
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA structure–property study of fluoranthene-cored hole-transporting materials enables 19.3% efficiency in dopant-free perovskite solar cells†
Xianglang
Sun‡
a,
Fei
Wu‡
b,
Cheng
Zhong
c,
Linna
Zhu
 *b and
Zhong'an
Li
*a
*b and
Zhong'an
Li
*a
aKey Laboratory for Material Chemistry of Energy Conversion and Storage, Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan, 430074, P. R. China. E-mail: lizha@hust.edu.cn
bChongqing Key Laboratory for Advanced Materials and Technologies of Clean Energy, Faculty of Materials & Energy, Southwest University, Chongqing, 400715, P. R. China. E-mail: lnzhu@swu.edu.cn
cDepartment of Chemistry, Wuhan University, Wuhan, 430072, P. R. China
First published on 4th June 2019
Abstract
To date, most of the prevailing organic hole-transporting materials (HTMs) used in perovskite solar cells (PVSCs), such as spiro-OMeTAD and PTAA, generally require a sophisticated doping process to ensure their reasonable hole-transporting properties. Unfortunately, the employed dopants/additives and the associated oxidation reactions have been shown to deteriorate the long-term device stability seriously. The exploitation of efficient and stable dopant-free HTMs is thus strongly desired for PVSCs. However, effective molecular design strategies for dopant-free HTMs are still lacking. Thus far, only a few of them yielded comparable performance to their doped counterparts, while their synthetic costs are still high. In this work, a new class of cost-effective small molecule dopant-free HTMs have been developed using readily available fluoranthene as the structural framework. The structure–property correlation of the fluoranthene-based HTMs was carefully investigated by tuning their structural geometry (linear vs. branched), connection between electron-donating and electron-withdrawing moieties (single bond vs. ethylene), and the substitution position of the methoxy side-groups (para- vs. meta-). As a result, the optimized molecule, FBA3, was demonstrated to serve as an efficient dopant-free HTM in a conventional PVSC to deliver an impressive power conversion efficiency of 19.27%, representing one of the best cost-effective dopant-free organic HTMs reported thus far.
Introduction
Since their debut in 2009,1 organic–inorganic hybrid perovskite solar cells (PVSCs) have made an impressive achievement and the record power conversion efficiency (PCE) has exceeded 23% recently.2 With such great performance, more recent research focus pertaining to PVSCs has been shifted to their long-term operational stability in order to fulfill further commercial applications,3,4 because perovskites have shown to possess poor stability under external stimuli such as light, moisture, oxygen, and heat. Besides using device encapsulation to address such a disadvantage, more fundamental approaches including composition engineering and interface engineering have been vigorously developed in recent years aiming to intrinsically enhance the materials' robustness.5–8It is worth noting that the employed charge-transporting layers (CTLs) also play a critical role in affecting the overall stability of the derived PVSCs.3–9 For example, the commonly used electron-transporting material (ETM), titanium dioxide (TiO2), has been shown to engender photodegradation of perovskite materials under ultraviolet light, thereby deteriorating the device performance.10 Meanwhile, the prevailing organic/polymeric hole-transporting materials (HTMs), such as 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD, Fig. 1) and poly(triarylamine) (PTAA), have also been demonstrated to be unfavorable for long-term operational device stability owing to the required ionic doping process.3,4,9 Although the doping indeed improves the charge transport properties of HTMs, the used dopants/additives, like bis(trifluoromethane)sulfonimide lithium salt (LiTFSI) and 4-tert-butylpyridine (tBP), are very hygroscopic and deliquescent and the oxidation process is generally associated with undesired ion migration/interactions. All these factors contribute to an inferior long-term operational stability of the derived PVSCs.11,12
 | ||
| Fig. 1 Chemical structures of spiro-OMeTAD and the studied fluoranthene-based HTMs. | ||
On one hand, using inorganic HTMs to replace the organic counterparts seems to be a feasible solution to address this shortage since they generally possess higher hole mobility and chemical stability than the latter.13 Nevertheless, most of the inorganic HTMs require harsh processing conditions that are incompatible with perovskites, which impedes the fabrication of efficient PVSCs especially for the n–i–p device configuration. Compared to inorganic materials, organic semiconductors generally possess milder processing conditions and have a much better flexibility in the molecular design.14–16 Therefore, their optoelectronic properties could be effectively tailored through synthetic manipulation. It is worth noting that some recent studies have developed HTM-free PVSCs based on a carbon electrode; however, all of them seem to compromise the resultant efficiency.17–19 As a result, the development of dopant-free HTMs is urgently desired to solve the stability issue caused by the doped HTMs. To date, only a few dopant-free HTMs have been reported to show comparable PCEs to doped spiro-OMeTAD,20,21 which reveals that effective molecular design strategies for dopant-free HTMs are still lacking.
The dopant-free HTMs exploited thus far, including both small molecules22–28 and polymers,29–34 are mainly based on the donor–acceptor (D–A) type design. For example, Sun et al.25,26 and Yang et al.27,28 have independently reported a series of A–D–A type small molecule dopant-free HTMs using two dimensional benzo[1,2-b:4,5-b′]dithiophene derivatives (2D-BDT) as the donor moiety to realize efficient PVSCs (PCE > 17%). Meanwhile, Park et al. prepared a new class of 2D-BDT-based D–A type conjugated polymers to serve as efficient dopant-free HTMs.29–31 On the other hand, the branched structure has also been successfully adopted in designing efficient dopant-free HTMs.35–39 By integrating these two molecular strategies, Nazeeruddin et al. developed a new class of star-shaped D–A type dopant-free HTMs using quinolizino acridine (FA) and triazatruxene as the core moiety to yield a high PCE of 19.03%.40,41 Note that, despite these achievements, all of those top-performing dopant-free HTMs developed so far are derived from complicated π-conjugated scaffolds requiring tedious synthesis and purification, such as BDT,25–31 quinolizino acridine (FA),40 triazatruxene41 and so on, making their synthetic costs too high to be used for widespread applications. Therefore, it motivates us to develop high-performance dopant-free HTMs with low synthetic complexity for scale-up to afford a good balance between efficiency, stability, and materials cost.
To find out the inherent regularity of designing low-cost but high-performance dopant-free HTMs, we herein develop a new class of fluoranthene-cored HTMs (Fig. 1) and systematically investigate their structure–property correlation. The fluoranthene unit is selected as the main structural framework due to its several promising advantages including low price, highly planar structure, easy functionality, and good thermal/electrochemical stability.42–44 Moreover, the unique central cyclopenta-fused ring endows the fluoranthene with an electron-deficient character,45 which thereby can be used as an electron-withdrawing core to construct various D–A type HTMs.46,47 Herein, we functionalize the fluoranthene moiety with methoxy-substituted triphenylamine (TPA) units on the 3-, 8- positions or 3-, 8-, 9- positions to form a linear and star-shaped structure, respectively. In addition, the connection between the TPA units and the central fluoranthene core (single bond vs. ethylene) as well as the substitution positions of methoxy groups in the TPA units (para- vs. meta-) are also tuned to elucidate how these structural variations affect HTM properties and the resultant performance in PVSCs.
Results and discussion
Notably, we have previously used fluoranthene as the core structure to synthesize BTF2 (Fig. 1), which exhibits a very low lab synthetic cost of 11.41$ per g.46 Unfortunately, owing to its high-lying highest occupied molecular orbital (HOMO) level (−4.8 eV) and low hole mobility of 2.13 × 10−5 cm2 V−1 s−1, BTF2 cannot serve as an efficient dopant-free HTM to realize high-performance PVSCs. On this basis, we further designed a new synthetic strategy based on the typical Diels–Alder reaction (Scheme S1†), by which electron-withdrawing cyano groups were introduced into the fluoranthene core that not only down-shifts the HOMO level (∼5.02 eV) but also improves the hole mobility (>10−4 cm2 V−1 s−1). Consequently, while serving as the dopant-free HTM in an n–i–p planar PVSC, the dicyanofluoranthene-cored BTF4 (Fig. 1) could deliver a much higher PCE of 18.03% than BTF2 (10.45%).46Note that replacing diphenylamine with TPA units could be an alternative approach to down-shift the HOMO level by weakening the material's donating ability.48 This molecular design has also been proven as an effective approach to increase the hole mobility as a result of enhanced π–π interactions.49,50 In this manner, we prepared FBA1 and FTA1 with linear and branched structures, respectively, wherein the para-methoxy substituted TPA units are connected with fluoranthene through a single bond. Meanwhile, the ethylene connection between TPA units and the fluoranthene core was also employed to afford linear FBA2 and branched FTA2, with the purpose of increasing the molecular planarity to improve the intermolecular π–π interactions. Besides the linking bridge, the meta-methoxy substituted TPA units were also utilized to prepare FBA3 in parallel with FBA2 consisting of the para-methoxy substituents. As compared to the para-methoxy substituted TPA, the meta-methoxy substituted TPA could further lower the HOMO level of the derived HTMs according to an inductive effect.51,52 Through these different structural modifications, we thus are able to conduct a systematic structure–property investigation to provide valuable insights for designing efficient dopant-free HTMs.
The synthetic route of FBA1–3 and FTA1–2 is shown in Scheme S2, while the experimental details are described in the ESI.† The synthesis is really straightforward, only involving two- or three-reaction steps under mild reaction conditions. As shown, by controlling the bromination of fluoranthene, 3,8-dibromofluorene (1) and 3,8,9-tribromofluoranthene (2) can be readily obtained, which then underwent a typical Pd-catalyzed Suzuki or Heck-coupling reaction with different functionalized TPA units (3, 4 and 7) to yield the desired linear HTMs (FBA1–3) and branched HTMs (FTA1–2), respectively. All these new molecules were well-characterized by spectroscopic methods, and gave satisfactory data (see the ESI for the details†). The material costs are estimated following the referenced method53 (see details in Tables S1–S5†). As shown in Table 1, most of them exhibit relatively low lab synthetic costs from 12.55$ per g (FBA1) to 86.1$ per g (FTA2), which are much lower than the commercial price of spiro-OMeTAD (170–475$ per g),54 attributed to the facile synthesis with simple purification processes. The highest synthetic cost of FTA2 is mainly due to its low isolated yield (15.7%) caused by a large loss during purification.
| HTMs | Costa ($ per g) | λ sol (nm) | λ fil (nm) | E g (eV) | HOMOd (eV) | T g (°C) | T d (°C) | Mobilityg (cm2 V−1 s−1) | τ 1 [ns] | τ 2 [ns] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Synthetic costs. b Absorption maxima of low-energy bands in dichloromethane solutions and as thin films. c Optical bandgaps calculated from solution absorption edges. d Measured from electrochemistry experiments, EHOMOs are calculated according to an equation of EHOMO = −(4.8 + Eox) eV. e Glass transition temperature detected by DSC analyses under nitrogen with a heating rate of 10 °C min−1. f The 5% weight loss temperature detected by the TGA analyses under nitrogen at a heating rate of 10 °C min−1. g Hole mobilities measured by the SCLC method. h τ 1 and τ2 correspond to the fitted fast and slow decay lifetime, respectively, based on time-resolved PL measurements of bi-layered MAPbIxCl3−x/non-doped HTM films. | ||||||||||
| BTF2 | 11.41 | 482 | 493 | 2.21 | −4.80 | — | 411 | 2.89 × 10−5 | 2.45 | 16.37 |
| FBA1 | 12.55 | 430 | 436 | 2.48 | −5.00 | 85 | 371 | 8.91 × 10−5 | 1.56 | 11.39 |
| FBA2 | 40.50 | 473 | 474 | 2.24 | −4.98 | 95 | 429 | 1.36 × 10−4 | 1.67 | 10.44 |
| FBA3 | 14.75 | 455 | 456 | 2.29 | −5.07 | 78 | 429 | 2.12 × 10−4 | 1.10 | 8.90 |
| FTA1 | 19.39 | 417 | 426 | 2.49 | −5.00 | 110 | 435 | 4.83 × 10−5 | 1.71 | 12.35 |
| FTA2 | 86.10 | 463 | 466 | 2.21 | −4.99 | 100 | 376 | 1.07 × 10−4 | 1.51 | 9.80 |
Fig. 2 illustrates the density functional theory (DFT)-optimized structures of these new fluoranthene-based HTMs. After introducing ethylene linkages, the torsional angles between the fluoranthene core and the attached phenyl groups can be significantly decreased. This is particularly pronounced for the angle setting on the 8 position of the fluoranthene core, from 32.9° (FBA1) to 2.4° (FBA2) and 3.0° (FBA3), respectively. These results thus suggest that the ethylene connection between the TPA units and fluoranthene core could make the structure more coplanar, which would be beneficial to hole transport. On the other hand, larger torsional angles on the 8 and 9 positions of fluoranthene are observed after forming the branched structure. As seen, from FBA1 to FTA1, such a torsional angle increases from 32.9° to 51.2°, while from FBA2 to FTA2, it increases from 2.4° to 23.1°. We speculate that this possibly arises from the steric interactions between the neighboring TPA units on the 8 and 9-positions of fluoranthene.
 | ||
| Fig. 2 The DFT-optimized geometrical structure of the studied HTMs. | ||
The thermal properties of these new fluoranthene-cored HTMs were measured using differential scanning calorimetry (DSC, Fig. 3a) and thermogravimetric analysis (TGA, Fig. S1a†), and the related data are summarized in Table 1. All the HTMs show high thermal stabilities, with the 5% weight loss temperatures (Tds) over 370 °C. For an ideal HTM, a high glass transition temperature (Tg) is often required to maintain a robust film morphology during device fabrication and operation.15 As seen, most of our prepared HTMs show high Tgs with the highest one of 110 °C, being comparable to the value of spiro-OMeTAD. It is interesting to note that there is a clear correlation between the molecular structure and the resulting Tgs. The Tgs were effectively increased from 85 °C (FBA1) and 95 °C (FBA2) to 110 °C (FTA1) and 100 °C (FTA2), respectively. This result indicates that the branched structure possesses better thermal stability. The linking bridge between the TPA units and fluoranthene core play a less important role in affecting the resultant Tg. FBA2 exhibits a higher Tg than FBA1 possibly due to its more planar structure. However, the meta-methoxy substitutions were found to decrease the Tg of FBA3 by ∼17 °C in comparison with FBA2, possibly due to the more asymmetric structure.
 | ||
| Fig. 3 (a) DSC curves under nitrogen with a heating rate of 10 °C min. (b) The absorption spectra in DCM solutions. (c) Corresponding energy levels relative to spiro-OMeTAD and perovskite. (d) Color variation of pristine fluoranthene-cored HTM films on a glass substrate before (top) and after (bottom) exposure to I2 vapor for 5 min. | ||
The absorption spectra of these HTMs in dichloromethane solutions and as thin films are presented in Fig. 3b and S1b,† respectively, with relevant data listed in Table 1. The lowest-energy absorption band around 400–600 nm is ascribed to the intramolecular charge transfer (ICT) interactions between the TPA units (D) and fluoranthene core (A), while the localized π–π* transition of TPA units corresponds to other higher energy absorption bands. As mentioned earlier, the diphenylamine unit has a stronger electron-donating ability than the TPA unit. Consequently, BTF2 shows the most red-shifted absorption band compared to others. Due to the extended conjugation of the ethylene linking bridge, the absorption maxima (λabs) of both FBA2 and FTA2 exhibit a red-shift of ∼40 nm when compared with FBA1 and FTA1. Interestingly, when the linear structures (FBA1/FBA2) are converted to the branched structures (FTA1/FTA2), there will be a blue-shift in λabs, possibly due to the more twisted structure of the latter that will make ICT interaction less efficient. On the other hand, FBA3 exhibits a blue-shifted λabs compared with FBA2, which confirms the weaker electron donating ability of the meta-methoxy substituted TPA unit relative to the para-methoxy substituted one as discussed earlier. The optical bandgap (Eopt, Table 1) was calculated from the absorption edges in DCM solutions, with the values from 2.21 eV to 2.49 eV. Moreover, all the materials' film absorption spectra exhibit similar features to their solution spectra albeit with a slightly red-shifted, broadened absorption band, indicating the absence of aggregation.
The energy levels of these fluoranthene-cored HTMs were characterized using electrochemical cyclic voltammetry (CV). Their corresponding CV curves measured from the DCM solutions are given in Fig. S3,† wherein most of them display reversible oxidative processes, except for FBA3. Their HOMO levels versus Fc/Fc+ were determined based on the average oxidation potentials or onset ones and are summarized in Table 1. Fig. 3c illustrates their energy-level alignment relative to spiro-OMeTAD and perovskite. As expected, by replacing the diphenylamine unit in BTF2 with the TPA unit, the resultant HOMO level of FBA1 can be efficiently decreased by ∼0.20 eV, which is comparable to that of BTF4 with cyano-substitutions on the fluoranthene core.46 While changing the linear structure to the branched structure or changing the single bond linking bridge to the ethylene linking bridge, the resultant HOMO levels show a negligible variation. It is interesting to note that FBA3 possesses a deeper-lying HOMO level than FBA2, which is more compatible with the valence band (VB) of the perovskite. This result is similar to those reported in the literature51,52 and could be attributed to the decreased electron-donating ability resulting from meta-methoxy substituents.
The HOMO and lowest unoccupied molecular orbital (LUMO) levels of these molecules were also simulated by DFT calculations, with results shown in Fig. S4.† The LUMOs of all HTMs are located mainly on the central fluoranthene core, while the relevant HOMOs are almost distributed over the entire π-conjugated system, particularly for linear molecules. These results reveal a substantial overlap between the LUMO and HOMO that will be beneficial to the generation of excitons and hole transport.55,56 In addition, the trend of the calculated HOMO levels is also in good accordance with those from CV measurements. By introducing ethylene linkage or forming branched structures, the calculated HOMOs of the corresponding HTMs can be increased accordingly, while meta-methoxy substituents are found to effectively reduce the calculated HOMO of FBA3 with a value of −4.86 eV.
The HTM's antioxidant capacity is also an important parameter for the long-term stability of its derived device, which is closely related to its HOMO level.57 To clarify this, we have tested the chemical stability of these fluoranthene-cored HTMs using I2 vapor, given that the HOMO levels of ∼5.0 eV for these compounds are close to the oxidation potential of I−/I3− and the reaction with I2 from perovskites might occur at the associated interface. As displayed in Fig. 3d, after exposure to I2 vapor for 5 min, a notable color change from light orange to brown was observed for the films of BTF2, FBA2, and FTA2 owing to their higher-lying HOMO levels. In contrast, FBA1 and FTA1 films only showed a slight color change. By considering that FBA2 and FTA2 only possess slightly higher HOMO levels (<0.02 eV) than FBA1 and FTA1, the ready oxidation with I2 of the former could be partially due to the existence of unstable ethylene groups. However, despite containing the ethylene groups, the film of FBA3 remained its original color, showing the best chemical stability among the studied HTMs. This result strongly underlines the importance of the deep-lying HOMO level for an ideal HTM.
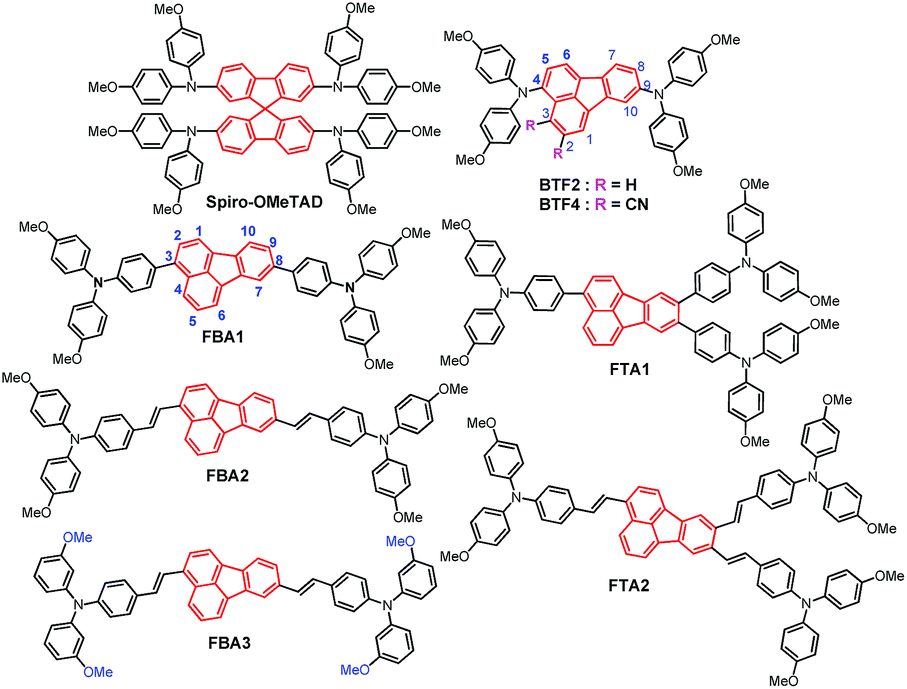
The hole-transporting properties of these fluoranthene-cored HTMs were evaluated by the space-charge-limited-current (SCLC) method without adding any dopants in a device configuration of ITO/PEDOT:PSS/HTMs/MoOx/Ag. The characterized curves are shown in Fig. 4a, with the estimated hole mobilities given in Table 1. As seen, the hole mobility (μh) of the studied compounds can be significantly improved by rational structural modifications. In addition to BTF2 and FTA1, all the compounds show respectable μh close to or beyond 10−4 cm2 V−1 s−1, exceeding the typical value of non-doped spiro-OMeTAD (∼2 × 10−5 cm2 V−1 s−1).46 This result indicates their promising potential to serve as dopant-free HTMs in n–i–p PVSCs. Note that, from BTF2 to FBA1 and FBA2, the μhs gradually increased from 2.91 × 10−5 cm2 V−1 s−1 to 8.91 × 10−5 cm2 V−1 s−1 and 1.36 × 10−4 cm2 V−1 s−1. Meanwhile, FTA2 also exhibits a much higher μh than FTA1. These observations therefore strongly manifest that both the replacement of diphenylamine with the TPA unit and the introduction of the ethylene linkage can effectively improve the hole-transporting capability of the derived HTMs, largely due to enhanced π–π interactions. Furthermore, the meta-methoxy substituted TPA units endow FBA3 with the highest μh among the studied HTMs (2.12 × 10−4 cm2 V−1 s−1), which is even comparable to that of doped spiro-OMeTAD (4.87 × 10−4 cm2 V−1 s−1, Fig. S6†). Unexpectedly, we noticed that both FTA1 and FTA2 exhibit decreased μhs compared to FBA1 and FBA2; this might be due to their weak intermolecular interactions in the solid-state caused by their three-dimensional branched structure. These data thus are consistent with those theoretical calculation results. As demonstrated in Fig. 2, the ethylene linking bridge makes the structure more coplanar to enhance intermolecular interactions, while the steric effect derived from the substituents on the neighboring 8 and 9 positions of the fluoranthene core potentially reduces the intermolecular interactions of the branched materials (FTA1 and FTA2).
 | ||
| Fig. 4 (a) The hole injection characteristics measured by the SCLC method based on a device structure of ITO/PEDOT:PSS/HTM/MoOx(10 nm)/Ag. (b) Steady PL spectra and (c) time-resolved PL spectra of bare perovskite (PV) films and bi-layered perovskite films capped with different fluoranthene-cored HTMs. | ||
To assess the hole transfer efficiency between the studied compounds and perovskite, the steady-state photoluminescence (PL) spectra of the bi-layered MAPbIxCl3−x/non-doped HTM films were examined as shown in Fig. 4b. As shown, the PL of perovskites at ∼780 nm was effectively quenched when capped with different HTM layers. This result suggests that the designed HTMs are capable of extracting holes from the perovskite even in the absence of dopants. Notably, the quenching efficiency follows a trend of FBA3 ≈ FBA2 > FTA2 > FBA1 > FTA1 > BTF2, which is highly consistent with mobility. Time-resolved PL measurements were also conducted to study their hole-extraction efficiency. As shown in Fig. 4c and Table 1, after introducing HTM layers, the decay time of the corresponding bi-layered films was significantly shortened in comparison with that of the bare perovskite film (∼49 ns). Besides, the declining trend of the decay time is quite similar to that of the PL quenching efficiency, and FBA3 shows the shortest decay time of 8.9 ns. All these results confirm an efficient hole transfer from the perovskite to the HTMs.
To realize an efficient n–i–p PVSC, besides respectable hole-transporting capability, the HTMs need to form homogeneous films atop the perovskite layer since a high quality HTM layer can effectively reduce the interfacial charge recombination loss and prevent the contact between the perovskite layer and metal electrode.9,14–16 To confirm their thin-film formation capabilities, atomic force microscopy (AFM) images of the prepared fluoranthene-cored HTM films atop the perovskite layer were investigated as shown in Fig. 5. In principle, the morphology is highly related to the molecular structure of a molecule, in addition to solubility and processability.58 As seen, from BTF2 to FBA1 and FBA2, the film quality was improved accordingly, accompanied by a reduction of pin holes. On the other hand, the films of FBA3, FTA1, and FTA2 showed a more uniform morphology. For FTA1 and FTA2, it can be easily rationalized by their more amorphous state confirmed by their high Tgs and weak intermolecular interactions. Besides, their branched structure might also contribute to the improved film-forming ability. As for FBA3, the improved film morphology may be attributed to the fact that in comparison with FBA2, the molecular symmetry can be further destroyed due to meta-methoxy substitutions on the TPA units.59
 | ||
| Fig. 5 AFM images of fluoranthene-cored HTM films atop perovskite layers. | ||
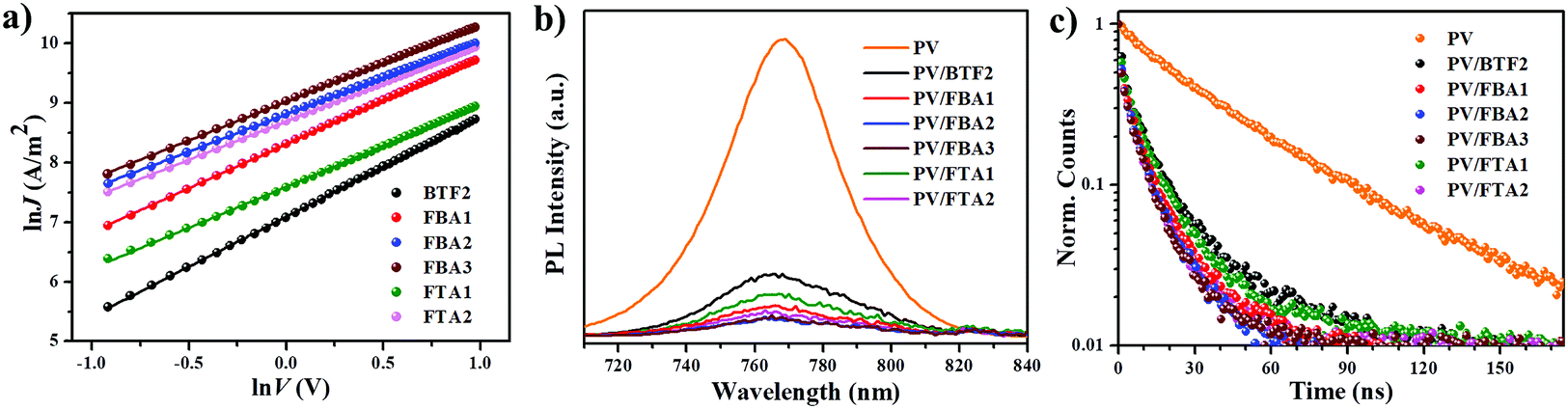
To test the effectiveness of these prepared fluoranthene derivatives as dopant-free HTMs, a typical n–i–p planar PVSC was fabricated in a device configuration of ITO/C60/perovskite/HTL/MoO3/Ag. The device fabrication details are described in the ESI.† Shown in Fig. 6a is the cross-sectional scanning electron microscopy (SEM) image of the FBA3-based device, in which the stratified device configuration can be seen obviously. MAPbIxCl3−x (MA: CH3NH3+) was selected as the photoactive layer instead of (FAPbI3)0.85(MAPbBr3)0.15 by considering that its VB of 5.4 eV matches with the HOMO levels of our designed HTMs much better. For comparison, a control device using regularly doped spiro-OMeTAD as the HTM was also fabricated with the same fabrication procedures.
 | ||
| Fig. 6 (a) Cross-sectional SEM image of the complete PVSC device. (b) J–V curves of the champion PVSCs with different dopant-free HTMs and doped spiro-OMeTAD. (c) Histograms of photovoltaic parameters. Environmental stability of PVSCs based on dopant-free FBA2, FBA3 and FTA2 and doped spiro-OMeTAD tested under simulated sunlight under nitrogen (d) or in ambient air with a humidity of 20–30% (e). | ||
The current density–voltage (J–V) curves of the champion PVSCs measured under AM 1.5 G irradiation at 100 mW cm−2 are shown in Fig. 6b and S7,† and the related photovoltaic parameters are summarized in Table 2. The processing solution concentration of each HTM was carefully optimized to achieve the best performance (Fig. S8†). Based on this new device structure, BTF2 yielded a higher PCE of 13.44% than the previously reported value of 10.45%. Encouragingly, the PVSCs using the newly prepared HTMs all show improved photovoltaic performance compared to the control BTF2 device. This thus reveals the success of rational molecular modifications in our design of dopant-free HTMs. Impressively, the best performing PVSC with a promising PCE of 19.27% was derived from FBA3, outperforming the control devices (17.57%) using the doped spiro-OMeTAD HTM. This result is also among the best for dopant-free HTMs reported thus far.20–41 In addition, FBA2 and FTA2 also delivered respectable PCEs of 18.70% and 17.73%, respectively. The stabilized PCE and photocurrent of the champion PVSCs using these dopant-free HTMs near the maximum power point were also tested as shown in Fig. S9.† These results clearly manifest the high reliability of our J–V curves and the absence of current hysteresis, combined with results from those J–V curves in forward and reverse scanning directions (Fig. S7†). Fig. S10† displays their corresponding incident photon-to-electron conversion efficiency (IPCE) spectra, in which the integrated short-circuit current density (Jsc) values show good consistency with the values obtained in the J–V measurements, affirming the good reliability of these device results.
| HTMs | V oc (V) | J sc (mA cm−2) | FF | PCE (%) |
|---|---|---|---|---|
| BTF2 | 0.96(0.96 ± 0.02) | 20.19(20.01 ± 0.32) | 0.693(0.667 ± 0.021) | 13.44(12.76 ± 0.52) |
| FBA1 | 1.05(1.04 ± 0.01) | 21.57(21.36 ± 0.29) | 0.742(0.729 ± 0.014) | 16.80(16.24 ± 0.46) |
| FBA2 | 1.06(1.06 ± 0.01) | 22.32(22.03 ± 0.33) | 0.790(0.773 ± 0.019) | 18.70(17.97 ± 0.55) |
| FBA3 | 1.09(1.08 ± 0.01) | 22.12(21.88 ± 0.32) | 0.799(0.781 ± 0.019) | 19.27(18.46 ± 0.59) |
| FTA1 | 1.01(1.01 ± 0.01) | 20.76(20.52 ± 0.30) | 0.723(0.703 ± 0.017) | 15.15(14.52 ± 0.47) |
| FTA2 | 1.03(1.02 ± 0.01) | 22.04(21.83 ± 0.33) | 0.781(0.765 ± 0.015) | 17.73(17.12 ± 0.57) |
| Doped spiro-OMeTAD | 1.07(1.06 ± 0.01) | 21.24(20.95 ± 0.37) | 0.773(0.758 ± 0.019) | 17.57(16.83 ± 0.65) |
The histograms of device parameters of PVSCs are shown in Fig. 6c. In general, the resulting Vocs are closely related to the HOMO levels of HTMs.14–16 As discussed earlier, by replacing diphenylamine with the TPA units, the resulting HOMO levels can be downshifted to be more compatible with the VB of perovskites. This thereby enables significantly enhanced Vocs from 5.2% improvement (FTA1) to 13.5% improvement (FBA3). As seen, despite exhibiting a high PCE of 18.7%, the FBA2-based PVSC shows a limited Voc value of 1.06 V. To further down-shift the HOMO level and thus to increase the resultant Voc, a slight structural regulation on FBA2 was then carried out by changing the para- into meta- methoxy-substitutions. As shown in Table 1, the derived FBA3 shows a deeper-lying HOMO level than FBA2, thereby enabling an increased Voc value up to 1.09 V without sacrificing other parameters to deliver the highest PCE of 19.27%. Our results therefore highlight the importance of the relative energy level between the perovskite layer and HTMs for achieving high Voc.
The improvement in the Jsc and fill factor (FF) has also been demonstrated by fine-tuning the molecular structure. From BTF2 to FBA1 and FBA2, the Jsc value was enhanced from 20.19 mA cm−2 to 21.57 mA cm−2 (6.8% improvement) and 22.32 mA cm−2 (10.5% improvement), respectively, while the FF value was enhanced from 0.693 to 0.742 (7.1% improvement) and 0.79 (11.4% improvement), respectively. This certainly originates from their gradually improved hole mobility/hole extraction ability and better film morphology as discussed earlier. After forming a branched structure, the corresponding hole mobility/hole extraction ability is reduced, and thus the obtained Jscs and FFs are lower as compared to those of the linear HTMs. Nonetheless, HTMs consisting of ethylene linking bridges show better performance than other HTMs, indicating the effectiveness of structural design towards dopant-free HTMs.
We finally compared the stability of PVSCs derived from dopant-free HTMs (FBA2, FBA3, and FTA2) and doped spiro-OMeTAD under the same aging conditions to check whether the removal of ionic dopants can improve device stability. First, all the un-encapsulated devices were tested under nitrogen to exclude the influence of external moisture and oxygen. Therefore, the effect of dopants on device stability can be the study focus. As shown in Fig. 6d, after exposure to the simulated sunlight for 500 h, over 90% of the initial PCEs can be retained for all the dopant-free devices, while the PCE of doped spiro-OMeTAD decayed to 75% of its initial value. Furthermore, we also checked the effect of dopant-free HTMs with different Tgs on the thermal stability of the fabricated PVSCs under nitrogen. As shown in Fig. S11,† the dopant-free device based on FTA2 with a Tg of 100 °C clearly shows an enhanced thermal stability compared to that of FBA3 with a Tg of 78 °C; when successively heating the devices at 80 °C, 90 °C, 100 °C and 110 °C for 20 min, the final PCE of the FTA2-based device can retain 94.9% of the original PCE, while that of the FBA3-based device is only 91.9%. Moreover, we found that both dopant-free devices show much better thermal stability than the doped spiro-OMeTAD control device during heating treatments, indicating that the removal of dopants/additives can also effectively enhance the thermal stability of resulting PVSCs given the fact that the t-BP additive is a low boiling point solvent.
We further evaluated the stability of these un-encapsulated devices in ambient air with a relative humidity of 20–30%. As shown in Fig. 6e, all the tested devices showed a faster degradation than the case where they were kept under nitrogen. After being stored for 180 h, the PCEs of the devices using dopant-free HTMs and doped spiro-OMeTAD were degraded by 20% and 43%, respectively. Furthermore we have tested the device stability in ambient air with a high humidity of 80% as shown in Fig. S12.† After being stored for 144 h, the PCE of the doped spiro-OMeTAD device decayed to only 7% of its original PCE, while the dopant-free devices can still retain 33–49% of their original PCEs after being stored for 168 h. These observations therefore indicate that the removal of dopants indeed improves the operational stability of PVSCs. Nonetheless, we have to note that our device stability against moisture is still imperfect. This is because the device stability is not only related to the dopants/additives in the HTMs, but also highly dependent on the HTM film quality atop the perovskite layer, film morphology stability, and the interface between the perovskite and HTM.60 Actually, the dopant-free devices based on FBA3 and FTA2 showed an enhanced stability under high humidity conditions compared to that of FBA2, which, we think, could be due to their better film quality atop the perovskite layer as evidenced by AFM measurements (Fig. 5).
Conclusions
In summary, we developed a new class of small molecule dopant-free HTMs using readily available fluoranthene as the structural framework. By tuning the structural geometry, the connection between the donor and acceptor and the substitution position of methoxy groups, the material structure was carefully regulated and optimized with the purpose of studying their impacts on molecular packing behaviors, thermal properties, photophysical properties and device performance. Our results demonstrate the following important points:(1) Replacing diphenylamine with triphenylamine as the capping unit of D–A type HTMs can effectively down-shift the HOMO levels and increase the hole mobilities of derived HTMs.
(2) Introducing ethylene as the connecting π-bridge can marginally raise the HOMO levels but effectively increase the hole mobilities of derived HTMs due to enhanced π–π interactions. However, it might result in a poor antioxidant capacity.
(3) In comparison with the linear structure, the branched materials can considerably increase the Tgs by over 20 °C and enable better film quality atop the perovskite layer. However, it might result in lower hole mobilities.
(4) meta-Methoxy substitution can be used as an effective structural design strategy to down-shift the HOMO levels and improve the hole mobilities of derived HTMs; however, this design would decrease the thermal stability.
Based on this systematic structure–property study, the inherent regularity governing the structure of dopant-free HTMs has been demonstrated preliminarily. Our designed fluoranthene-cored molecules can work as efficient dopant-free HTMs to realize high-performance n–i–p planar PVSCs associated with improved device stability. In particular, FBA3 with a low lab synthetic cost of 14.75$ per g can exhibit an impressive PCE of 19.27%, representing one of the best cost-effective dopant-free organic HTMs reported thus far.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the National Science Foundation of China (Grant No. 21704030, 51703183, 51873160). We would also like to thank the Analytical and Testing Center of HUST for access to their facilities.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
.
- https://www.nrel.gov/pv/assets/images/efficiency-chart.png .
- L. Meng, J. You and Y. Yang, Nat. Commun., 2018, 9, 5265 CrossRef PubMed
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418–3451 CrossRef CAS PubMed
- N. H. Tiep, Z. Ku and H. J. Fan, Adv. Energy Mater., 2016, 6, 1501420 CrossRef
- A. Sharenko and M. F. Toney, J. Am. Chem. Soc., 2016, 138, 463–470 CrossRef CAS PubMed
- Q. Fu, X. Tang, B. Huang, T. Hu, L. Tan, L. Chen and Y. Chen, Adv. Sci., 2018, 5, 1700387 CrossRef PubMed
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed
- B. Roose, Q. Wang and A. Abate, Adv. Energy Mater., 2019, 9, 1803140 Search PubMed
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 CrossRef PubMed
- J. P. Bastos, U. W. Paetzold, R. Gehlhaar, W. Qiu, D. Cheyns, S. Surana, V. Spampinato, T. Aernouts and J. Poortmans, Adv. Energy Mater., 2018, 8, 1800554 CrossRef
- S. Wang, Z. Huang, X. Wang, Y. Li, M. Günther, S. Valenzuela, P. Parikh, A. Cabreros, W. Xiong and Y. S. Meng, J. Am. Chem. Soc., 2018, 140, 16720–16730 CrossRef CAS PubMed
- Z. Yu and L. Sun, Small Methods, 2018, 2, 1700280 CrossRef
- Z. Yu and L. Sun, Adv. Energy Mater., 2015, 5, 1500213 CrossRef
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef PubMed
- J. Urieta-Mora, I. García-Benito, A. Molina-Ontoria and N. Martín, Chem. Soc. Rev., 2018, 47, 8541–8571 RSC
- A. Mei, X. Li, L. Liu, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Grätzel and H. Han, Science, 2014, 345, 295–298 CrossRef CAS PubMed
- X. Meng, J. Zhou, J. Hou, X. Tao, S. H. Cheung, S. K. So and S. Yang, Adv. Mater., 2018, 30, 1706975 CrossRef PubMed
- Z. Wu, Z. Liu, Z. Hu, Z. Hawash, L. Qiu, Y. Jiang, L. K. Ono and Y. Qi, Adv. Mater., 2019, 31, 1804284 CrossRef PubMed
- X. Sun, D. Zhao and Z. a. Li, Chin. Chem. Lett., 2018, 29, 219–231 CrossRef CAS
- W. Zhou, Z. Wen and P. Gao, Adv. Energy Mater., 2018, 8, 1702512 CrossRef
- J. H. Heo, S. Park, S. H. Im and H. J. Son, ACS Appl. Mater. Interfaces, 2017, 9, 39511–39518 CrossRef CAS PubMed
- J. H. Yun, S. Park, J. H. Heo, H.-S. Lee, S. Yoon, J. Kang, S. H. Im, H. Kim, W. Lee, B. Kim, M. J. Ko, D. S. Chung and H. J. Son, Chem. Sci., 2016, 7, 6649–6661 RSC
- P. Qin, H. Kast, M. K. Nazeeruddin, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, Energy Environ. Sci., 2014, 7, 2981–2985 RSC
- M. Cheng, K. Aitola, C. Chen, F. Zhang, P. Liu, K. Sveinbjörnsson, Y. Hua, L. Kloo, G. Boschloo and L. Sun, Nano Energy, 2016, 30, 387–397 CrossRef CAS
- C. Chen, M. Cheng, P. Liu, J. Gao, L. Kloo and L. Sun, Nano Energy, 2016, 23, 40–49 CrossRef CAS
- Y. Liu, Z. Hong, Q. Chen, H. Chen, W. H. Chang, Y. M. Yang, T. B. Song and Y. Yang, Adv. Mater., 2016, 28, 440–446 CrossRef CAS PubMed
- Y. Liu, Q. Chen, H.-S. Duan, H. Zhou, Y. Yang, H. Chen, S. Luo, T.-B. Song, L. Dou, Z. Hong and Y. Yang, J. Mater. Chem. A, 2015, 3, 11940–11947 RSC
- G.-W. Kim, G. Kang, J. Kim, G.-Y. Lee, H. I. Kim, L. Pyeon, J. Lee and T. Park, Energy Environ. Sci., 2016, 9, 2326–2333 RSC
- J. Lee, M. M. Byranvand, G. Kang, S. Y. Son, S. Song, G. W. Kim and T. Park, J. Am. Chem. Soc., 2017, 139, 12175–12181 CrossRef CAS PubMed
- G.-W. Kim, J. Lee, G. Kang, T. Kim and T. Park, Adv. Energy Mater., 2018, 8, 1701935 CrossRef
- Y. S. Kwon, J. Lim, H.-J. Yun, Y.-H. Kim and T. Park, Energy Environ. Sci., 2014, 7, 1454–1460 RSC
- H.-C. Liao, T. L. D. Tam, P. Guo, Y. Wu, E. F. Manley, W. Huang, N. Zhou, C. M. M. Soe, B. Wang, M. R. Wasielewski, L. X. Chen, M. G. Kanatzidis, A. Facchetti, R. P. H. Chang and T. J. Marks, Adv. Energy Mater., 2016, 6, 1600502 CrossRef
- K. Kranthiraja, K. Gunasekar, H. Kim, A. N. Cho, N. G. Park, S. Kim, B. J. Kim, R. Nishikubo, A. Saeki, M. Song and S. H. Jin, Adv. Mater., 2017, 29, 1700183 CrossRef PubMed
- M. Franckevičius, A. Mishra, F. Kreuzer, J. Luo, S. M. Zakeeruddin and M. Grätzel, Mater. Horiz., 2015, 2, 613–618 RSC
- F. Zhang, C. Yi, P. Wei, X. Bi, J. Luo, G. Jacopin, S. Wang, X. Li, Y. Xiao, S. M. Zakeeruddin and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600401 CrossRef
- C. Huang, W. Fu, C. Z. Li, Z. Zhang, W. Qiu, M. Shi, P. Heremans, A. K. Jen and H. Chen, J. Am. Chem. Soc., 2016, 138, 2528–2531 CrossRef CAS PubMed
- C. Yin, J. Lu, Y. Xu, Y. Yun, K. Wang, J. Li, L. Jiang, J. Sun, A. D. Scully, F. Huang, J. Zhong, J. Wang, Y.-B. Cheng, T. Qin and W. Huang, Adv. Energy Mater., 2018, 8, 1800538 CrossRef
- C. Shen, Y. Wu, H. Zhang, E. Li, W. Zhang, X. Xu, W. Wu, H. Tian and W. H. Zhu, Angew. Chem., Int. Ed., 2019, 58, 3784–3789 CrossRef CAS PubMed
- S. Paek, P. Qin, Y. Lee, K. T. Cho, P. Gao, G. Grancini, E. Oveisi, P. Gratia, K. Rakstys, S. A. Al-Muhtaseb, C. Ludwig, J. Ko and M. K. Nazeeruddin, Adv. Mater., 2017, 29, 1606555 CrossRef PubMed
- K. Rakstys, S. Paek, P. Gao, P. Gratia, T. Marszalek, G. Grancini, K. T. Cho, K. Genevicius, V. Jankauskas, W. Pisula and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 7811–7815 RSC
- S. Kumar and S. Patil, J. Phys. Chem. C, 2015, 119, 19297–19304 CrossRef CAS
- Y. Q. Zheng, Y. Z. Dai, Y. Zhou, J. Y. Wang and J. Pei, Chem. Commun., 2014, 50, 1591–1594 RSC
- N. Kapoor and K. R. J. Thomas, New J. Chem., 2010, 34, 2739–2748 RSC
- A. Palmaerts, M. V. Haren, L. Lutsen, T. J. Cleij and D. Vanderzande, Macromolecules, 2006, 39, 2438–2440 CrossRef CAS
- X. Sun, Q. Xue, Z. Zhu, Q. Xiao, K. Jiang, H. L. Yip, H. Yan and Z. Li, Chem. Sci., 2018, 9, 2698–2704 RSC
- Y. Li, K. R. Scheel, R. G. Clevenger, W. Shou, H. Pan, K. V. Kilway and Z. Peng, Adv. Energy Mater., 2018, 8, 1801248 CrossRef
- K. Liu, Y. Yao, J. Wang, L. Zhu, M. Sun, B. Ren, L. Xie, Y. Luo, Q. Meng and X. Zhan, Mater. Chem. Front., 2017, 1, 100–110 RSC
- A. Molina-Ontoria, I. Zimmermann, I. Garcia-Benito, P. Gratia, C. Roldan-Carmona, S. Aghazada, M. Graetzel, M. K. Nazeeruddin and N. Martin, Angew. Chem., Int. Ed., 2016, 55, 6270–6274 CrossRef CAS PubMed
- R. Grisorio, B. Roose, S. Colella, A. Listorti, G. P. Suranna and A. Abate, ACS Energy Lett., 2017, 2, 1029–1034 CrossRef
- N. J. Jeon, H. G. Lee, Y. C. Kim, J. Seo, J. H. Noh, J. Lee and S. I. Seok, J. Am. Chem. Soc., 2014, 136, 7837–7840 CrossRef CAS PubMed
- R. Azmi, S. Y. Nam, S. Sinaga, Z. A. Akbar, C.-L. Lee, S. C. Yoon, I. H. Jung and S.-Y. Jang, Nano Energy, 2018, 44, 191–198 CrossRef CAS
- T. P. Osedach, T. L. Andrew and V. Bulović, Energy Environ. Sci., 2013, 6, 711–718 RSC
- H. D. Pham, T. T. Do, J. Kim, C. Charbonneau, S. Manzhos, K. Feron, W. C. Tsoi, J. R. Durrant, S. M. Jain and P. Sonar, Adv. Energy Mater., 2018, 8, 1703007 CrossRef
- A. Krishna, D. Sabba, H. Li, J. Yin, P. P. Boix, C. Soci, S. G. Mhaisalkar and A. C. Grimsdale, Chem. Sci., 2014, 5, 2702–2709 RSC
- F. Zhang, S. Wang, H. Zhu, X. Liu, H. Liu, X. Li, Y. Xiao, S. M. Zakeeruddin and M. Grätzel, ACS Energy Lett., 2018, 3, 1145–1152 CrossRef CAS
- Y. Hou, X. Du, S. Scheiner, D. P. McMeekin, Z. Wang, N. Li, M. S. Killian, H. Chen, M. Richter, I. Levchuk, N. Schrenker, E. Spiecker, T. Stubhan, N. A. Luechinger, A. Hirsch, P. Schmuki, H.-P. Steinrück, R. H. Fink, M. Halik, H. J. Snaith and C. J. Brabec, Science, 2017, 358, 1192–1197 CrossRef CAS PubMed
- D. E. M. Rojas, K. T. Cho, Y. Zhang, M. Urbani, N. Tabet, G. de la Torre, M. K. Nazeeruddin and T. Torres, Adv. Energy Mater., 2018, 8, 1800681 CrossRef
- X. Wang, J. Zhang, S. Yu, W. Yu, P. Fu, X. Liu, D. Tu, X. Guo and C. Li, Angew. Chem., Int. Ed., 2018, 57, 12529–12533 CrossRef CAS PubMed
- Y. Wang, Y. Yue, X. Yang and L. Han, Adv. Energy Mater., 2018, 8, 1800249 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01697j |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2019 |