
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent probes for organelle-targeted bioactive species imaging
Peng
Gao
 ,
Wei
Pan
,
Na
Li
* and
Bo
Tang
*
,
Wei
Pan
,
Na
Li
* and
Bo
Tang
*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Institute of Molecular and Nano Science, Shandong Normal University, Jinan 250014, P. R. China. E-mail: lina@sdnu.edu.cn; tangb@sdnu.edu.cn
First published on 24th May 2019
Abstract
Bioactive species, including reactive oxygen species (ROS, including O2˙−, H2O2, HOCl, 1O2, ˙OH, HOBr, etc.), reactive nitrogen species (RNS, including ONOO−, NO, NO2, HNO, etc.), reactive sulfur species (RSS, including GSH, Hcy, Cys, H2S, H2Sn, SO2 derivatives, etc.), ATP, HCHO, CO and so on, are a highly important category of molecules in living cells. The dynamic fluctuations of these molecules in subcellular microenvironments determine cellular homeostasis, signal conduction, immunity and metabolism. However, their abnormal expressions can cause disorders which are associated with diverse major diseases. Monitoring bioactive molecules in subcellular structures is therefore critical for bioanalysis and related drug discovery. With the emergence of organelle-targeted fluorescent probes, significant progress has been made in subcellular imaging. Among the developed subcellular localization fluorescent tools, ROS, RNS and RSS (RONSS) probes are highly attractive, owing to their potential for revealing the physiological and pathological functions of these highly reactive, interactive and interconvertible molecules during diverse biological events, which are rather significant for advancing our understanding of different life phenomena and exploring new technologies for life regulation. This review mainly illustrates the design principles, detection mechanisms, current challenges, and potential future directions of organelle-targeted fluorescent probes toward RONSS.
1. Introduction
Revolutions in science and technology have greatly enhanced our lives. As a prominent example, in the past few decades fluorescent imaging technology has made great achievements, which have brought increasingly fascinating news for diverse fields including biomedicine and bioanalysis.1 Extensive evidence shows that diverse bioactive species are critical for conducting physiological processes throughout the cell cycle, and numerous major diseases are associated with the abnormal expression of these species.2 Meanwhile, the concentrations of these molecules are inhomogeneous in different subcellular environments at different times.3 Therefore, a better understanding of diverse physiological and pathological processes can be obtained via accurate monitoring of subcellular molecular concentrations of these molecules, which is beneficial for exploring novel strategies for regulating biological phenomena and improving diagnosis, treatment and prognosis of different diseases.4Subcellular microenvironments are maintained by the cell endomembrane system, typically known as organelles, including the nucleus, mitochondria, lysosomes, endoplasmic reticulum (ER), Golgi apparatus, lipid droplets and so on. These organelles with complex but ordered structures play important roles in ensuring the smooth progress of cell growth, proliferation, fission, signal transduction and apoptosis. They are also the main sources as well as targets of diverse bioactive species. However, the disruption of intracellular homeostasis can result in various disorders.2g,3a,c,d For example, owing to the extremely rapid respiration rate, reactive oxygen species (ROS) are usually overexpressed in diverse organelles of cancer cells, which are also associated with lower intracellular pH. To maintain cell proliferation, the contents of antioxidants in malignant cells are elevated accordingly. Therefore, detecting the real status of subcellular microenvironments is critical in understanding different pathogeneses of a broad spectrum of major diseases.
Unfortunately, the real-time, in situ, dynamic, accurate, and reversible monitoring of bioactive molecules in subcellular microenvironments has been a great challenge due to theoretical and technological limitations, and many related investigations have remained at the cellular level during the last few decades.1e,g,3c,d,5 Notably, over the past several years, significant efforts have been devoted to the design and development of organelle-targeted fluorescent probes for precise monitoring of subcellular microenvironments, including pH,6 polarity,7 ions,8 viscosity,9 ROS (including O2˙−, H2O2, HOCl, 1O2, ˙OH, HOBr, etc.),10 reactive nitrogen species (RNS, including ONOO−, NO, NO2, HNO, etc.),11 reactive sulfur species (RSS, including GSH, Hcy, Cys, H2S, H2Sn, SO2 derivatives, etc.),1e,f,12 ATP,13 HCHO14 and so on. Among these targets, ROS, RNS and RSS (termed RONSS) have attracted increasing interest because they are closely related to the intracellular redox homeostasis. In other words, they are capable of buffering the subcellular redox balance during different processes, which can further determine a number of biological events. Therefore, organelle-targeted RONSS fluorescent probes are of great significance for explaining and exploring the physiological and pathological functions of these molecules, and could offer theoretical bases for clinical diagnosis as well as drug design.2g,3b–d,4c
This review mainly focuses on the latest achievements of organelle-targeted fluorescent probes for RONSS imaging. The design principles, detection mechanisms and biological applications of these fluorescence tools are carefully illustrated. Moreover, the current challenges and the potential future directions, which would evoke more research interest and open new avenues for biological analysis, are included as well.
2. General rules for functional probe design
The advancement of microscopic imaging technology has introduced new opportunities for visualizing subtle intracellular structures. To investigate the critical events in living cells on the organelle and even more subtle levels, elaborately designed fluorescent probes are indispensable.15 Currently, benefiting from their easy design, preparation and functionalization, fluorescent molecular and nanoprobes are being widely employed for biological imaging.1a,b,f,2c Organelle-targeted fluorescent probes mainly contain three moieties: targeting groups, fluorophores, and recognition units.3a,d Benefiting from the different physiochemical properties of diverse organelles, probes with diverse targeting groups can localize in specific organelles.3b After reaching subcellular locations of interest, they can subsequently react with diverse bioactive species through specific reaction mechanisms and further make detectable signal changes via different response mechanisms, which enables us to readily and effectively monitor these species in different organelles. In this part, diverse targeting approaches, reaction mechanisms and response mechanisms of organelle-targeted RONSS probes will be elaborated.2.1. Approaches for delivering fluorescent probes into organelles
Typically, the nucleus is enveloped by a double-layered membrane, which contains numerous nuclear pores as well as ribosomes. Some small molecules and ions can permeate through the nuclear membrane freely, while large biomolecules, such as ribosomes and RNA, can pass through the nuclear pores via energy related pathways. By targeting the minor grooves in negatively charged DNA double-strands, small cationic fluorescent probes with two or more cationic centers and hydrophobic planar aromatic structures can preferentially label DNA molecules, and some of them have been commercialized (Fig. 1A).16a,18 In addition to labelling DNA, decorating probes with nuclear localization signal peptides, which enables binding of probes with importins and further actively delivering them into the nucleus through nuclear pores, is also an established protocol.16b,19
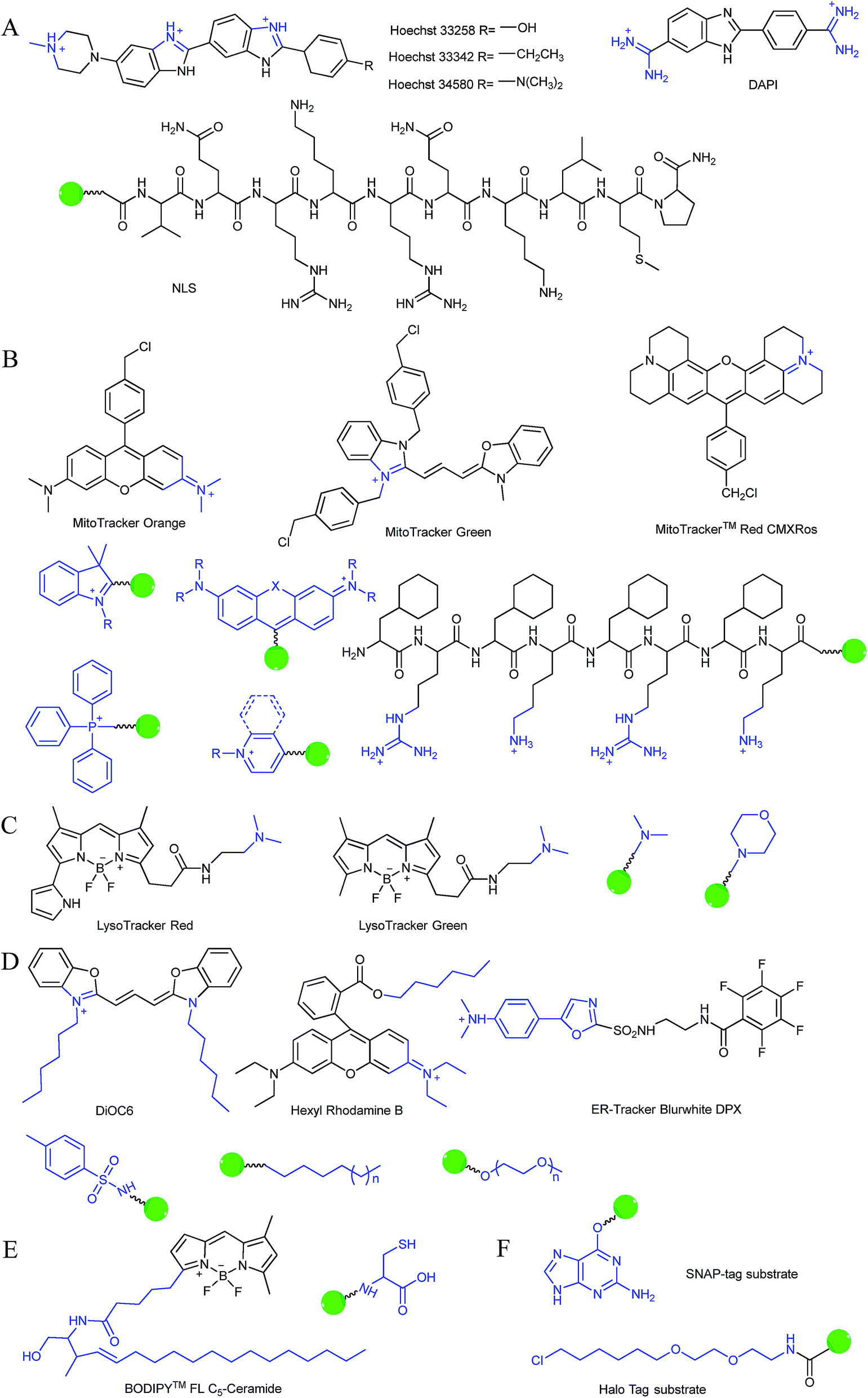 | ||
| Fig. 1 Structures of diverse commercialized organelle-labelling dyes and common groups for organelle-targeted probe design: (A) for nucleus, (B) for mitochondria, (C) for lysosomes, (D) for endoplasmic reticulum, (E) for Golgi apparatus and (F) substrates for protein tagging probes. | ||
During mitochondrial respiration, the proton pumps in the inner mitochondrial membrane transport protons to the mitochondrial intermembrane space, which results in the formation of a highly negative mitochondrial transmembrane potential (MMP, ∼−180 mV).21 Inspired by this process, many delocalized lipophilic cations have been proved to exhibit mitochondrion targeting capabilities.22 Fluorescent probes with intrinsic or post-functionalized cationic aromatic structures can be used for mitochondrial imaging. Typical targeting ligands including triphenylphosphonium (TPP), positively charged pyridine and quinoline derivatives, cyanine and rhodamine are popular fluorophores for designing mitochondrion-targeted probes (Fig. 1B).3d,7a,23 In addition to MMP, mitochondrial transport proteins have also been used as targets. Some functional groups, such as peptides and pyruvate, have also been developed for constructing mitochondrion-targeted probes owing to their affinities for specific mitochondrial proteins.24
Currently, modification of probes with lipophilic amines is the most widely employed strategy for driving probes into lysosomes/endosomes.6a,26 The protonated amines in lysosomes are membrane impermeable, which results in the lysosomal/endosomal selective entrapment of the probes. 4-(2-Hydroxyethyl) morpholine and N,N-dimethylethylenediamine are the most popular groups for constructing lysosome-targeted fluorescent probes (Fig. 1C).26a,b,27 Interestingly, fluorescent probes without lysosomotropic groups can also be used to detect species in lysosomes specifically, and they are usually logic molecules and can only release fluorescence signals in acidic environments after reacting with analytes.28
The Golgi apparatus is another important intracellular compartment for the modification, storage and transportation of lipids, carbohydrates and protein. After modification and labelling, cargoes in the Golgi apparatus can be transferred into their final destinations, such as lysosomes and the cytoplasmic membrane, and subsequently exert their biological functions. It has been proved that a stress-signalling overload in the Golgi apparatus can induce a series of disorders, which can further cause diverse diseases as well as cell death. Therefore, development of efficient Golgi apparatus specific probes has also attracted certain interest in recent years.3b,31 However, the intrinsic rules of Golgi apparatus-targeted probes are still vague, which has severely hampered the development of related fluorescent tools. Very recently, inspired by the rich cysteine residues in Golgi apparatus localized galactosyltransferase and protein kinase D, Huang and co-workers demonstrated L-cysteine (Fig. 1E) as an efficient Golgi apparatus targeting ligand, and several probes have been developed using this strategy.31a–c
2.2. Detection mechanisms toward RONSS
As the most reactive bioactive species in living cells, RONSS are responsible for intracellular redox homeostasis. Any uncontrolled expression of RONSS could cause oxidative, nitrative or reductive stress owing to the capabilities of these molecular species in reacting with diverse biomolecules, such as DNA, lipids and enzymes.3c Such imbalances can hamper the smooth progress of different biological activities and further result in a series of major diseases, such as cardiovascular disease, neurodegenerative disease, diabetes, inflammation and cancer.1e,f,2a,10a,b Many other pathological processes, like drug induced liver injury (DILI), ischemia reperfusion injury (IRI) and so on, are also accompanied by the overproduction of RONSS.3c More importantly, an increasing number of research studies have revealed that RONSS can be potential messengers for cell signalling, which further spurred investigations into the development of efficient tools for tracing subcellular concentrations of diverse RONSS.3a,dAmong the diverse detection strategies, fluorescence imaging represents a simple, sensitive and non-invasive technique, which enables real-time, dynamic and selective imaging of numerous targets in living cells, and has greatly advanced biological analysis, clinical drug discovery and so on.1b,g,3a,7a,10a,21b Intracellular RONSS feature short life span, low concentration, high reactivity and environmental sensitivity, and they are non-emissive. Therefore the specific imaging of RONSS cannot leave out efficient fluorescent probes.1d,e,4b,11 Up to now, a series of probes based on different reaction mechanisms have been developed for selective labelling of RONSS in living cells. A summary of the detection mechanisms of organelle targeted fluorescent probes toward RONSS is shown in Table 1.
