. Zhang and T. Rovis, J. Am. Chem. Soc., 2004, 126, 15964–15965 CrossRef CAS PubMed.
- P. H. Gehrtz, P. Kathe and I. Fleischer, Chem.–Eur. J., 2018, 24, 8774–8778 CrossRef CAS PubMed.
- (a) M. Onaka, Y. Matsuoka and T. Mukaiyama, Chem. Lett., 1981, 10, 531–534 CrossRef; (b) C. Cardellicchio, V. Fiandanese, G. Marchese and L. Ronzini, Tetrahedron Lett., 1985, 26, 3595–3598 CrossRef CAS; (c) W. Oppolzer, C. Darcel, P. Rochet, S. Rosset and J. De Brabander, Helv. Chim. Acta, 1997, 80, 1319–1337 CrossRef CAS; (d) B. Li, R. A. Buzon, C. K. F. Chiu, S. T. Colgan, M. L. Jorgensen and N. Kasthurikrishnan, Tetrahedron Lett., 2004, 45, 6887–6890 CrossRef CAS; (e) K. Kunchithapatham, C. C. Eichman and J. P. Stambuli, Chem. Commun., 2011, 47, 12679–12681 RSC; (f) A. H. Cherney and S. E. Reisman, Tetrahedron, 2014, 70, 3259–3265 CrossRef CAS; (g) R. Haraguchi, S.-g. Tanazawa, N. Tokunaga and S.-i. Fukuzawa, Org. Lett., 2017, 19, 1646–1649 CrossRef CAS PubMed.
- (a) S. Bernhardt, G. Manolikakes, T. Kunz and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9205–9209 CrossRef CAS PubMed; (b) C. I. Stathakis, S. Bernhardt, V. Quint and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9428–9432 CrossRef CAS PubMed; (c) J. R. Colombe, S. Bernhardt, C. Stathakis, S. L. Buchwald and P. Knochel, Org. Lett., 2013, 15, 5754–5757 CrossRef CAS PubMed; (d) S. M. Manolikakes, M. Ellwart, C. I. Stathakis and P. Knochel, Chem.–Eur. J., 2014, 20, 12289–12297 CrossRef CAS PubMed; (e) M. Ellwart and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 10662–10665 CrossRef CAS PubMed; (f) Y.-H. Chen, C. P. Tüllmann, M. Ellwart and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 9236–9239 CrossRef CAS PubMed; (g) C. P. Tüllmann, Y.-H. Chen, R. J. Schuster and P. Knochel, Org. Lett., 2018, 20, 4601–4605 CrossRef PubMed; (h) M. S. Hofmayer, F. H. Lutter, L. Grokenberger, J. M. Hammann and P. Knochel, Org. Lett., 2019, 21, 36–39 CrossRef CAS PubMed.
- (a) J. M. Hammann, F. H. Lutter, D. Haas and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 1082–1086 CrossRef CAS PubMed; (b) M. S. Hofmayer, J. M. Hammann, F. H. Lutter and P. Knochel, Synthesis, 2017, 49, 3925–3930 CrossRef CAS; (c) J. M. Hammann, L. Thomas, Y.-H. Chen, D. Haas and P. Knochel, Org. Lett., 2017, 19, 3847–3850 CrossRef CAS PubMed; (d) Y.-H. Chen, S. Graßl and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 1108–1111 CrossRef CAS PubMed; (e) L. Thomas, F. H. Lutter, M. S. Hofmayer, K. Karaghiosoff and P. Knochel, Org. Lett., 2018, 20, 2441–2444 CrossRef CAS PubMed; (f) F. H. Lutter, S. Graßl, L. Grokenberger, M. S. Hofmayer, Y.-H. Chen and P. Knochel, ChemCatChem, 2019 DOI:10.1002/cctc.201900070.
- B. P. Bandgar, P. E. More, V. T. Kamble and S. S. Sawant, Aust. J. Chem., 2008, 61, 1006–1010 CrossRef CAS.
- (a) T. Endo, S. Ikenaga and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1970, 43, 2632–2633 CrossRef CAS; (b) T. Mukaiyama, M. Araki and H. Takei, J. Am. Chem. Soc., 1973, 95, 4763–4765 CrossRef CAS; (c) T. Hofmann and P. Schieberle, J. Agric. Food Chem., 1998, 46, 616–619 CrossRef CAS PubMed.
- World market prices: Pd ca. 33938 € per kg and 3594 € per mol, Co ca. 49 € per kg and 3 € per mol; http://www.infomine.com/; retrieved April 2019.
- A new stirring bar and flask was used for the reaction, see: E. O. Pentsak, D. B. Eremin, E. G. Gordeev and V. P. Ananikov, ACS Catal., 2019, 9, 3070–3081 CrossRef CAS.
- For further details, see ESI.†.
- The yield was determined by iodometric titration, see: A. Krasovskiy and P. Knochel, Synthesis, 2006, 890 CAS.
- P. A. Todd and A. Ward, Drugs, 1988, 36, 314–339 CrossRef CAS PubMed.
- For unsuccessful acylation reactions with (hetero)arylzinc pivalates including several electron poor N-heterocyclic and sterically hindered organozinc reagents see ESI.†.
- (a) G. T. Crisp and T. P. Bubner, Synth. Commun., 1990, 20, 1665–1670 CrossRef CAS; (b) G. Cahiez and E. Metais, Tetrahedron Lett., 1995, 36, 6449–6452 CrossRef CAS; (c) T. Fukuyama and H. Tokuyama, Aldrichimica Acta, 2004, 37, 100–101 Search PubMed; (d) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed; (e) R. Oost, A. Misale and N. Maulide, Angew. Chem., Int. Ed., 2016, 55, 4587–4590 CrossRef CAS PubMed.
- (a) H. Li, H. Yang and L. S. Liebeskind, Org. Lett., 2008, 10, 4375–4378 CrossRef CAS PubMed; (b) L. S. Liebeskind, H. Yang and H. Li, Angew. Chem., Int. Ed., 2009, 48, 1417–1421 CrossRef CAS PubMed.
- G. M. Keating and K. F. Croom, Drugs, 2007, 67, 121–153 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01817d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |