
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of ligand encapsulation on cobalt-59 chemical-shift thermometry†
Tyler M.
Ozvat‡
 ,
Manuel E.
Peña‡
and
Joseph M.
Zadrozny
*
,
Manuel E.
Peña‡
and
Joseph M.
Zadrozny
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: joe.zadrozny@colostate.edu
First published on 5th June 2019
Abstract
Thermometry via magnetic resonance imaging (MRI) would provide a powerful noninvasive window into physiological temperature management. Cobalt-59 nuclear spins demonstrate exceptional temperature dependence of their NMR chemical shifts, yet the insight to control this dependence via molecular design is lacking. We present the first systematic evidence that encapsulation of this spin system amplifies the temperature sensitivity. We tested the temperature dependence of the 59Co chemical shift (Δδ/ΔT) in a series of five low-spin cobalt(III) complexes as a function of increasing encapsulation within the 1st coordination sphere. This study spans from [Co(NH3)6]Cl3, with no interligand connectivity, to a fully encapsulated dinitrosarcophagine (diNOsar) complex, [Co(diNOsar)]Cl3. We discovered Δδ/ΔT values that span from 1.44(2) ppm °C−1 in [Co(NH3)6]Cl3 to 2.04(2) ppm °C−1 in [Co(diNOsar)]Cl3, the latter among the highest for a molecular complex. The data herein suggest that designing 59Co NMR thermometers toward high chemical stability can be coincident with high Δδ/ΔT. To better understand this phenomenon, variable-temperature UV-Vis, 59Co NMR relaxation, Raman spectroscopic, and variable-solvent investigations were performed. Data from these measurements highlight an unexpected impact of encapsulation – an increasingly dynamic and flexible inner coordination sphere. These results comprise the first systematic studies to reveal insight into the molecular factors that govern Δδ/ΔT and provide the first evidence of 59Co nuclear-spin control via vibrational means.
Introduction
The structural flexibilities of metal complexes are key design principles for applications in the areas of reactivity,1,2 medicine,3 photophysical properties,4 and magnetic information storage.5 Flexibility engenders stimuli-dependent changes in the coordination geometry of a metal, hence impacting d-orbital energies and any properties stemming from electronic structure.6 Thus, the control of flexibility is potentially a powerful way for targeting applications for metal complexes. One such application is biomedical thermometry by magnetic resonance imaging (MRI),7,8 where the temperature-dependent structure of a flexible complex induces highly temperature-dependent spin-Hamiltonian parameters or relaxation times. If this variation could be harnessed to develop an imaging technique, such an application would circumvent many of the challenges associated with invasive thermometry, e.g. the point-like nature of the measurement.One promising system for such thermometry by magnetic resonance is the cobalt-59 nucleus in low-spin cobalt(III) complexes. This NMR-active nucleus is 100% naturally abundant, I = 7/2, and has a receptivity of ca. 30% that of 1H. Furthermore, the nucleus displays a wide reported chemical shift (δ) window (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) as a result of a paramagnetic contribution to δ that is directly tied to the ligand field splitting, Δo.9–11 For complexes that contain cobalt-59 nuclei, changes in solution structure, such as lengthening Co–ligand bonds, can impact Δo, imparting changes in δ, and providing a mechanism for thermometry. In principle, then, the 59Co chemical shift could be used to spatially map temperature through a technique known as chemical shift imaging.12–14 Initial studies reveal sensitivities (Δδ/ΔT) on the order of 1–3 ppm °C−1,15–20 order-of-magnitude upgrades to the possibilities for conventional 1H NMR thermometry.21,22 Hence, these species may be useful to develop as new probes for chemical-shift imaging of temperature.12 However, fundamental insight about the factors to govern that sensitivity is lacking (this manuscript explores one factor – encapsulation – as depicted in Fig. 1). As a result, design principles for enhancing Δδ/ΔT values are absent, and the true potential of cobalt-59 NMR thermometers for MRI remains unrealized.
000 ppm) as a result of a paramagnetic contribution to δ that is directly tied to the ligand field splitting, Δo.9–11 For complexes that contain cobalt-59 nuclei, changes in solution structure, such as lengthening Co–ligand bonds, can impact Δo, imparting changes in δ, and providing a mechanism for thermometry. In principle, then, the 59Co chemical shift could be used to spatially map temperature through a technique known as chemical shift imaging.12–14 Initial studies reveal sensitivities (Δδ/ΔT) on the order of 1–3 ppm °C−1,15–20 order-of-magnitude upgrades to the possibilities for conventional 1H NMR thermometry.21,22 Hence, these species may be useful to develop as new probes for chemical-shift imaging of temperature.12 However, fundamental insight about the factors to govern that sensitivity is lacking (this manuscript explores one factor – encapsulation – as depicted in Fig. 1). As a result, design principles for enhancing Δδ/ΔT values are absent, and the true potential of cobalt-59 NMR thermometers for MRI remains unrealized.
 | ||
| Fig. 1 Tested design parameter in this manuscript. Molecular structures of (left) [Co(diNOsar)]3+ (diNOsar = dinitrosarcophagine) and (right) [Co(NH3)6]3+ are taken from crystal structures in ref. 23 and 24. Anions and hydrogens omitted for clarity where necessary. Purple, blue, red, grey, and light grey spheres correspond to cobalt, nitrogen, oxygen, carbon, and hydrogen atoms, respectively. | ||
This manuscript details the first systematic exploration of the molecular factors that govern Δδ/ΔT in a family of cobalt-59 NMR thermometers (Fig. 1 and 2). As a first step, we sought to explore the role of ligand encapsulation on the temperature sensitivity of the ligand field and cobalt-59 NMR properties. Encapsulation is known to afford enhanced stability for metal complexes via the chelate and related macrocyclic effects.25 Such stability is an important property for any imaging agent, as release of the metal can both induce toxicity and deactivate the magnetic species being used as a sensor. At the same time, a rigid, encapsulated ion can be readily envisioned to lack the flexibility needed for thermometry via structural change. Hence, encapsulation, while affording significant chemical stability, might simultaneously subdue the ability to sense temperature via cobalt-59 NMR.
 | ||
| Fig. 2 Bond-line representations of complexes 1–5, the systems studied in this manuscript. Anions and hydrogens bound to carbons are omitted for clarity. | ||
To test this hypothesis, we investigated Δδ/ΔT for the 59Co nuclei of the low-spin Co(III) complexes (Fig. 2) [Co(NH3)6]Cl3 (1),26 [Co(en)3]Cl3 (2, en = ethylenediamine),27 [Co(pn)3]Cl3 (3, pn = 1,3-diaminopropane),28 [Co(tame)2]Cl3 (4, tame = triaminomethylethane),29 [Co(diNOsar)]Cl3 (5, = dinitrosarcophagine),23 and K3[Co(CN)6], the 59Co NMR standard. This series of complexes was selected to enable an investigation of both molecular and electronic structure on Δδ/ΔT. First, we hypothesized that the increasing connectivity between the nitrogen donor atoms in 1–5 would engender an increasingly rigid coordination environment and, hence, suppress Δδ/ΔT. Thus, we expected that sensitivity to temperature would be compromised in favor of chemical stability. Indeed, the fully-encapsulating sarcophagine scaffold23,25,30 will only surrender its NMR-active Co(III) ion under harsh conditions – heating in concentrated cyanide solution or acidic media.31,32 The second investigation enabled by this set of complexes is the test of whether Δδ/ΔT directly correlates with Δo. The 59Co chemical shift is proportional to 1/Δo,10 hence, δ should be more sensitive to tiny fluctuations in Δo at lower Δo.10 These studies are the first to reveal three key facts about Δδ/ΔT. Firstly, in contrast to our expectations, encapsulation enhances Δδ/ΔT. That is – the “rigid” ligand frameworks in 5 and 4 induce a stronger temperature-dependence in Δo (and δ) than the less-encapsulated species 1–3. Indeed, variable-temperature UV-Vis and 59Co spin-lattice relaxation studies indicate that encapsulation counterintuitively supports higher temperature dependence in the coordination geometry. Second, our studies show that Δo alone does not correlate to the magnitude of Δδ/ΔT. Finally, third, Raman spectroscopy studies suggest molecular vibrational lifetimes – prolonged by high interconnectivity among donor atoms – are important factors governing Δδ/ΔT. Together, the data highlight a new implication for rigidity in molecular magnetism.
Results and discussion
Understanding the temperature sensitivity of the chemical shift requires first establishing the electronic structures of the cobalt(III) ions in 1–5. UV-Vis electronic absorption spectra of compounds 1–5 and K3[Co(CN)6] in H2O reproduce reported results for the individual complexes (Fig. 3), wherein the lowest energy peak indicates the 1A1g → 1T1g transition and the higher energy peak indicates the 1A1g → 1T2g transition.10,23 The energies of these two peaks and a Tanabe-Sugano diagram permit quantitation of Δo, which increases from 3 (22376 cm−1) to 5 (22754 cm−1) and 1 (23018 cm−1) to 4 (23276 cm−1) to 2 (23321 cm−1) (Table S1†). These values are consistent with literature values for 1–5 and stand in contrast to the strong ligand field of K3[Co(CN)6] that engenders a Δo of 38000 cm−1.3359Co resonant frequencies were observed for 1–5 over the range of 6800 to 8400 ppm (referenced to K3[Co(CN)6]). According to the 59Co chemical shifts, the magnitude of Δo increases in the order 3 < 1 < 4 < 2 < 5 < K3[Co(CN)6]. This order is at odds with the trend obtained from electronic absorption spectroscopy measurements (see Fig. S1†). However, reported correlations between UV-Vis peak position and δ are only approximate, not quantitative.9,10 Nevertheless, these measurements provide (i) two points of reference to test for a correlation between Δδ/ΔT and Δo, and (ii) the location of the 59Co NMR resonances for variable-temperature analyses.
 | ||
| Fig. 3 Characterization of Co(III) electronic structure in 1–5. Top: electronic absorption (UV-Vis) spectra for 1–5 and K3[Co(CN)6] in H2O at room temperature. The lower-energy peak is the 1A1g to 1T1g transition while the higher-wavenumber peak is 1A1g to 1T2g. Bottom: 500 MHz 59Co NMR spectra for 1–5 in H2O at room temperature. | ||
Variable-temperature 59Co NMR spectra were collected for 1–5 and K3[Co(CN)6] in H2O from 10–60 °C (see Fig. 4, 5 and S2–S7†) to explore the temperature dependence of δ. With increasing temperature, peaks for 1–5 and K3[Co(CN)6] shift downfield to higher δ. This temperature-dependent shift of peaks is consistent with varying coordination geometry in solution.18,34 As temperature is increased, energy is introduced into the vibrational modes of the cobalt complex, expanding M–L bond distances and engendering generally weaker Δo.18,35
 | ||
| Fig. 4 Variable-temperature 59Co NMR for 4 at 500 MHz in H2O, collected in increments of 5 °C. The system was allowed to equilibrate for at least 5 minutes between each temperature point prior to measurement. | ||
 | ||
| Fig. 5 Chemical shift values for 1–5 and K3[Co(CN)6] as a function of temperature. Solid lines are the result of linear regression – see main text for further details. | ||
Precise determination of the sensitivity of the 59Co NMR peak to temperature (Δδ/ΔT) was achieved via linear regression of the temperature-dependent data (see Fig. 5, S8, and Table S2†). These analyses revealed Δδ/ΔT values for K3[Co(CN)6] and 1–5, respectively, of 1.44(1), 1.44(2), 1.38(1), 1.30(2), 1.71(1), and 2.04(2) ppm °C−1. These values are within the ranges of sensitivity reported for the few 59Co NMR thermometers,15–18,20 but it's worth noting that, to the best of our knowledge, the Δδ/ΔT of 5 is eclipsed only by Co(acac)3, a molecule that is completely unsuitable for aqueous (e.g. physiological) applications.16,20 Most importantly (and surprisingly), these data indicate that the highest sensitivity to changes in temperature is held by the completely encaged complex 5.
The values of Δδ/ΔT follow an opposing trend to the initial hypothesis, in that 5, with the highest degree of encapsulation, displays the strongest Δδ/ΔT. Complex 4, with the second highest degree of encapsulation, displays the second highest sensitivity of our studied complexes. Yet, a comprehensive trend for all complexes on the basis of encapsulation is not indicated by these data. For example, in 1 and K3[Co(CN)6], the ligand donor atoms are not connected in any manner. Yet, these species demonstrate higher Δδ/ΔT than both 2 and 3, which contain bidentate chelates. Furthermore, the collected data show that electronic structure considerations alone (specifically, Δo) do not govern sensitivity. Here, neither the trend in Δo extracted from UV-Vis (3 < 5 < 1 < 4 < 2 < K3[Co(CN)6]) nor that from the 25 °C 59Co NMR (3 < 1 < 4 < 2 < 5 < K3[Co(CN)6]) reproduce the trend in Δδ/ΔT (see Fig. S9 and Table S3†).
The foregoing results highlight the need for deeper studies to derive fundamental insight. An important implication of the foregoing results is the concept that the encaged complex counterintuitively demonstrates the highest fluxionality in the inner-coordination structure. Four key experiments were applied to further test this rationale.
If the molecular structure of [Co(diNOsar)]Cl3 is truly more temperature-dependent than 1–4, then Δo for 5 should show the greatest temperature dependence. Variable-temperature UV-Vis spectra for 1–5 show slight shifts to lower energy with increasing temperature (Fig. 6 and S10–S12†). Analyses of these data reveal a change in Δo as a function of temperature, ΔΔo/ΔT. Over 1–5, ΔΔo/ΔT assumes values of −2.78(4), −1.36(17), −2.91(5), −3.70(17), and −5.65(32) cm−1 °C−1 for 1–5 respectively. These spectral changes are consistent with studies probing temperature-dependent UV-Vis spectra for metal complexes wherein spin-state changes are absent36,37 (versus systems displaying spin-crossover38 or valence tautomerization39). These data trend in a manner (particularly for 2–5) that seems opposed to an association between encapsulation and increased rigidity of the coordination environment. Indeed, 5 exhibits the largest change in temperature, followed by 4, then 3 and 1, and finally 2. Hence, these data point to a more dynamic inner coordination sphere.
 | ||
| Fig. 6 Variable temperature UV-Vis analysis. Top: normalized variable-temperature spectra for 5 in H2O at 4.5 mM concentration. Bottom: temperature dependence of Δo for 1–5. Linear regression yielded the grey lines and ΔΔo/ΔT – see main text for more detail. | ||
If the inner coordination sphere is less rigid upon encapsulation, then cobalt-59 spin lattice relaxation times should reflect that point. Indeed, the I = 7/2 cobalt-59 nucleus is quadrupolar, and, hence, its spin-lattice relaxation rate (1/T1) is dominated by fluctuations in the local electric field gradient.40 Hence, the anticipated higher fluxionality in the CoN6 environment of 4 and 5versus1–3 should correspondingly engender shorter T1. Analysis of the inversion recovery traces for 1–5 (Fig. 7) reveal T1 parameters for 1–5 that follow a trend with encapsulation, wherein [Co(NH3)6]Cl3 (1) displays the longest T1 (48.47(5) ms), followed by 2 (9.09(2) ms) and 3 (2.73(1) ms). In contrast, the species of highest encapsulation, 4 and 5, have the shortest T1 values (346(1) and 323(1) μs, respectively). For 1 and 2, these values match previously reported results.40–42 Quadrupolar relaxation is also enhanced in systems with a higher 59Co quadrupole coupling constant, and this constant is smaller for high-symmetry complexes.9 Compound 1 is clearly higher symmetry (Oh) than 2–5 (D3). This symmetry difference is likely an important contributor to the T1 of 1versus2–5, but quadrupolar couplings in this latter set of compounds are similar (when known).42,43 Moreover, solution-phase rotational rates for the series extrema, 1 and 5, are similar,42 suggesting rotational correlation is also not driving the difference in T1 across the series. Together, these points suggest that considerations beyond symmetry/rotation define T1 for these compounds. In light of the other data in this paper, we propose that the T1 trend evidences a more dynamic coordination environment upon encapsulation, though deeper investigations are needed to test this hypothesis.
 | ||
| Fig. 7 Spin-lattice relaxation data for 1–5. (a) 59Co NMR spectrum as a function of delay time for 3 following the 22.5 μs inversion pulse. (b) Specific inversion recovery sequence used, and intensity of the peaks as a function of the delay time. Grey lines are exponential fits to the data, yielding T1. See main text for more information on the fits. All data were collected at 25 °C and 33 mM concentration. | ||
Vibrational spectra in the 100–650 cm−1 window, wherein metal–ligand vibrations typically occur, ought to vary with rigidity as well.44 To test this concept, microcrystalline powders of 1–5 were analyzed via Raman spectroscopy. As the probed molecules increase in structural complexity, so do the Raman spectra, with compound 1 exhibiting 8, 2 displaying 12, and 5 producing 21 bands below 650 cm−1 (Fig. 8). Previous reports identify symmetric Co–N bond stretches at 500 and 486 cm−1 for 1, and 526, 444, and 476 cm−1 for 2.45,46 For 3–5, in contrast, no Raman spectra are reported to the best of our knowledge. Closer inspection of the Raman spectra reveals a general sharpening of transitions with increasing encapsulation. This sharpening is most noticeable when comparing the spectra of lesser encapsulated compounds 1–3 with the completely encapsulated species 5. Linewidth analyses of the observed vibrations for 1–5 permitted relative quantitation of the general degree of sharpness of these spectra (Fig. S13–S17 and Table S4†). The averages of the peak linewidths for the spectra are ordered from 5 < 2 < 4 < 1 < 3, where the fully encapsulated species, 5, exhibits the smallest average peak width.
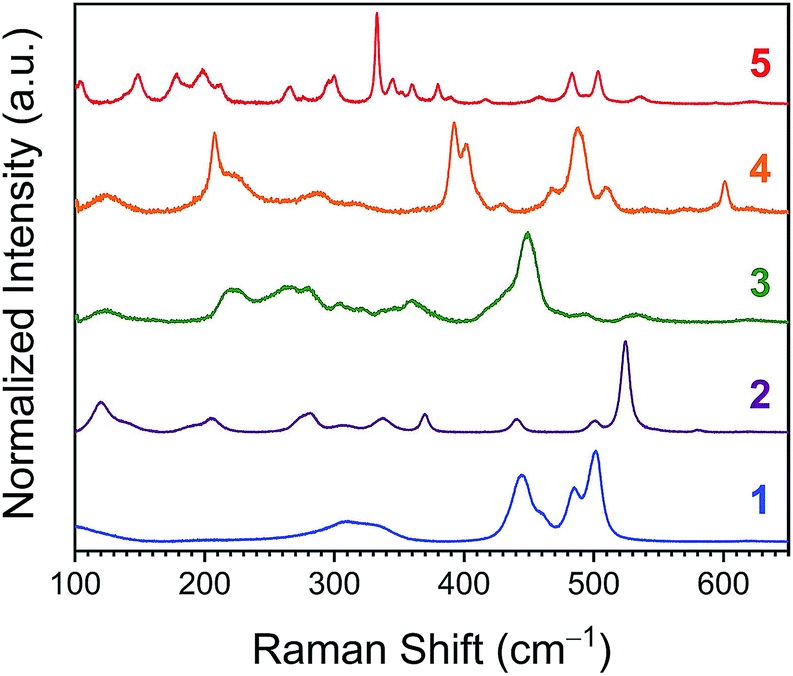 | ||
| Fig. 8 Room-temperature Raman spectra collected on pure powder samples of 1–5. All spectra are baseline corrected. | ||
On the basis of the variable-temperature NMR and UV-Vis data, the highest flexibility is observed for 5. However, the lifetime of the NMR experiment is much longer than that of vibrational spectroscopy.47 Hence, one may therefore expect the greater structural variability from the NMR/UV-Vis analyses to result in greater inhomogeneous broadening of the vibrational peaks for 5. The observations from the Raman spectra are in contrast to this expectation, as 5 demonstrates the sharpest peaks. One alternative mechanism that governs peak linewidths is homogeneous broadening, which causes sharper peaks for excitations that have longer lifetimes.48 This mechanism is acknowledged as dominant in studies of M(CO)n at room temperature in solution.49,50 If operative and dominant in powders of 1–5, this admittedly simplistic model of broadening would suggest that the lifetimes of the vibrations of the coordination sphere are enhanced by encapsulation. Translation of the average linewidths of 1–5 into average vibrational, spectroscopic lifetimes (via the relationship FWHM = 1/πτ) yields lifetimes of 0.4(2), 0.6(3), 0.4(2), 0.8(4), and 1.3(4) ps for 1–5, respectively.
The foregoing linewidth interpretation should be treated with caution owing to three specific factors. First, differences in microcrystalline environment can have an important impact on Raman linewidths (e.g.ref. 51 and 52). We note, however, that a preliminary powder diffraction analysis of the same samples measured by Raman spectroscopy did not reveal a noticeable trend of crystallinity correlating to the observed lifetimes (Fig. S18†). Second, modes of differing symmetries can yield different linewidths,49 as is likely evidenced here in the spread of linewidths in the deconvoluted peaks. Third, true elucidation of the vibration lifetimes requires time-resolved methods, which would also help differentiate inhomogeneous versus homogeneous broadening mechanisms.50 These data clearly motivate further solution-phase, time-resolved vibrational studies, a critical component of planned follow up work. Nevertheless, the obtained lifetimes are in the general picosecond range expected for metal complexes.49,50
If encapsulation affects Δδ/ΔT via modulating vibration lifetimes, that insight would provide a new design principle for vibrational control of molecular spin. Variable-solvent studies of 2 were performed as one final test of this concept. In particular, as the polar N–H bonds of the coordinated nitrogen atoms in 1–5 likely interact with the aqueous environment, this interaction should mediate the vibrations and structure of the [Co(en)3]3+ moiety, potentially imparting large differences to Δδ/ΔT. Indeed, such hydrogen bonding interactions are demonstrated to enable modulation of M–N and M–O bonds in other molecular systems.53,54 Here, this concept is being tested for temperature-dependent magnetic effects.
Initial studies focused on one member of the series, [Co(en)3]Cl3 (2), dissolved in four additional solvents: dimethylformamide (DMF), hexamethylphosphoramide (HMPA),§ dimethylsulfoxide (DMSO) and d6-dimethylsulfoxide (d6-DMSO). The solvents DMF, HMPA, and DMSO were selected to test polarity, and d6-DMSO chosen to test the impact of environmental deuteration. While solvent/deuteration impacts on 59Co δ are reported,55 their role on thermometry is not yet understood. 59Co NMR spectra collected at 25 °C reveal a peak position that shifts over a range of 200 ppm as a function of solvent identity (see Fig. 9). This solvent-dependent effect is known for the ClO4− salt of the [Co(en)3]3+ cation, stemming from modulation of the N-atom ligand field via hydrogen bonding between the solvent and N–H protons.56 Furthermore, only a tiny shift in δ is observed between DMSO and d6-DMSO, also in line with expected results.55
 | ||
| Fig. 9 Variable-solvent 59Co NMR studies of [Co(en)3]Cl3 (2). Top: 59Co NMR spectra of 2 in solvents of differing polarity. All data are reported at 25 °C from a 500 MHz NMR. Middle: temperature dependence of peak position as a function of solvent for 2. Solid lines are the result of linear regression to yield Δδ/ΔT (see main text). Bottom: sensitivity of 59Co δ to changes in temperature as a function of solvent π* polarity index. Error bars are less than the symbol size, except for DMF. The solid line is the result of linear regression with R2 = 0.9. | ||
Variable-temperature analyses tested the impact of these differing solvent cages on Δδ/ΔT (Fig. S19–S22 and Table S5†). Here, analysis of the variable-temperature 59Co NMR peak positions as a function of solvent demonstrate a noticeable impact of solvent identity on Δδ/ΔT (Fig. 9). As in H2O, all 59Co NMR chemical shifts move downfield with increasing temperature. Linear regression of these temperature-dependent data reveal Δδ/ΔT values of 1.19(2), 1.23(1), 1.27(1), and 1.28(1) ppm °C−1, respectively, for DMF, HMPA, DMSO, and d6-DMSO. These values are all lower than in H2O (Δδ/ΔT = 1.38(1) ppm °C−1) and indicate nearly no role for solvent deuteration on Δδ/ΔT in the present compound.
Comparison of the solvent-dependent Δδ/ΔT results for 2 against measures of solvent–solute interaction potentially provide deeper insight into the role of the solvent cage (Fig. 9, S23 and Table S5†). In particular, the trend in Δδ/ΔT was contrasted against (1) the solvent acceptor and donor numbers,57,58 (2) the π* solvent polarity scale,59,60 and (3) the β and α hydrogen-bonding donor/acceptor scales.60–62 There may be an approximate correlation between Δδ/ΔT and acceptor number, whereas there is clearly none for donor number and little, if any with β (see Fig. S23†). Analysis with α values is complicated as α is 0 for all solvents here except H2O.60 However, the π* scale clearly reveals a correlation (R2 = 0.9, Fig. 9, bottom).
The foregoing data point toward a coordination environment that is counterintuitively more flexible and dynamic with increasing encapsulation. Electron transfer studies of sarcophagine-like ligands provided the first assertions of rigidity in encapsulated Co(III) complexes based on a conformationally inflexible environment.63–67 We propose that this conformation-based description of rigidity is insufficient for understanding the trend of Δδ/ΔT. Instead, we tentatively posit an alternative, spin-relevant interpretation in this context. Here, the enhanced connectivity in 4 and 5 ensures a higher rigidity in the coordination environment, except in this case the rigidity permits vibrations of the encapsulated coordination environment to persist longer. Such longer lifetimes ultimately sustain a change in the coordination sphere by lengthening the equilibrium Co–N bond distances. Hence, there is a temperature dependence of Δo and δ in 4 and 5 that is larger than 1–3. This tentative interpretation of the data is also consistent with the solvent dependence of Δδ/ΔT in 2, as the solvent cage is known for impacting vibration lifetimes in coordination complexes.49,68,69 The fundamental argument we propose here is an analogue to the justification of long phonon lifetimes in materials like diamond,70,71 except here related to the molecular vibrations of a complex in solution. These studies clearly motivate future investigations to evaluate the validity of this picture of vibration-controlled spin properties.
Furthermore, the solvent-dependent data hint at a rich area of inquiry into the role of the second coordination sphere and counterions. When considering the [Co(en)3]3+ unit, interactions with the solvent are most easily intuited via the N–H protons accepting electron density from solvent molecule lone pairs. The association of a higher Δδ/ΔT with a higher π* index of solvent polarity59 would mesh with this intuited picture. This model would also be consistent with the match between a lower β value and a higher Δδ/ΔT, as a low β occurs when a solute will only weakly accept a proton.62 Thus, these data suggest that the N–H interactions are key to understanding Δδ/ΔT. However, the interpretation isn't without some uncertainty. To the extent that there is any correlation of Δδ/ΔT with solvent properties, it is with their acceptor number, not donor number, meaning that [Co(en)3]3+ acts as a donor. This argument only makes sense if one also considers the lone pairs of a bound Cl− counterion, not the N–H bonds. Indeed, earlier studies of [Co(en)3]3+ and [Co(diNOsar)]3+ demonstrate a close association between these species and their Cl− counterions that persists in solution.43,67,72 Noted reservations about generalizing the acceptor/donor number scale lend caution to the second explanation of the solvent-dependent data.73 Nevertheless, the conflict between these two interpretations underlines the necessity of further investigations into the role of the counterion and solvent cage on Δδ/ΔT.
Conclusions and outlook
The foregoing results are the first evidence of synergy between ligand encapsulation and enhanced temperature-dependent magnetic changes in metal-ion nuclear spins. Such knowledge is of broad impact, as exploiting molecular rigidity to control magnetism is an emerging trend in designing molecules for other spin-based technologies, e.g. molecular quantum bit development.74–76 Importantly, the presented arguments potentially tie vibration lifetimes to nuclear magnetism – necessitating future time-resolved measurements to test the validity of this analysis. Finally, our studies reveal that in addition to the ligand, manipulations of the counterion and solvent cage are the next stage for understanding the mechanisms that control Δδ/ΔT. Beyond the targeted applications in thermometry, the concepts herein could be extended to understanding the impacts of molecular rigidity on other spin-based applications, for example, designing electron paramagnetic resonance imaging probes,77 rigid systems for dynamic nuclear polarization78–80 (particularly with metal ions),81,82 or molecular quantum sensors.83,84Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Mr B. Gerold, Ms L. Beck, Dr C. Rithner and E. Ellison, and Profs. M. Nippe and E. Johnston-Halperin for useful discussions and experimental assistance. We are further grateful to some exceptionally helpful reviewers. This research was performed with the support of Colorado State University (CSU) and the NIH (R21-EB027293). NMR experiments were performed at the CSU Central Instrument Facility, which is supported by an NIH-SIG award (1S10OD021814-01) and the CSU-CORES Program. A portion of this work was performed at the Raman Microspectroscopy Laboratory in the Department of Geological Science at the University of Colorado-Boulder.Notes and references
- S. Bestgen, N. H. Rees and J. M. Goicoechea, Organometallics, 2018, 37, 4147–4155 CrossRef CAS.
- G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217–219 CrossRef PubMed.
- L. Riccardi, V. Genna and M. De Vivo, Nat. Rev. Chem., 2018, 2, 100–112 CrossRef CAS.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed.
- B. Figgis and M. Hitchman, Ligand field theory and its applications, Wiley-VCH, New York, 2000 Search PubMed.
- V. Rieke and K. B. Pauly, J. Magn. Reson. Imaging, 2008, 27, 376–390 CrossRef PubMed.
- D. Townsend, Z. Cheng, D. Georg, W. Drexler and E. Moser, Frontiers in Physics, 2013, 1, 1–6 CrossRef.
- J. C. C. Chan and S. C. F. Au-Yeung, Annu. Rep. NMR Spectrosc., 2000, 41, 1–54 CrossRef CAS.
- R. Bramley, M. Brorson, A. M. Sargeson and C. E. Schaeffer, J. Am. Chem. Soc., 1985, 107, 2780–2787 CrossRef CAS.
- A. Yamasaki, J. Coord. Chem., 1991, 24, 211–260 CrossRef CAS.
- L. Brateman, Am. J. Roentgenol., 1986, 146, 971–980 CrossRef CAS PubMed.
- A. Haase, J. Frahm, W. Hanicke and D. Matthae, Phys. Med. Biol., 1985, 30, 341–344 CrossRef CAS PubMed.
- T. R. Brown, B. M. Kincaid and K. Ugurbil, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 3523–3526 CrossRef CAS PubMed.
- A. G. Webb, M. Wong, M. Niesman, K. J. Kolbeck, L. J. Wilmess, R. L. Magin and K. S. Suslick, Int. J. Hyperthermia, 1995, 11, 821–827 CrossRef CAS PubMed.
- G. C. Levy, T. J. Bailey and D. A. Wright, J. Magn. Reson., 1980, 37, 353–356 CAS.
- D. G. Gillies, L. H. Sutcliffe and A. J. Williams, Magn. Reson. Chem., 2002, 40, 57–64 CrossRef CAS.
- G. B. Benedek, R. Englman and J. A. Armstrong, J. Chem. Phys., 1963, 39, 3349–3363 CrossRef CAS.
- M. Kanakubo, H. Ikeuchi and G. P. Satô, J. Magn. Reson., Ser. A, 1995, 112, 13–16 CrossRef CAS.
- M. Kanakubo, T. Uda, H. Ikeuchi and G. P. Satô, J. Solution Chem., 1998, 27, 645–653 CrossRef CAS.
- J. De Poorter, C. De Wagter, Y. De Deene, C. Thomsen, F. Ståhlberg and E. Achten, Magn. Reson. Med., 1995, 33, 74–81 CrossRef CAS PubMed.
- D. L. Carter, J. R. MacFall, S. T. Clegg, X. Wan, D. M. Prescott, H. C. Charles and T. V Samulski, Int. J. Radiat. Oncol., Biol., Phys., 1998, 40, 815–822 CrossRef CAS.
- R. J. Geue, T. W. Hambley, J. M. Harrowfield, A. M. Sargeson and M. R. Snow, J. Am. Chem. Soc., 1984, 106, 5478–5488 CrossRef CAS.
- X. Wang, R. Justice and S. C. Sevov, Inorg. Chem., 2007, 46, 4626–4631 CrossRef CAS PubMed.
- Y. Voloshin, I. Belaya and R. Krämer, The Encapsulation Phenomenon: Synthesis, Reactivity and Applications of Caged Ions and Molecules, Springer International Publishing, Switzerland, 2016 Search PubMed.
- J. Bjerrum and J. P. McReynolds, Inorg. Synth., 1946, II, 216–221 CAS.
- J. B. Work, Inorg. Synth., 1946, II, 221–222 CAS.
- J. C. Bailar and J. B. Work, J. Am. Chem. Soc., 1946, 68, 232–235 CrossRef CAS.
- R. J. Geue and M. R. Snow, Inorg. Chem., 1977, 16, 231–241 CrossRef CAS.
- L. R. Gahan and J. M. Harrowfield, Polyhedron, 2015, 94, 1–51 CrossRef CAS.
- H. Cai, J. Fissekis and P. S. Conti, Dalton Trans., 2009, 5395 RSC.
- G. A. Bottomley, I. J. Clark, I. I. Creaser, L. M. Engelhardt, R. J. Geue, K. S. Hagen, J. M. Harrowfield, G. A. Lawrance, P. A. Lay, A. M. Sargeson, A. J. See, B. W. Skelton, A. H. White and F. R. Wilner, Aust. J. Chem., 1994, 47, 143–179 CrossRef CAS.
- J. J. Alexander and H. B. Gray, J. Am. Chem. Soc., 1968, 90, 4260–4271 CrossRef CAS.
- C. J. Jameson, D. Rehder and M. Hoch, J. Am. Chem. Soc., 1987, 109, 2589–2594 CrossRef CAS.
- R. Englman, Mol. Phys., 1960, 3, 23–34 CrossRef CAS.
- L. V. Koplitz, K. Kim and D. S. McClure, Inorg. Chem., 1994, 33, 702–704 CrossRef CAS.
- O. G. Holmes and D. S. McClure, J. Chem. Phys., 1957, 26, 1686–1694 CrossRef CAS.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- R. Ader and A. Loewenstein, J. Magn. Reson., 1971, 5, 248–261 CAS.
- S. C. F. F. Au-Yeung, R. J. Buist and D. R. Eaton, J. Magn. Reson., 1983, 55, 24–38 CAS.
- C. W. Kirby, C. M. Puranda and W. P. Power, J. Phys. Chem., 1996, 100, 14618–14624 CrossRef CAS.
- Y. Masuda and H. Yamatera, J. Phys. Chem., 1988, 92, 2067–2071 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Hoboken, NJ, 2009 Search PubMed.
- Y. Chen, D. H. Christensen, G. O. Sørensen, O. F. Nielsen and E. Pedersen, J. Mol. Struct., 1993, 299, 61–72 CrossRef CAS.
- P. Stein, V. Miskowski, W. H. Woodruff, J. P. Griffin, K. G. Werner, B. P. Gaber and T. G. Spiro, J. Chem. Phys., 1976, 64, 2159–2167 CrossRef CAS.
- R. S. Drago, Physical Methods for Chemists, Surfside Scientific Publishers, Gainesville, 2nd edn, 1992 Search PubMed.
- J. L. McHale, Molecular Spectroscopy, CRC Press, 2nd edn, 2017 Search PubMed.
- J. J. Turner, Coord. Chem. Rev., 2002, 230, 213–224 CrossRef CAS.
- A. Tokmakoff and M. D. Fayer, Acc. Chem. Res., 1995, 28, 437–445 CrossRef CAS.
- H. Inada, K. Ohnishi, S. Nomura, A. Higuchi, H. Nakano and Y. Shirota, J. Mater. Chem., 1994, 4, 171 RSC.
- J. Petzelt, S. Kamba and I. Gregora, Phase Transitions, 1997, 63, 107–145 CrossRef CAS.
- A. S. Borovik, Acc. Chem. Res., 2005, 38, 54–61 CrossRef CAS PubMed.
- R. L. Lucas, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2005, 127, 11596–11597 CrossRef CAS PubMed.
- H. Grahn, U. Edlund and T. A. Holak, Magn. Reson. Chem., 1987, 25, 497–502 CrossRef CAS.
- G. Gonzalez, U. Mayer and V. Gutmann, Inorg. Nucl. Chem. Lett., 1979, 15, 155–159 CrossRef CAS.
- W. R. Fawcett, J. Phys. Chem., 1993, 97, 9540–9546 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- M. H. Abraham, Chem. Soc. Rev., 1993, 22, 73 RSC.
- R. W. Taft, W. J. Shuely, R. M. Doherty and M. J. Kamlet, J. Org. Chem., 1988, 53, 1737–1741 CrossRef CAS.
- R. J. Geue, M. G. McCarthy and A. M. Sargeson, J. Am. Chem. Soc., 1984, 106, 8282–8291 CrossRef CAS.
- P. V. Bernhardt, A. M. T. Bygott, R. J. Geue, A. J. Hendry, B. R. Korybut-Daszkiewicz, P. A. Lay, J. R. Pladziewicz, A. M. Sargeson and A. C. Willis, Inorg. Chem., 1994, 33, 4553–4561 CrossRef CAS.
- A. M. Sargeson and P. A. Lay, Aust. J. Chem., 2009, 62, 1280–1290 CrossRef CAS.
- A. G. Algarra, Eur. J. Inorg. Chem., 2015, 2015, 503–511 CrossRef CAS.
- I. Ling, A. N. Sobolev, R. Hashim and J. M. Harrowfield, CrystEngComm, 2014, 16, 11058–11063 RSC.
- G. M. Sando, Q. Zhong and J. C. Owrutsky, J. Chem. Phys., 2004, 121, 2158–2168 CrossRef CAS PubMed.
- J. T. King, M. R. Ross and K. J. Kubarych, J. Phys. Chem. B, 2012, 116, 3754–3759 CrossRef CAS PubMed.
- L. Bergman and R. J. Nemanich, Annu. Rev. Mater. Sci., 1996, 26, 551–579 CrossRef CAS.
- K. C. Lee, B. J. Sussman, J. Nunn, V. O. Lorenz, K. Reim, D. Jaksch, I. A. Walmsley, P. Spizzirri and S. Prawer, Diamond Relat. Mater., 2010, 19, 1289–1295 CrossRef CAS.
- M. Iida, T. Nakamori, Y. Mizuno and Y. Masuda, J. Mol. Liq., 1995, 65–66, 269–272 CrossRef.
- C. Laurence and J.-F. Gal, Lewis Basicity and Affinity Scales, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- M. Atzori, S. Benci, E. Morra, L. Tesi, M. Chiesa, R. Torre, L. Sorace and R. Sessoli, Inorg. Chem., 2018, 57, 731–740 CrossRef CAS PubMed.
- M. Atzori, L. Tesi, S. Benci, A. Lunghi, R. Righini, A. Taschin, R. Torre, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2017, 139, 4338–4341 CrossRef CAS PubMed.
- L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2018, 9, 3265–3275 RSC.
- In Vivo EPR (ESR), ed. L. J. Berliner, Springer US, Boston, MA, 2003, vol. 18 Search PubMed.
- M. K. Kiesewetter, B. Corzilius, A. A. Smith, R. G. Griffin and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 4537–4540 CrossRef CAS PubMed.
- A. Zagdoun, G. Casano, O. Ouari, G. Lapadula, A. J. Rossini, M. Lelli, M. Baffert, D. Gajan, L. Veyre, W. E. Maas, M. Rosay, R. T. Weber, C. Thieuleux, C. Coperet, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2012, 134, 2284–2291 CrossRef CAS PubMed.
- E. L. Dane, B. Corzilius, E. Rizzato, P. Stocker, T. Maly, A. A. Smith, R. G. Griffin, O. Ouari, P. Tordo and T. M. Swager, J. Org. Chem., 2012, 77, 1789–1797 CrossRef CAS PubMed.
- B. Corzilius, V. K. Michaelis, S. A. Penzel, E. Ravera, A. A. Smith, C. Luchinat and R. G. Griffin, J. Am. Chem. Soc., 2014, 136, 11716–11727 CrossRef CAS PubMed.
- B. Corzilius, A. A. Smith, A. B. Barnes, C. Luchinat, I. Bertini and R. G. Griffin, J. Am. Chem. Soc., 2011, 133, 5648–5651 CrossRef CAS PubMed.
- G. Balasubramanian, A. Lazariev, S. R. Arumugam and D. Duan, Curr. Opin. Chem. Biol., 2014, 20, 69–77 CrossRef CAS PubMed.
- R. Schirhagl, K. Chang, M. Loretz and C. L. Degen, Annu. Rev. Phys. Chem., 2014, 65, 83–105 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01689a |
| ‡ Denotes equal contribution to manuscript. |
| § HMPA is a Class 1B carcinogen and mutagen and should be handled with extreme care. |
| This journal is © The Royal Society of Chemistry 2019 |