| Bioactive species | Detection mechanisms | Ref. | |
|---|---|---|---|
| ROS | O2˙− | Nucleophilic substitution | 24a and 34 |
| Oxidation of benzothiazoline | 35 | ||
| H2O2 | Oxidation of phenylboronic acid | 36 | |
| Oxidation of a diketone | 36c | ||
| Oxidation of phenol | 37 | ||
| HOCl | Oxidation of double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC and CS) N, CC and CS) |
38 | |
| Oxidation of a lactam or thiolactone | 39 | ||
| Oxidation of a thioether or selenide | 40 | ||
| 1O2 | Formation of an endoperoxide | 41 | |
| ˙OH | Oxidation of phenothiazine | 42 | |
| HOBr | Catalytic cyclization between the amino and S-methyl group | 43 | |
| RNS | ONOO− | Oxidation of phenylhydrazine | 44 |
| Oxidation of an unsaturated bond | 45 | ||
| Deboronation | 46 | ||
| Decomposition of an α-ketoamide group | 47 | ||
| NO | Formation of a triazole with o-phenylenediamine | 48 | |
| N-Nitrosation of an aromatic secondary amine | 49 | ||
| HNO | Staudinger reaction | 50 | |
| RSS | GSH | Michael addition | 51 |
| Nucleophilic substitution | 52 | ||
| Cys | Michael addition | 53 | |
| H2S | Reduction of an aryl azide/nitro group | 26b and 54 | |
| Thiolysis | 55 | ||
| Cu2+ precipitation | 56 | ||
| Nucleophilic addition | 57 | ||
| H2Sn | Nucleophilic aromatic substitution | 58 | |
| SO2 | Nucleophilic addition | 59 | |
2.3. Response mechanisms of RONSS fluorescent probes
After reacting with RONSS, the signal outputs of the fluorescent probes can give appropriate changes via specific mechanisms that can subsequently be detected by a detector. In general, fluorescence resonance energy transfer (FRET), photo-induced electron transfer (PET), intramolecular charge transfer (ICT) and excited-state intramolecular proton transfer (ESIPT) are the most popular response mechanisms for organelle-targeted fluorescent probes.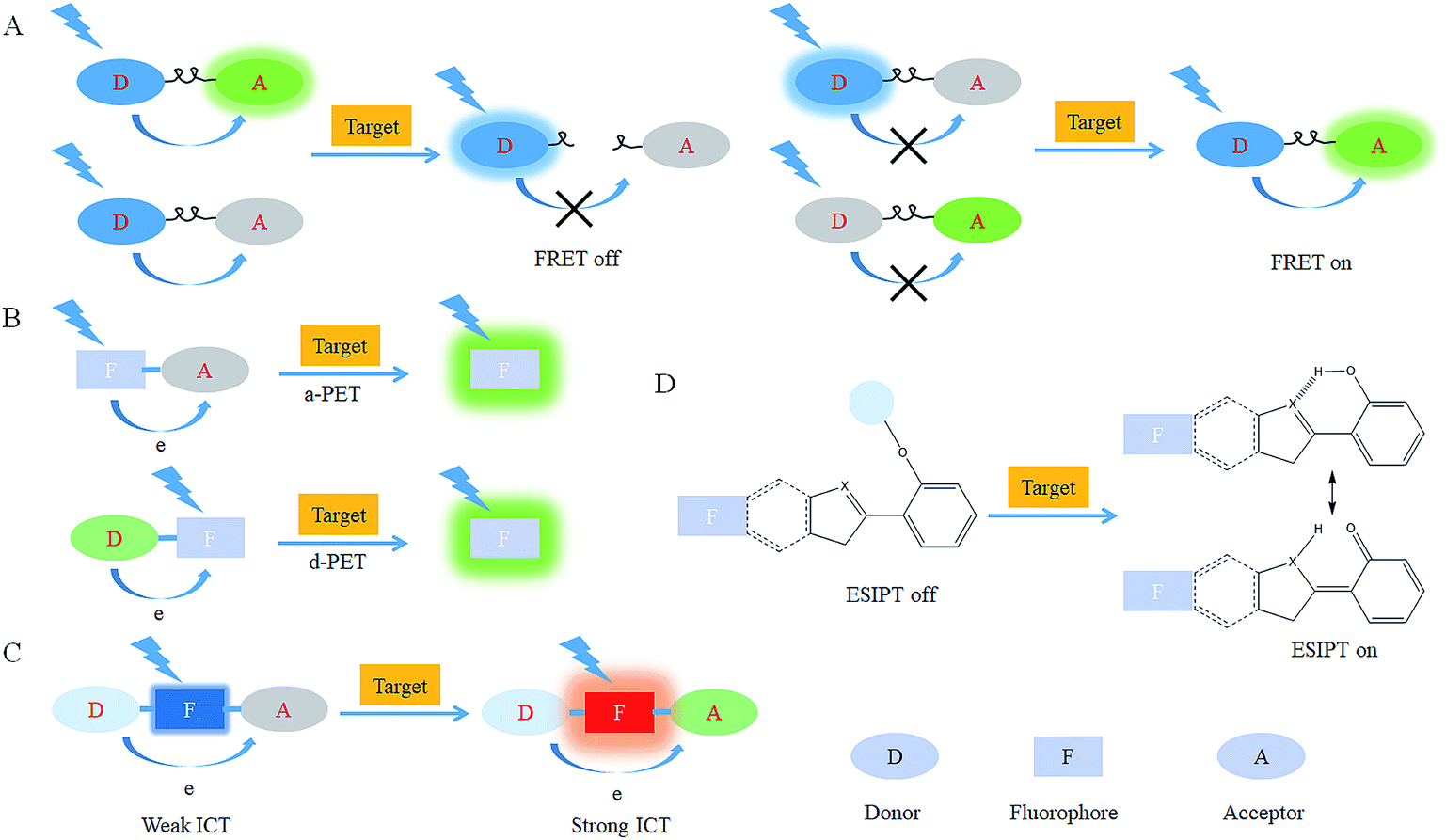 | ||
| Fig. 2 Response mechanisms for organelle-targeted probe design: (A) FRET, (B) PET, (C) ICT and (D) ESIPT. | ||
3. Nucleus-targeted probes
As discussed above, the nucleus is a crucial organelle of eukaryotic cells. An increasing number of reports have proved that RONSS can be important messengers during gene expression. However, only a limited number of nucleus-targeted probes have been devised up to now, though they are favourable for further revealing/verifying the potential biological functions of these molecules (Fig. 3).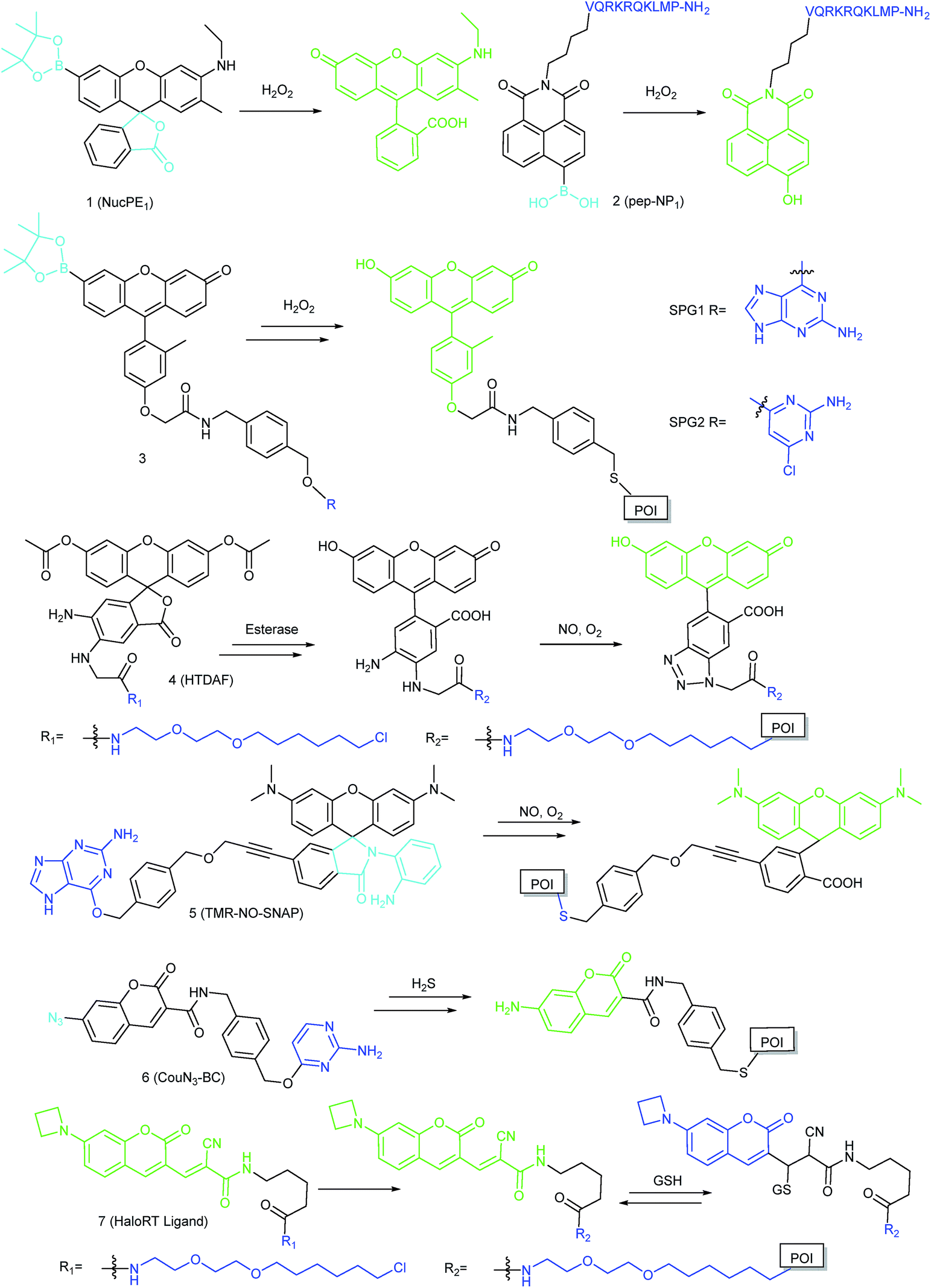 | ||
| Fig. 3 Fluorescent probes for imaging RONSS in the nucleus. | ||
3.1. Imaging of ROS in the nucleus
ROS can diffuse into the nucleus and regulate numerous critical physiological processes, while excess ROS can cause DNA damage/mutation and disrupt nuclear homeostasis.3c Thus tracing the nuclear ROS concentration holds promise for revealing the molecular mechanisms of some related biological events. However, there are only limited reports on nucleus-targeted ROS imaging up to now, most likely because it is challenging to drive probes into the nucleus effectively and selectively image the bioactive molecules of interest at ultralow concentrations.By screening an arylboronate based probe, Chang's research group discovered a nucleus-targeted H2O2 probe, 1 (NucPE1).17 Interestingly, although without a specific targeting moiety, this probe can accumulate in the nucleus of several mammalian cell lines and C. elegans. Moreover, once 1 reacted with H2O2 in the cytoplasm, it lost its targeting capability, which enables the nucleus-selective H2O2 imaging. Although the targeting mechanism remains to be further clarified, this probe can serve as a novel fluorescent tool for monitoring the nucleus H2O2 fluxes. Importantly, they revealed the relationships between sir-2.1 overexpression and enhanced regulation of nuclear ROS pools with this probe, which proved that the longevity is closely related to the ROS regulation capacity.
Using nuclear-localization signal peptides (NLS, detailed structure shown in Fig. 1A) as the targeting moiety, Yi et al. successfully prepared a naphthalimide based probe, 2 (pep-NP1), for nucleus-targeted H2O2 imaging.19 Confocal laser scanning microscope (CLSM) experiments indicated the successful nuclear localization of 2, while probe NP1 without NLS peptide functionalization was retained in the cytoplasm. 2 was used for ratiometric imaging of the intra-nuclear H2O2 in HeLa cells (Fig. 4).
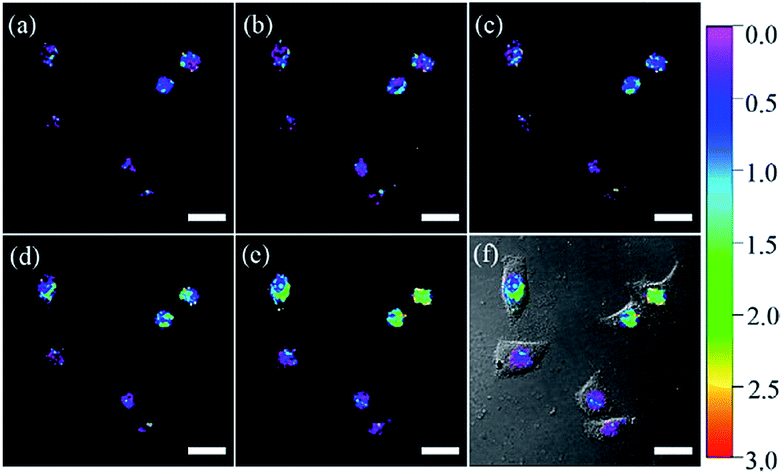 | ||
| Fig. 4 CLSM ratio images of HeLa cells loaded with 50 μM 2 and stimulated with 200 μM H2O2 for (a) 0, (b) 15, (c) 30, (d) 60, and (e) 75 min at 37 °C. (f) The overlay image of (e) and the brightfield image. Adapted with permission from ref. 19. Copyright 2014 American Chemical Society. | ||
Protein tagging is another effective strategy for delivering probes into the nucleus. Chang's group reported SNAP-tag based H2O2 probes 3 (SPG1, membrane-impermeable, and SPG2, membrane-permeable), which can specifically bind with AGT fused protein.65 Upon coming into contact with the localized H2O2, 32-fold turn-on fluorescence can be detected due to the deprotection of boronate groups. By transfecting different plasmids, H2O2 in the nucleus, mitochondria, plasma membrane and ER of living cells was detected with the probes, which offered a versatile fluorescent platform for tracing the subcellular H2O2 level.
3.2. Imaging of RNS in the nucleus
In addition to ROS, RNS are also critical bioactive species for living organisms. Due to the ultrashort lifespan and diffuse distance of RNS, the action sites of these molecules are usually determined by their locations. Therefore, precise imaging of the subcellular RNS distribution will accelerate our understanding of the biological functions of these molecules.NO is an important signalling molecule that can exert multiple biological effects on living cells, and it has also been widely investigated for cancer treatment. Clarifying the function and identifying the target sites of NO can facilitate related research studies. By taking advantage of the HaloTag technology, Yang and Hu et al. developed an organelle-targetable probe, 4 (HTDAF-2DA).66 The probe has excellent cell permeability due to the introduction of diacetate groups. After entering living cells, the ester groups were hydrolyzed and the probe was subsequently immobilized by HaloTag protein. Upon the addition of exogenous NO, the fluorescence of probes can be turned on due to the inhibition of the PET process after formation of a triazole product. Based on different plasmids, the probe was specifically delivered into the nucleus, cytosol, mitochondria and plasma membrane. The target site of exogenous NO was successfully revealed to be the plasma membrane with 4.
By connecting the rhodamine derivative with the SNAP-tag substrate, Xiao's group reported another organelle-targetable genetically encoded NO probe, 5 (TMR-NO-SNAP), which has been used for imaging the endogenous subcellular NO generation in LPS/IFN-γ stimulated RAW264.7 cells and etoposide treated COS-7 cells.67
3.3. Imaging of RSS in the nucleus
RSS are another group of bioactive species closely related to cellular redox homeostasis. Benefiting from the reductive activity of these species, RSS can interact with diverse proteins within the nucleus, which can further influence the gene expression. Therefore, mapping the intra-nuclear concentration and distribution of RSS is another significant task for investigating the redox biology in the nucleus.In 2016, Wang's group reported a genetically encoded coumarin based probe, 6 (CouN3-BC), for nucleus- and mitochondrion-targeted H2S imaging.68 After transfecting pCLIP-H2B and pCLIP-COX8A plasmids,69 the tag protein was successfully fused with H2B protein (in the nucleus) and a cytochrome c oxidase subunit (in the mitochondrial inner membrane and matrix), respectively. Subsequently, probe CouN3-BC was selectively anchored to the nucleus or mitochondria and further lit up localized H2S. H2S accumulation in the nucleus of HeLa cells under NO donor sodium nitroprusside stimulation was successfully tracked with this probe. In 2018, they proposed another genetically encoded probe, 7 (HaloRT Ligand), for reversible GSH imaging.70 Based on the HaloTag technique, 7 was successfully labelled in the nucleus, mitochondria, ER, and cytosol. And they revealed for the first time that under various stimulations, the GSH concentrations in the nucleus and cytosol remained nearly the same, which suggested that there is no GSH pool in the nucleus.
3.4. Challenges for nucleus-targeted fluorescent probes
Several reports have suggested that the conjugation of molecular or nanomaterials with NLS peptides can offer them the capability for nucleus specific accumulation, and nucleus-targeted cancer therapy has been widely investigated.16a,18,71 However, novel nucleus-targeted fluorescent probes are still rare, and due to the fact that the targeting efficiency of those materials is unsatisfactory, many probes require long incubation time for nuclear localization. Moreover, direct conjugation of probes with nucleus-targeted molecular scaffolds or NLS peptides is usually unsuccessful, most likely because minor structural alterations can reduce the affinity of the targeting ligands.3a Protein tagging seems to be an ideal strategy for anchoring probes into almost any organelles including the nucleus. But the synthetic difficulty and complexity, and loss of solubility of probes after binding with the substrates can hinder the application of genetically encoded probes. Excess un-tagged probes during imaging must be washed out several times to reduce the background fluorescence, which can hamper the activity and change the subcellular microenvironments of cells. Moreover, only the RONSS near the fusion protein can be detected by the probes, which further diminished the applications of genetically encoded probes. Thus, more attention should be paid to establish novel methodologies for nucleus-targeted ultrasensitive probe design.4. Mitochondrion-targeted probes
Mitochondria are the energy center of living cells, and they produce the majority of ATP for numerous biological events, including cell proliferation, division, biomolecule fabrication and so on.24d Therefore, the mitochondrial function and homeostasis determine the physiological and pathological processes of living cells. RONSS are closely related to the signal conducting and mitochondrial microenvironments.72 The past decade has witnessed significant progress of mitochondrion targeted fluorescent probes for localized RONSS imaging, owing to its potential in offering theoretical bases for disease diagnosis and therapy (Fig. 5).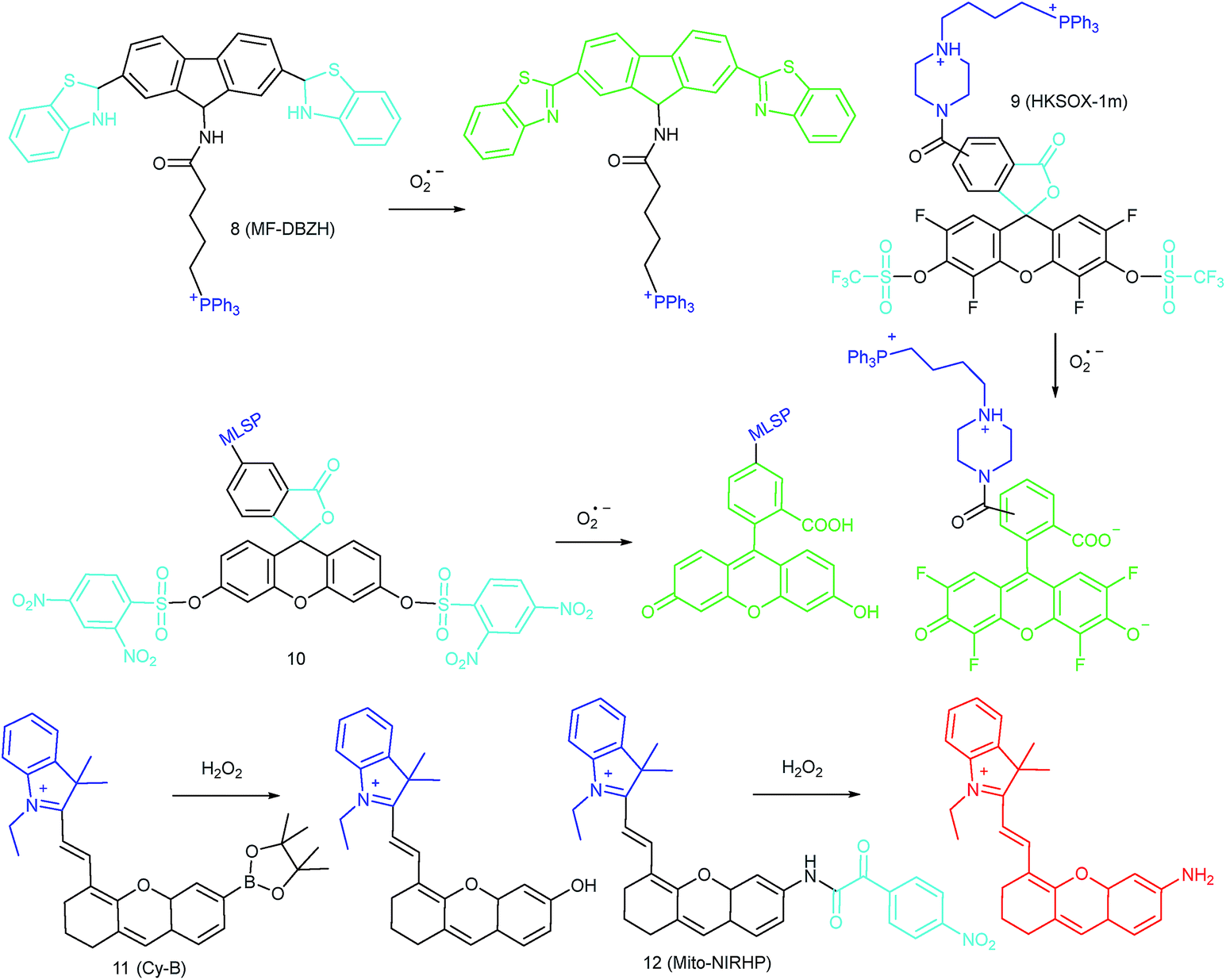 | ||
| Fig. 5 Mitochondrion-targeted probes for O2˙− and H2O2 imaging. | ||
4.1. Imaging of ROS in mitochondria
To ensure intracellular energy supply, mitochondria perform diverse electron-transport chain (ETC) reactions for ATP synthesis. Once the electrons leak from the ETC, they can react with oxygen molecules to generate O2˙−. Under the catalysis of diverse enzymes, O2˙− can be further transformed into H2O2, ONOO−, ˙OH, 1O2 and so on.23a,b,35,43a,44 These bioactive species can serve not only as oxides to maintain the mitochondrial redox homeostasis, but also as signal molecules to regulate the cellular function. However, once overexpressed, they can cause serious oxidative stress and further hamper the mitochondrial function, resulting in diverse disorders as well as diseases.2g,3d,7a,38b,39a,43b In the past decade, a variety of mitochondrion specific fluorescent probes have been developed for tracing ROS fluctuations during diverse physiological and pathological processes.Inspired by the mitochondrion targeting capability of the cationic lipophilic group TPP, our group developed a two-photon (TP) fluorescent probe, 8 (MF-DBZH), for mitochondrial endogenous O2˙− detection.35 The probe is composed of TPP, O2˙− sensitive benzothiazoline and TP fluorophore fluorene. Upon the addition of O2˙− to a solution of 8, the specific benzothiazoline dehydrogenation reaction results in an extended conjugate plane, leading to TP fluorescence enhancement due to the disruption of the PET process. The limit of detection (LOD) of this probe was calculated to be 9.5 nM. Living cell co-staining revealed that 8 effectively localized into the mitochondria under TPP guidance. This pH-insensitive and photostable TP probe has been used to selectively image the mitochondrial endogenous O2˙− fluctuation during the inflammation process at cellular and living body levels with the assistance of TP microscopy (Fig. 6).
 | ||
| Fig. 6 TP microscopy images of mice with LPS-mediated abdomen injury using probe 8. (A) O2˙− was produced inside the peritoneal cavity of the mice during the LPS-mediated inflammatory response. (B) TP fluorescence in situ images of mice abdomen incubated with 8 (100 μM). Adapted with permission from ref. 35. Copyright 2013 American Chemical Society. | ||
Another TPP functionalized mitochondrion-targeted O2˙− probe, 9 (HKSOX-1m), was reported by Yang's group.349 employs fluorescein as the fluorophore and aryl trifluoromethanesulfonate as the recognition group. The cleavage of the aryl trifluoromethanesulfonate group and rearrangement reaction during the nucleophilic reaction between 9 and O2˙− resulted in dramatic fluorescence intensity enhancement. The emission intensity of 9 increased linearly upon the addition of O2˙− into the probe solution. The mitochondrial O2˙− concentration fluctuation in human THP-1 cells in the resting state or under antimycin A stimulation was monitored with this probe. 9 also showed promise for revealing other biological and pathological functions of O2˙−.
Apart from using TPP as the localization group, peptides have also been employed for mitochondrion-specific probe design.24a Zhong's group reported a mitochondrial localization signal peptide (MLSP) functionalized O2˙− probe, 10, which has been successfully used for monitoring mitochondrial O2˙− generation in RAW264.7 macrophages under the stimulation of chemical molecules or single-walled carbon nanotubes.24a
Generally speaking, aryl boronates and phenylboronic acid are the most widely employed groups for H2O2 specific probes. Peng's group reported a hemicyanine based H2O2 probe, 11 (Cy-B), which was prepared by direct modification of hemicyanine dye with a borate ester moiety.36a The cationic feature of 11 makes it specifically accumulate in the mitochondria (overlapped with rhodamine 123, Pearson's coefficient = 0.98), while the mitochondrial H2O2 can specifically oxidize the boronate moiety and cause the “turn-on” of fluorescence. 11 was successfully employed for imaging mitochondrial H2O2 during autophagy induced by rapamycin. Benefiting from its NIR emissive behavior, in vivo H2O2 imaging was also demonstrated.
IRI occurs in many cases, including traumatic shock, surgery, organ transplantation, frostbite, thrombosis and so on, and can cause deadly damage to organs. Recent research studies have revealed that ROS are associated with IRI. Fluorescent probes capable of monitoring the subcellular ROS level during IRI are critical for understanding the pathogenesis and may further accelerate the related drug discovery. By using α-ketoamide as the responsive moiety, Our group developed another hemicyanine-based mitochondrion-targeted H2O2 probe, 12 (Mito-NIRHP).36c Benefiting from the ICT mechanism as well as the specific response capability of α-ketoamide, 12 was used for H2O2 imaging under both in vitro and in vivo conditions (Fig. 7). Importantly, with this probe, the relationship between H2O2 and kidney IRI was elucidated in subcellular, organ, and living body levels.
 | ||
| Fig. 7 Fluorescence imaging (pseudocolor) of H2O2 in Kunming mice. (a) Only 12 (20 μM, 100 μL) was injected as a control. (b) Mouse pretreated with rotenone for 1 h and then injected with 12. (c) Mouse successively injected with rotenone, NAC, and 12. (d) Relative fluorescence intensities of (a–c). Adapted with permission from ref. 36c. Copyright 2013 American Chemical Society. | ||
N, CC, CS and NN), lactams, and thiolactones, and oxidation of thioethers and selenides (Fig. 8).38a,b,39,40c,74
 | ||
| Fig. 8 Mitochondrion-targeted probes for HOCl, HOBr, 1O2 and ˙OH imaging. | ||
Wang's group reported 13 (PMN-TPP) as a colorimetric and ratiometric probe for HOCl imaging.38c In the presence of HOCl, the specific oxidative cleavage of CC disrupts the conjugated structure and disturbs the ICT process, which results in a decrease in the fluorescence at 640 nm as well as the appearance of a new emission peak at 522 nm. Confocal imaging experiments revealed that 13 localized well in the mitochondria of macrophages due to the TPP functionalization. 13 has been successfully used for ratiometric imaging of endogenous HOCl in nude mice with acute inflammation (Fig. 9).
 | ||
| Fig. 9 In vivo dual channel imaging of LPS and PMA treated nude mice with probe 13. Images were taken after stimulation for 0, 5, 10, 15, 20, 30, 45 and 60 min, respectively. Adapted with permission from ref. 38c. Copyright 2015 Royal Society of Chemistry. | ||
Apart from double bonds, many other groups have also been introduced for mitochondrion-targeted HOCl probe design. By using rhodamine as the fluorophore and benzoyl acetohydrazide as the responsive group, Yu's group developed several mitochondrion specific HOCl probes.39a,75 Based on the FRET pair of coumarin and rhodamine, TPP modified probe 14 (RMClO-2) was facilely prepared.39a14 has excellent membrane permeability, mitochondrion localization capacity, and HOCl selectivity, with an ultralow LOD of 0.84 nM. Benefiting from these features, 14 was used to quantify the basal mitochondrial HOCl of cancer cells for the first time.
As important immune cells, macrophages play a critical role in the innate immune system. Though it has been reported that HOCl could be detected in the cytoplasm, it was unclear whether HOCl can appear in the mitochondria during infection before Ma and co-workers developed the rhodamine based probe 15 (RSTPP).39b The fluorescence quantum yield of 15 remained low (<0.01), but was elevated to 0.34 in the presence of HOCl, with a >200-fold fluorescence intensity enhancement due to the oxidative cleavage of the thiolactone ring and formation of a large conjugated plane. 15 exhibits high specificity toward HOCl over other ROS with a LOD of 9 nM. Confocal laser fluorescence experiments revealed that this TPP decorated rhodamine thiolactone probe localizes at the mitochondria of RAW264.7 cells specifically. The HOCl fluctuation in macrophages during microbial infection was visualized for the first time, which further proved the great potential of subcellular localized probes for bioanalysis.
Later, a mitochondrion-targeted HOBr probe, 16 (RhSN-mito), was developed. Rhodamine 110 was employed as the fluorophore owing to its high quantum yield, excellent solubility and mitochondrion-targeting capability.43a16 exhibits a large Stokes shift (∼100 nm) after reacting with HOBr. The quantum yield of the final product was determined to be 0.68, and the LOD of 16 was 20 pM. Endogenous HOBr in HepG2 cells and zebrafish were tracked with this novel probe (Fig. 10). Very recently, TPP decorated probe 17 (HAP)43b was devised by Tian's group for tracing mitochondrial HOBr burst in RAW264.7 cells during O2˙− triggered oxidative stress (Fig. 11).
 | ||
| Fig. 10 In vivo fluorescence imaging of HOBr in zebrafish embryos. (a) Zebrafish fed with 16 for 30 min (50.0 μM), (b) zebrafish fed with Br− (100 μM) for 30 min and then fed with 16 (50.0 μM) for 30 min, and (c) zebrafish incubated with NAC (20.0 μM) for 30 min and then with 16 (50.0 μM) for 30 min. (d)–(f) are the corresponding bright-field images of (a)–(c). Adapted with permission from ref. 43a. Copyright 2017 American Chemical Society. | ||
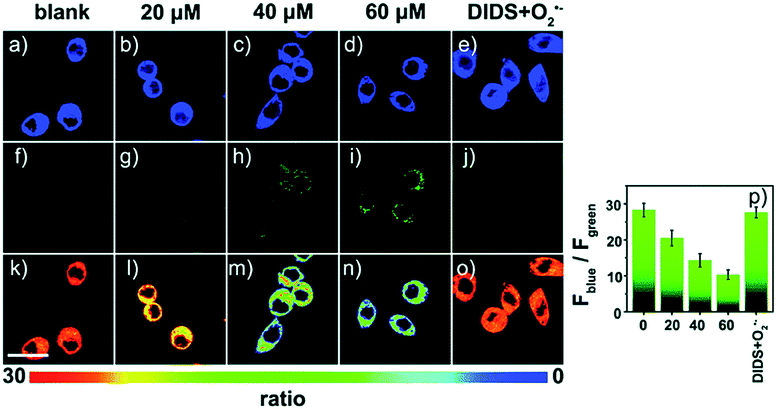 | ||
| Fig. 11 Confocal fluorescence images of RAW264.7 cells stained with 5 μM 17 under different treatments: (a, f and k) control; (b, g and l) 20 μM O2˙−; (c, h and m) 40 μM O2˙−; (d, i and n) 60 μM O2˙−; (e, j and o) 100 μM DIDS + 60 μM O2˙−. (p) The corresponding Fblue/Fgreen. Adapted with permission from ref. 43b. Copyright 2018 Royal Society of Chemistry. | ||
In 2014, Majima and co-workers reported a silicon-containing rhodamine-based probe, 18 (Si-DMA), for mitochondrial 1O2 imaging.41b In this probe, 9,10-dimethylanthracene (DMA) is employed as the 1O2 recognition moiety, and mitochondrion-specific cationic lipophilic silicon-containing rhodamine (Si-rhodamine) serves as the fluorophore. Initially, the fluorescence of rhodamine is quenched due to the PET effect between DMA and Si-rhodamine. During the reaction between 18 and 1O2, an endoperoxide is formed between anthracene and 1O2, resulting in the disruption of the PET process in the probe, accompanied by fluorescence intensity enhancement. Using this far-red fluorescent probe, the 1O2 generation in the mitochondria near the perinuclear region during PDT has been successfully recorded (Fig. 12). Using TPP as the targeting group and naphthalimide as the TP fluorophore, Zhang et al. reported 1O2 probe 19 (MNAH)41a and successfully visualized the mitochondrial 1O2 accumulation during PDT, which offered an alternative tool for tracing PDT in deeper tissues.
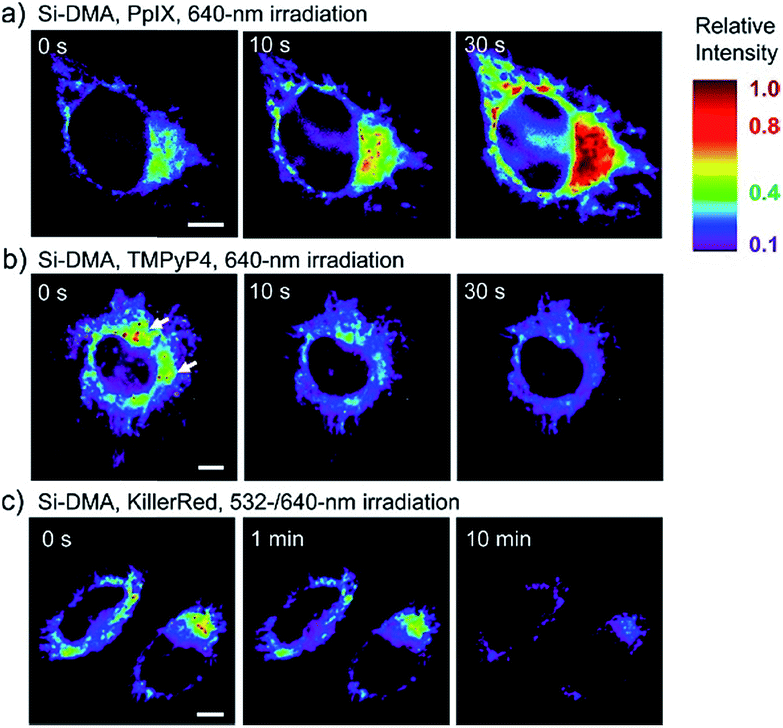 | ||
| Fig. 12 Fluorescence microscopy images of HeLa cells treated with 18 and (a) 5-ALA induced protoporphyrin IX (PpIX), (b) tetrakis(N-methyl-4-pyridyl)porphyrin (TMPyP4) and a lysosome marker (A647-dextran), and (c) mitochondrion-targeted KillerRed. (a and b) Cells were irradiated with a 640 nm laser to generate 1O2 and monitor the fluorescence induced by 18 simultaneously. (c) Two-color irradiation at 532 and 640 nm was used to excite 18 and KillerRed. Adapted with permission from ref. 41b. Copyright 2014 American Chemical Society. | ||
4.2. Imaging of RNS in mitochondria
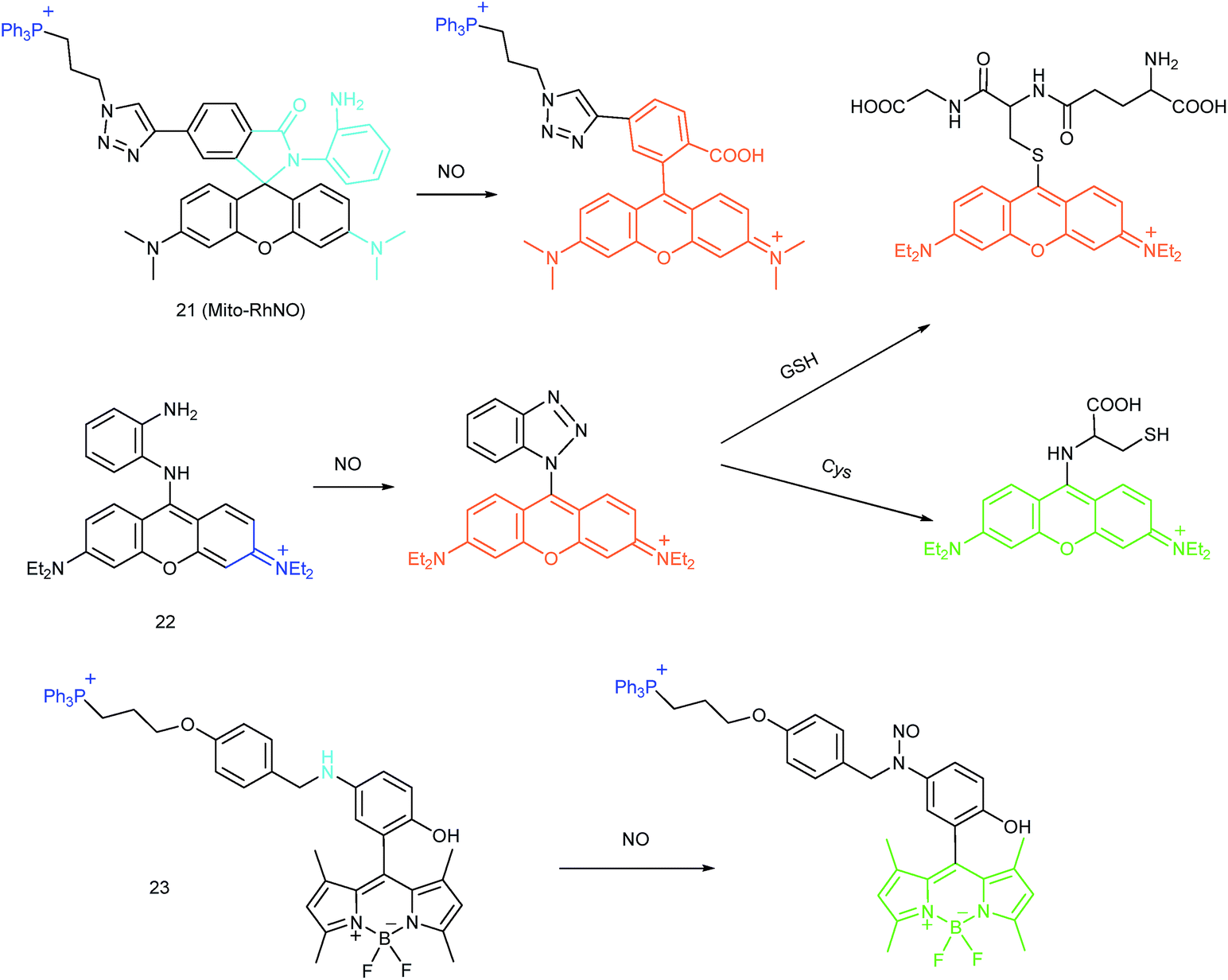
As another group of bioactive species for living cells, RNS also has critical impacts on mitochondrial functions.44,45,47,78 Tracing the real concentrations of mitochondrial RNS remains challenging, but it is vitally significant for investigating related physiological and pathological processes, which may offer new insights for chemical–biological bioanalysis and clinical disease theranostics.Jin et al. reported the first mitochondrion-targeted NO probe 21 (Mito-RhNO).48c This probe employed TPP as the targeting group, rhodamine as the fluorophore and o-phenylenediamine as the recognition moiety. The ring-opening of rhodamine spirolactam during the specific reaction between o-phenylenediamine and NO caused significant fluorescence enhancement at 585 nm. The LOD of 21 was determined to be 4.0 nM. Endogenous mitochondrial NO in the RAW264.7 macrophages under different stimulations was visualized with this biocompatible probe.
Guo et al. demonstrated another fluorescent probe (22) for mitochondrion-targeted NO imaging.48a22 was prepared by connecting o-phenylenediamine with cationic pyronin dye, which could effectively accumulate in the mitochondria. The fluorescence of 22 was dark due to the PET process. Addition of NO to the solution of 22 induced a 2743-fold fluorescence enhancement at 616 nm, attributed to the triazole generating reaction that lead to the disruption of the PET process. The LOD of 22 was calculated to be 12 nM. Interestingly, in the presence of Cys or GSH, a substitution–rearrangement reaction would occur and a bright green or red fluorescence could be generated, ensuring that the mitochondrial NO could be visualized in two distinct emission channels (Fig. 13).
 | ||
| Fig. 13 Fluorescent probes for imaging NO in mitochondria. | ||
Based on the N-nitrosation of aromatic secondary amines, Miao and coworkers developed mitochondrion-targeted NO probe 23.49 In this BODIPY based probe, TPP was introduced as the targeting group and an N-benzyl-4-hydroxyaniline moiety was the reaction site. The PET process in 23 was disrupted when the fast and specific N-nitrosation of the aromatic secondary amine occurred, resulting in a 92-fold fluorescence enhancement at 518 nm. The LOD of 23 was estimated to be 4.8 nM. With probe 23, the NO levels in macrophages in the resting state or under diverse stimulations were successfully visualized.
 | ||
| Fig. 14 Fluorescent probes for imaging ONOO− and HNO in mitochondria. | ||
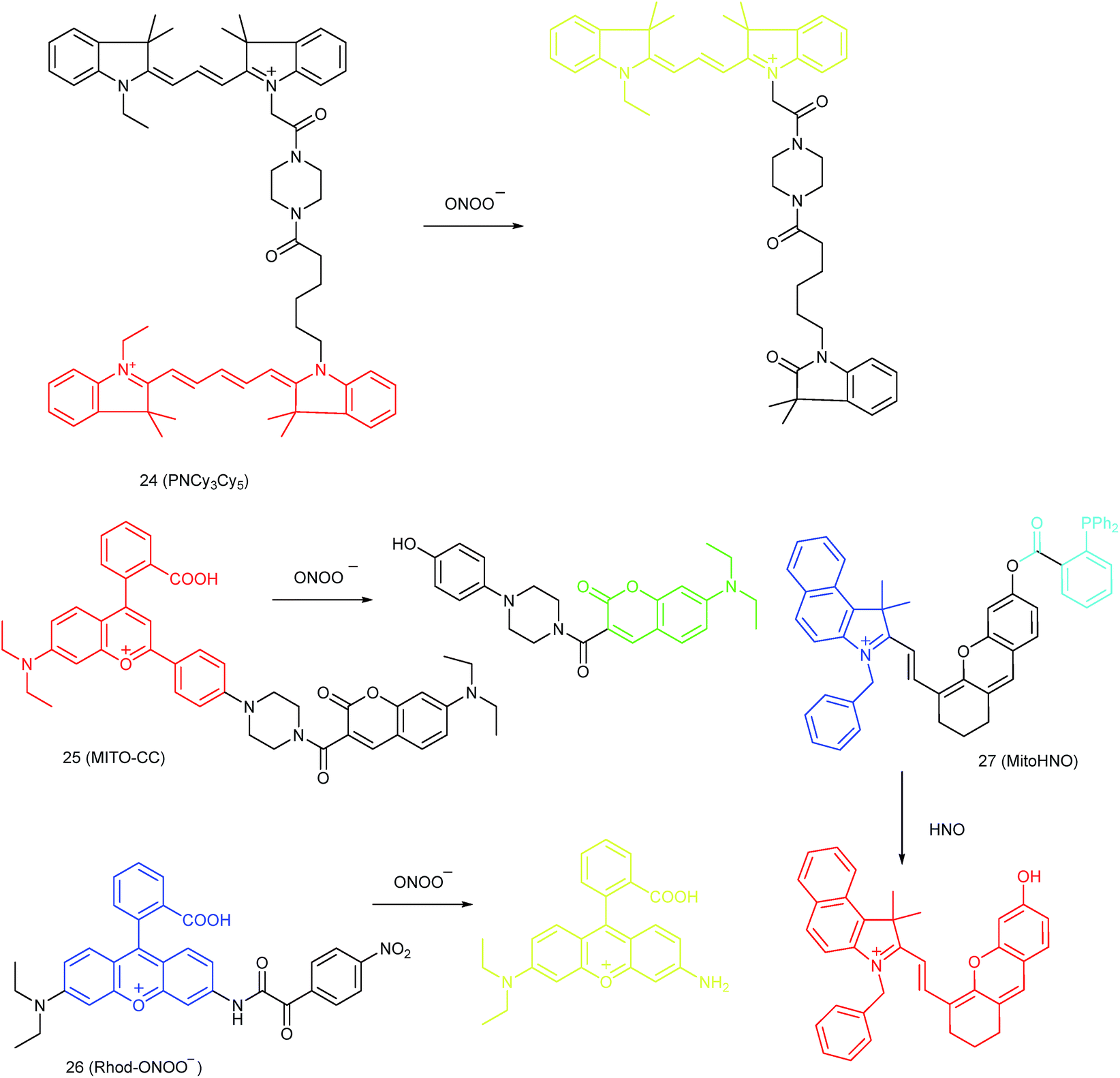
Qian and co-workers devised a ratiometric probe, 24 (PNCy3Cy5), for mitochondrial endogenous ONOO− imaging.4524 was prepared by covalent connection of the famous FRET pair, Cy3 and Cy5, through a 1.4 nm linker. In this probe, Cy5 could be readily oxidized by ONOO−, while Cy3 was stable due to the shorter polymethine chain. Under 530 nm laser excitation, 24 emitted fluorescence at 660 nm (belonging to Cy5) due to the FRET effect. Upon the addition of ONOO− to the probe solution, the fluorescence intensity at 660 nm decreased due to the oxidation of Cy5 and the emission band at 560 nm emerged as a result of disruption of FRET. Confocal imaging experiments revealed the excellent cellular permeability and mitochondrion-targeting capability of this probe, and the LOD of 24 was determined to be 0.65 nM. Endogenous ONOO− in macrophage cells under diverse stimulations was monitored in a ratiometric imaging manner, which suggested that 24 was promising for revealing the biological and pathological functions of ONOO−.
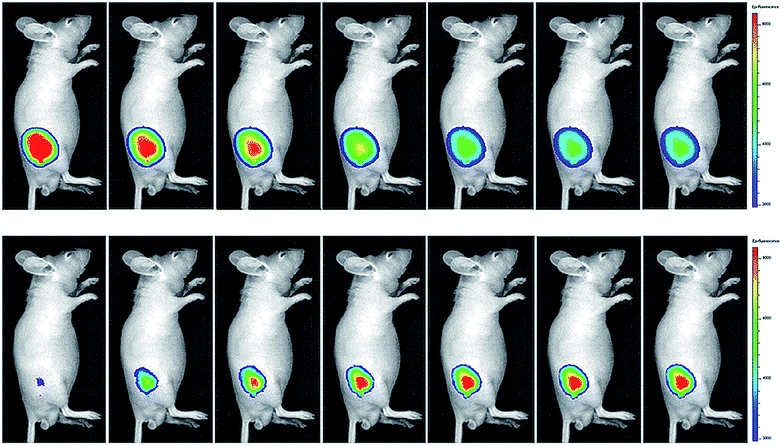
Yuan and co-workers reported TP probe 25 (MITO-CC) for mitochondrial ONOO− imaging.78 19 fluorescent compounds were screened under physiological conditions, and they obtained a mitochondrion-specific fluorophore with high sensitivity and selectivity toward ONOO−. By further connecting coumarin as the energy donor, a mitochondrion-targeted ratiometric probe with high fluorescence quantum yield, high molar absorbance, excellent photostability and solubility was obtained. 25 has a fast response rate (less than 20 s) and low LOD (1.13 × 10−8 M) toward ONOO−. Ratiometric imaging of endogenous mitochondrial ONOO− differences between normal and inflamed cells or tissues was realized with this probe (Fig. 15), and it suggested that 25 was favourable for tracing the small fluctuations of ONOO− in diverse biological processes.
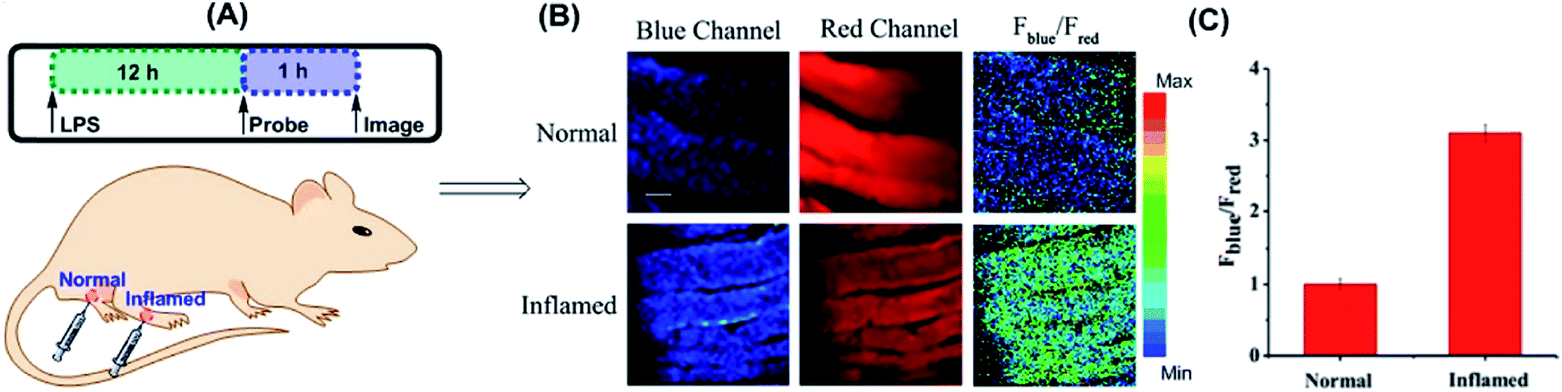 | ||
| Fig. 15 TP fluorescence imaging of ONOO− generation in inflamed tissues. (A) 200 μL of LPS (1 mg mL−1) was subcutaneously injected into the right leg of mice to cause inflammation. After 12 h, 20 μL of 500 μM 25 was subcutaneously injected in situ. After 1 h, the leg skin of mice was sectioned after being anaesthetized. (B) Fluorescence images of 25 in the normal and inflamed tissues. (C) Average Fblue/Fred intensity ratios in panel (B). Blue channel, λEm = 460–500 nm; red channel, λEm = 605–680 nm. λEx = 800 nm. Adapted with permission from ref. 78. Copyright 2016 American Chemical Society. | ||
DILI is a common threat for public health. It has been reported that ONOO− could be an important DILI indicator, so imaging of the ONOO− levels in living cells can be critical for drug toxicity testing. Yuan et al. further developed 26 (Rhod-ONOO−) by a combination of ICT and PET mechanisms, for mitochondrion-targeted ONOO− detection.47 Confocal imaging experiments in HepG2 cells revealed the excellent mitochondrion-targeting capacity of 26. Mitochondrial ONOO− generation during DILI was visualized with this probe, and the drug-induced hepatotoxicity pathway was successfully discriminated. Therefore, 26 is a powerful tool for DILI measurement and it also proved the great promise of subcellular localized fluorescent probes in bioanalysis and drug discovery.
In 2016, Yang et al. reported 27 (MitoHNO) as the first mitochondrion-targetable NIR HNO probe.81a A 2-(diphenylphosphino)benzoyl group was employed as the HNO recognition group, and a lipophilic indolium cation was used as the targeting moiety. During the reaction between 27 and HNO, 38-fold fluorescence intensity enhancement at 727 nm was observed due to the release of a fluorophore. The LOD of 27 was estimated to be 60 nM. Mitochondrial HNO in SMMC-7721 cells was specifically detected with this probe.
4.3. Imaging of RSS in mitochondria
As discussed above, mitochondria perform numerous respiration reactions to maintain cellular ATP synthesis, and this makes the mitochondrial ROS and RNS levels higher than those in other subcellular compartments. Reductive species are indispensable for signal transduction and protecting mitochondria from oxidative stress; therefore, they are also reliable biomarkers for diverse bioactivities as well as diseases.56,58,59,82 Fluorescent probes for tracing the localized RSS concentration in mitochondria hold great promise for advancing our understanding of the physiological and pathological functions of RSS, which should also be beneficial for the exploration of novel drugs toward diverse related diseases (Fig. 16). | ||
| Fig. 16 Fluorescent probes for imaging RSS in mitochondria. | ||
Kim's group reported a mitochondrion-targeted NIR probe, 28 (MitoGP), for GSH imaging.52 A heptamethine unit was used as the fluorophore owing to its NIR emissive and mitochondrion-targeting capacity, while a nitroazo group was employed as the GSH recognition group and fluorescence quencher. Significant fluorescence enhancement at 810 nm was observed in the presence of GSH, due to the specific 1,6-conjugate addition and subsequent elimination reaction triggered fluorophore release. 28 exhibited high selectivity toward GSH over other biothiols, and the LOD was estimated to be 10 μM. The mitochondrion-targeting capability of 28 was demonstrated to be comparable to that of the commercialized MitoTracker, and mitochondrial GSH in HeLa cells under different stimulations was easily monitored with this probe (Fig. 17).
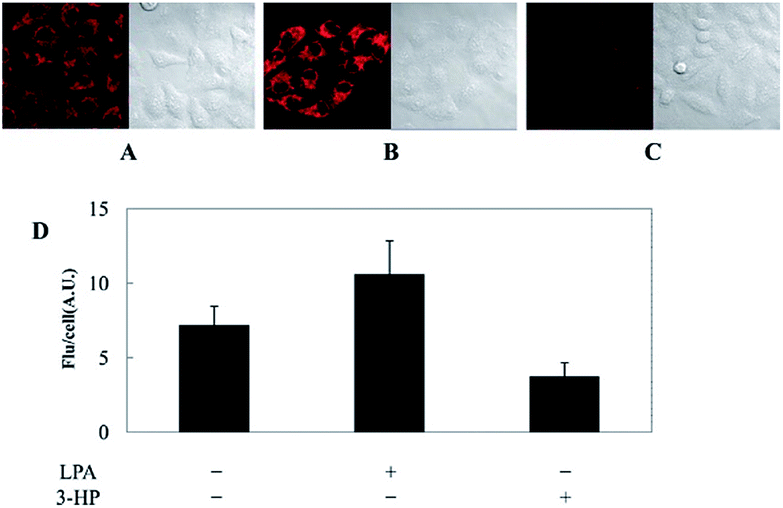 | ||
| Fig. 17 CLSM images of HeLa cells subjected to different treatments (A–C) with 28. (A) 28 only, (B) 28 + LPA (1.8 mM, 24 h), and (C) 28 + 3-HP (1 mM, 5 min then 2 h incubation); (D) their histograms of fluorescence intensities. Adapted with permission from ref. 52. Copyright 2014 American Chemical Society. | ||
Zhao et al. reported a BODIPY based probe (29) for monitoring mitochondrial GSH.83 The self-immolative group para-dinitrophenoxy benzyl pyridinium was introduced at the meso position of BODIPY, and it could simultaneously act as the targeting ligand and responsive group. Upon reaction with GSH, the dinitrophenyl moiety of 29 was first cleaved through an SNAr reaction; subsequently, the fluorophore BODIPY was released via a self-immolation reaction, which results a 32-fold fluorescence intensity enhancement at 599 nm. 29 was used for mitochondrion-targeted GSH imaging in HeLa cells.
Compared to traditional fluorescent probes, the design of molecular tools for bioactive species imaging based on a reversible reaction is still a challenge. However, reversible probes are promising for elucidating the dynamic states of analytes of interest in different processes, and would offer more information in cell imaging. Wang et al. introduced the first mitochondrion-targeted probe, 30 (MitoRT), for the reversible detection of GSH.51 TPP was linked with the fluorophore through the optimized 4-carbon linker. Benefiting from the reversibility, 30 responds to both increases and decreases of GSH levels and can be used for ratiometric imaging. The response time of this probe was less than 30 s. 30 has been successfully used to monitor mitochondrial GSH levels during redox perturbance. Importantly, with this probe, it was demonstrated that cells prioritize to maintain mitochondrial GSH levels during cell growth.
Li and Feng et al. reported a semiheptamethine derivative, 31 (NFL1), for selective detection of Cys in mitochondria.53b Based on the specific Michael addition and subsequent cyclization reaction, strong NIR emission intensity enhancement at 735 nm was observed in the presence of Cys, as a result of the disruption of the PET process. The LOD of 31 was estimated to be as low as 14.5 nM. 31 has been successfully applied for the assessment of mitochondrial oxidative stress. Cys imaging in a living nude mice model was also demonstrated owing to its NIR emissive features (Fig. 18).
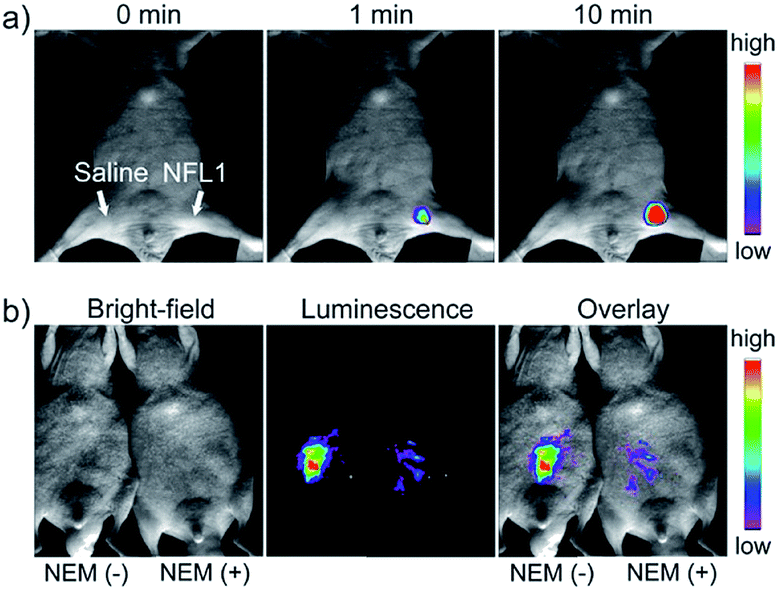 | ||
| Fig. 18 In vivo NIR imaging of living mice subjected to different treatments with probe 47. (a) The mice was treated with 0.1 mL of saline (left side) or 31 (10 μM, right side) solution and imaged at different times after subcutaneous injection of 47. (b) The left mice was injected with probe 31 (10 μM, 0.2 mL) only for 10 min. The right mice was preinjected with NEM (1 mM, 0.2 mL) for 30 min and then injected with 31 (10 μM, 0.2 mL) for 10 min. Adapted with permission from ref. 53b. Copyright 2015 American Chemical Society. | ||
Yoon et al. reported a naphthalimide-based probe (32) with the 7-nitro-1,2,3-benzoxadiazole (NBD) moiety for mitochondrial H2S detection.85 The fluorescence of 32 was dark due to the quenching effect of the NBD moiety, but it underwent 68-fold intensity enhancement at 532 nm in the presence of Na2S as a consequence of the cleavage of the NBD amine and release of a fluorophore. Confocal imaging experiments revealed that the probe localized in mitochondria well. The mitochondrial H2S of HeLa cells under different treatments could be monitored with this biocompatible probe.
Yang et al. devised a triarylboron based “turn-on” probe, 33 (TAB-2), for mitochondrial H2S imaging.56 The probe was prepared by functionalization of one diphenylamine and two cyclens at the para position of phenyl groups of triarylboron. After complex formation with Cu2+, the fluorescence of TAB-2 was quenched due to the inhibition of the ICT process. Upon exposure to H2S, dramatic fluorescence intensity recovery was observed; the LOD of 33 was 47 nM. Interestingly, the lipophilic cationic structure endowed 33 with the capability of mitochondrion-targeting. 33 has been employed for TP and fluorescence lifetime imaging of H2S in NIH/3T3 cells.
Taking advantage of the SNAP-tag technology, Pluth's group reported a probe, 34 (HSN2-BG), for discriminating the H2S release profiles of different H2S donor molecules.12b In view of the existence of H2S synthase in mitochondria, and some that the H2S donor can store in the lysosomes, they transfected and expressed AGT protein into mitochondria and lysosomes, respectively. Benefiting from the precise subcellular localization of the probes, they revealed the disparate H2S release profiles between different H2S donors in mitochondria and lysosomes, offering a new fluorescent tool for the investigation of subcellular H2S.
In 2015, Yuan and Chang et al. reported the first example of a mitochondrion-targeted ratio probe, 36 (Mito-Ratio-SO2).59a A mechanism study revealed that the nucleophilic addition of SO2 to 36 could generate two stable adducts and block the ICT process within the probe. The blocked ICT process between coumarin and indole groups resulted in a blue-shifted emission. Interestingly, the emission shift between the two bands was as large as 170 nm. 36 had high selectivity toward SO2 with a LOD of 0.09 μM. The generation of SO2 in the mitochondria of HeLa cells was detected with this ratiometric probe.
Zhao's group devised probe 37 (DSPT) based on the FRET mechanism, and it was constructed from the dansyl-piperazine-benzothiazolium conjugate platform.87 In this probe, the cationic benzothiazolium group was employed as the targeting group, while a neighbouring CC bond was the recognition moiety. The FRET process of the probe was “on” due to the spectral overlap, but was “switched off” in the presence of HSO3−, as a consequence of the fact that the Michael addition reaction disrupted the conjugated structure of the acceptor fluorophore. The LOD of 37 was 69 nM, which indicates that it was suitable for detecting SO2 derivatives in diverse real samples. The exogenous and endogenous SO2 derivatives in the mitochondria of HepG2 cells were visualized in a ratiometric imaging manner.
4.4. Challenges for mitochondrion-targeted probes
Benefiting from the critical biological function and high activity of mitochondria, fluorescent probes, taking mitochondria as the targeting sites, have been widely developed and employed for RONSS imaging in different cases. However, the majority of targeting ligands are lipophilic cationic structures that carry the risk of reducing MMP. Also, during diverse stimulations or fixation with chemical agents, these dyes can easily leak out due to the MMP lost, and cannot work in these situations. Genetically encoded probes are capable of covalently anchoring to mitochondria with high selectivity and may avoid the dye leakage. But the gene transfection efficiency cannot be guaranteed, while repeated washing may change the mitochondrial microenvironment. Also, the gene transfecting process makes the probes difficult to use for in vivo imaging (imaging mitochondria during liver/kidney IRI and so on). Moreover, there are hundreds of biomolecules in mitochondria, including enzymes, cations, anions, lipids, mitochondrial DNA, RNA and so on. In situ selective staining of bioactive molecules at ultralow concentrations (usually nanomolar amounts) using fluorescent probes is quite challenging. Many reported probes only work when large amounts of exogenous analytes are added or cells are stimulated by diverse chemical agents. Probes capable of selectively imaging bioactive species at basal concentrations are still rare, which hampers the further explorations of the physiological functions of these species. Ratiometric probes are promising for eliminating diverse interferences, but only limited mitochondrion-targeted ratiometric probes have been designed and constructed up to now.5. Lysosome-targeted probes
As the main digestive compartments of cells, lysosomes store numerous enzymes for degradation/recycling of diverse exogenous and endogenous biomolecules, and can effectively defend cells from infection, starvation and stress.3b,40a,b,88 The bioactivity of RONSS makes them feasible for regulating the enzyme activity in such organelle, and serious disorders can be caused when expressed in an uncontrolled manner. As people have gradually recognized the importance of lysosomes, more and more lysosome-targeted fluorescent probes have been explored for RONSS imaging in different study models.5.1. Imaging of ROS in lysosomes
Benefiting from the acidic and digestive lysosomal microenvironments, the activities of diverse ROS scavenging enzymes (such as SOD and peroxidase) in lysosomes are restricted, which facilitates the ROS accumulation in such single-layered membrane bounded organelle.28,89 The lysosomal ROS level is closely related to the lysosomal enzyme regulation, which can be critical for maintaining cellular homeostasis. Lysosomal oxidative stress caused by over-accumulation of ROS can result in lipid peroxidation, which will further cause lysosomal membrane permeabilization (LMP) and cell disorders.90 Therefore, monitoring the lysosomal ROS concentration has been recognized as an important task for molecular pathology. Novel fluorescent probes capable of tracing the lysosomal ROS fluctuation are highly significant for bioanalysis (Fig. 19).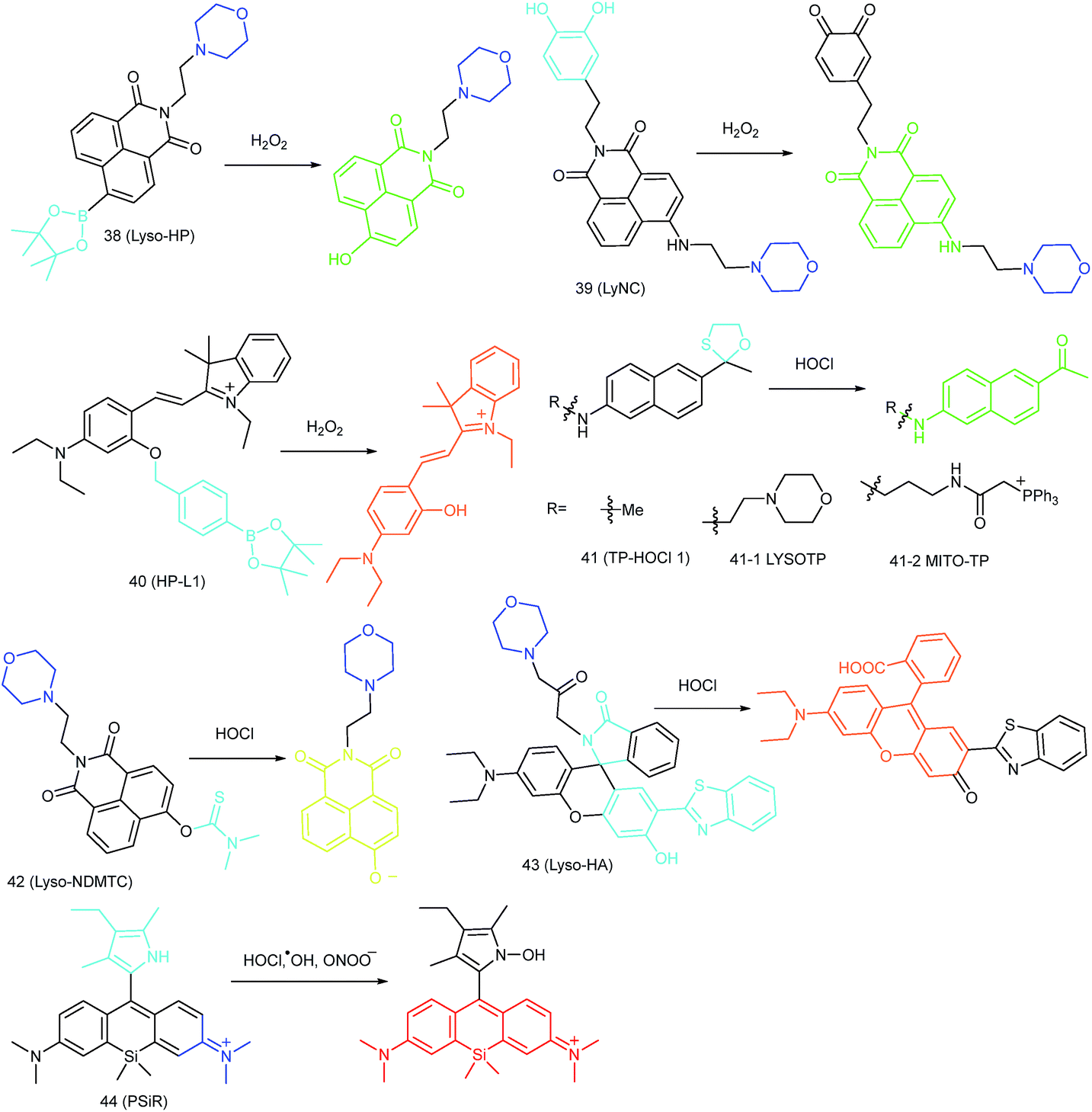 | ||
| Fig. 19 Fluorescent probes for imaging ROS in mitochondria. | ||
Kumar et al. reported another morpholine decorated naphthalimide based probe, 39 (LyNC), for lysosomal H2O2 imaging, and it employed catechol as the reaction site.37 In the absence of H2O2, the PET process between the catechol moiety and naphthalimide blocked the fluorescence; in the presence of H2O2, the probe emitted evidently due to the disruption of the PET process caused by oxidation of catechol into o-quinone. 39 was successfully used for tracing the lysosomal H2O2 in C6 glioma cells, BV-2 microglial cells and nematodes under different stimulations.
In view of the acidic lysosomal microenvironments, probes that can simultaneously respond to the pH and H2O2 should be promising for reducing the background during fluorescent imaging. Wu and Zhao et al. introduced an acidic pH activatable probe, 40 (HP-L1), for lysosomal H2O2 detection.28 In this probe, boronate was employed as the responsive moiety, and spirobenzopyran as the pH switchable fluorophore. This probe could only emit intensive fluorescence in lysosomes after reacting with H2O2, which made it a powerful tool for avoiding the background fluorescence during lysosomal H2O2 imaging.
In 2015, Chang's group reported an “off–on” HOCl probe, 41 (TP-HOCl 1), with a fast response rate (within seconds) and low LOD (16.6 nM).40b The response mechanism of 41 was the HOCl mediated oxidative deprotection of the oxathiolane/mercaptal group, which resulted in the recovery of the “push–pull” structure in acedan and production of fluorescence at 500 nm. 41 exhibited remarkable specificity toward HOCl among diverse ROS. Further modification of morpholine or TPP with 41 yielded lysosome- and mitochondrion-targeted probes (41-1 LYSOTP and 41-2 MITO-TP, respectively). They were successfully employed for TP imaging under both in vitro and in vivo conditions, and the subcellular distribution of HOCl was elucidated.
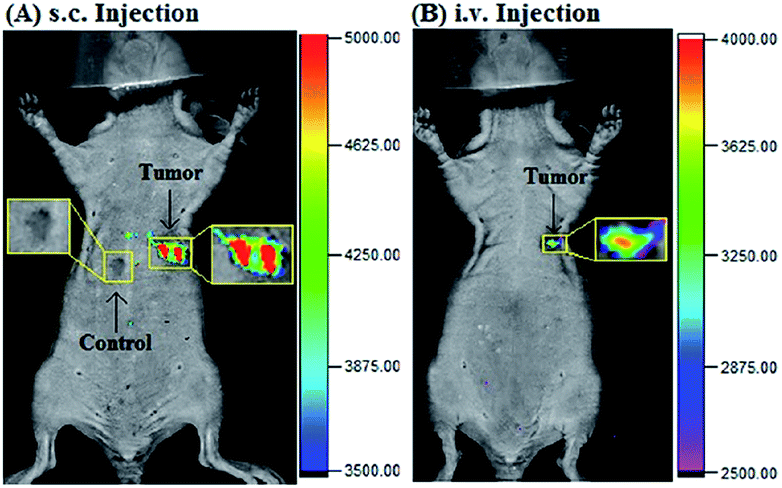
Our group prepared a thiocarbamate derivative based TP probe, 42 (Lyso-NDMTC), for lysosomal HOCl imaging.88 This probe was fabricated by decorating naphthalimide with morpholine and N,N-dimethylthiocarbamate (DMTC). 42 emitted faintly initially but highly after reacting with the analyte of interest, due to the electrophilic addition reaction of Cl+ induced cleavage of DMTC. Benefiting from the reaction reactivity of DMTC and sensitivity of the ICT process, the LOD of 42 was estimated to be 7.6 pM. 42 is the first probe capable of imaging the basal lysosomal HOCl at the picomolar level, and it has been successfully employed to distinguish tumor tissue from normal tissue in vivo with the assistance of TP microscopy (Fig. 20).
 | ||
| Fig. 20 Top: CLSM images of live HepG2 cells pretreated with 42 (20.0 μM) and Lyso-Tracker Red (100.0 nM) for 20 min: (a) bright-field image, (b) green channel for probe fluorescence, (c) red channel for Lyso-Tracker fluorescence, (d) merged image from panels (b) and (c); and (e) intensity correlation plot of 42 and Lyso-Tracker Red. Bottom: Two-photon fluorescence imaging at different depths and the 3D distribution of HOCl in the normal and cancerous breasts of mice pretreated with 42 (100.0 μM) for 30 min. Fluorescence images were obtained with an 800 nm light source. Adapted with permission from ref. 88. Copyright 2016 American Chemical Society. | ||
Lin's group devised a lysosome-targeted ratiometric HOCl probe, 43 (Lyso-HA), based on the ESIPT mechanism.92 Before coming into contact with HOCl, 43 adopted the spirocyclic form and the probe underwent the ESIPT process between the phenolic hydroxyl group and benzazole ring, which gave emission at 450 nm. Upon reaction with HOCl, the spirocyclic ring opened and there was no proton close to the benzazole ring. The blocking of the ESIPT process resulted in a decrease in the emission peak at 450 nm as well as the appearance of a new emission band at 586 nm. Benefiting from the remarkable emission wavelength red shift (136 nm), 43 is an ideal tool for exogenous and endogenous lysosomal HOCl imaging (Fig. 21).
 | ||
| Fig. 21 Fluorescent probes for imaging RNS in lysosomes. | ||
 | ||
| Fig. 22 Imaging tumor using 44 in the tumor-bearing mouse xenograft models prepared by subcutaneous inoculation of HeLa cells. (A) The mouse was subcutaneously injected with 44 (2 mM, 10 μL) in the tumor region and normal region, respectively, and then imaged immediately. (B) The mouse was intravenously injected with 44 (1.5 mg kg−1), and then imaged after 25 min. Adapted with permission from ref. 89. Copyright 2017 Elsevier Ltd. | ||
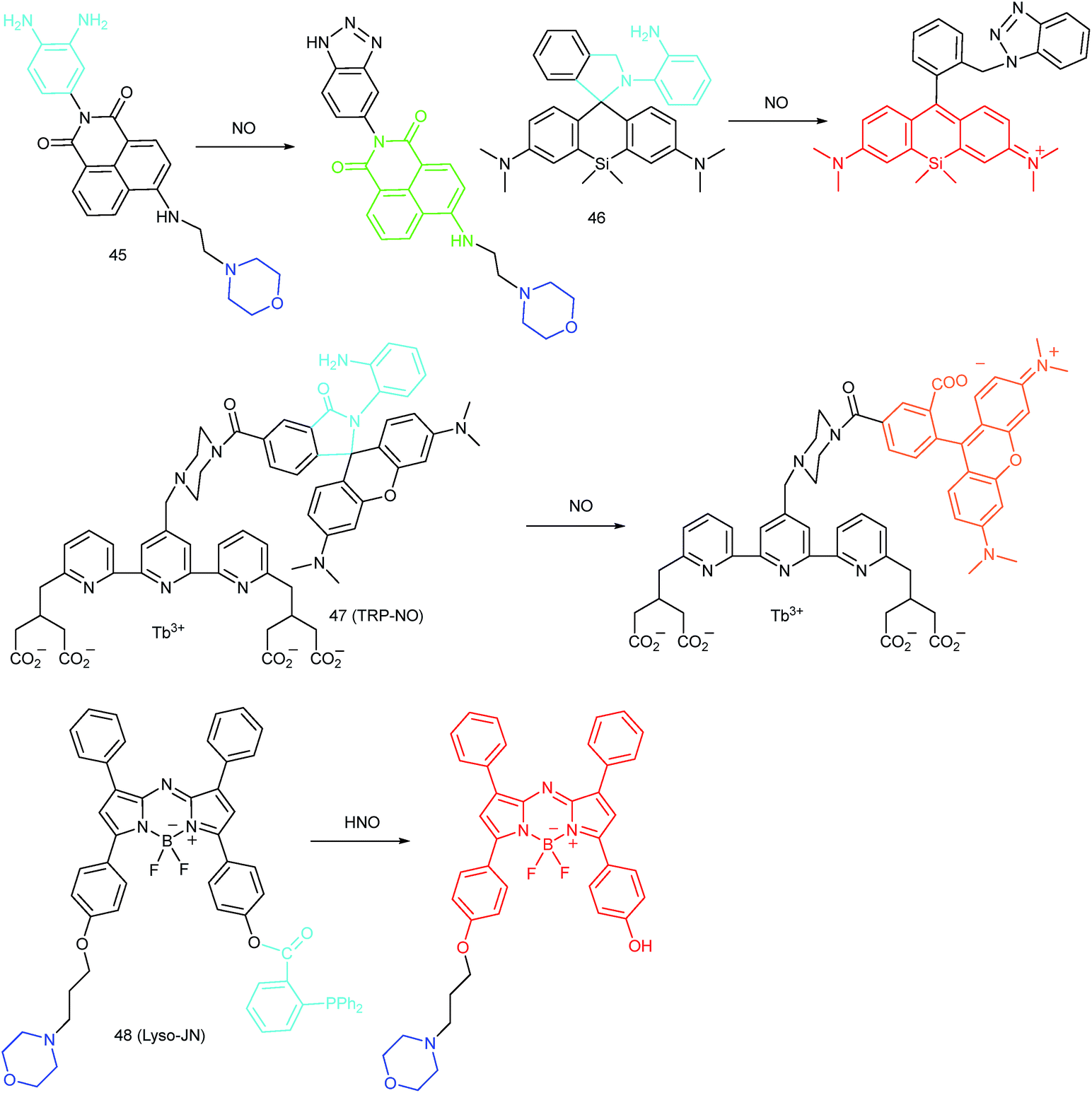
5.2. Imaging of RNS in lysosomes
RNS in lysosomes are important for lysosomal microenvironment regulation. The overproduction of RNS in lysosomes can cause lysosomal nitrative stress and diverse disorders.48b,d,50a,93 Therefore, lysosomal RNS can be promising biomarkers for diverse diseases. For example, overproduction of NO in the lysosomes was discovered in diverse cancerous and inflamed cells.93b Thus, lysosome targetable RNS probes are helpful for further understanding the biological and pathological functions of these molecules.In 2017, Guo's group developed a Si-rhodamine deoxylactam-based fluorescent probe (46) for the detection of lysosomal NO.48b Benefiting from the characteristics of the PET mechanism and the Si-rhodamine fluorophore, this probe had a large fluorescence “off–on” ratio (6300-fold), strong emission intensity, low detection limit (0.12 nM), fast response rate (within seconds) and high specificity. Interestingly, 46 localized in the lysosomes specifically owing to its easy protonation in mild acidic environments. Endogenous NO in a series of normal and inflamed cells as well as streptozotocin-treated mouse models was successfully visualized with this probe (Fig. 23).
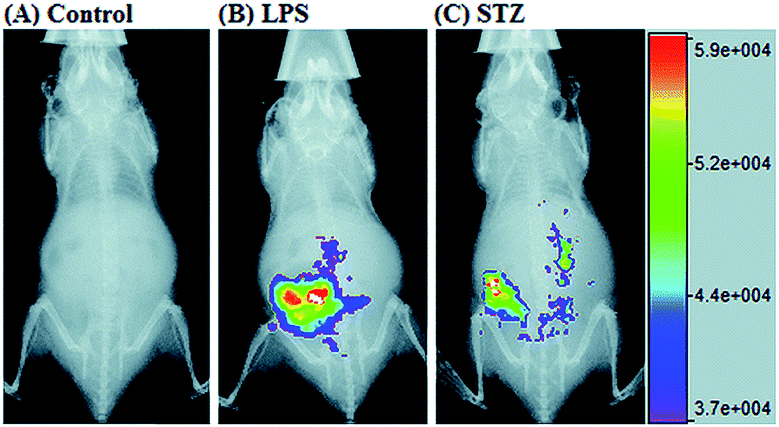 | ||
| Fig. 23 Overlay of the fluorescence and X-ray images of mice. (A) The mouse was intraperitoneally injected with 46 (100 μL, 2 μM) for 30 min. (B) The mouse was first intraperitoneally injected with LPS (1 mg mL−1, 100 μL) for 24 h, and then intraperitoneally injected with 46 (2 μM, 100 μL) for 30 min. (C) The mouse was first intraperitoneally injected with streptozotocin (40 mg kg−1) for 24 h, and then intraperitoneally injected with 46 (100 μL, 2 μM) for 30 min. Adapted with permission from ref. 48b. Copyright 2017 Royal Society of Chemistry. | ||
Song and Yuan et al. developed a time-gated luminescence probe, 47 (TRP-NO), for in vivo NO imaging.48d The luminescence resonance energy transfer process from the luminescent Tb3+ complex to rhodamine was activated when the non-emissive spirolactam derivative was opened upon reacting with NO. 47 could be used for ratiometric and luminescence lifetime imaging, and the LOD of this probe was determined to be 1.8 μM. Interestingly, dye co-localization studies revealed that 47 had high affinity toward lysosomes. NO imaging in HepG2 cells and Daphnia magna was realized with this probe (Fig. 24).
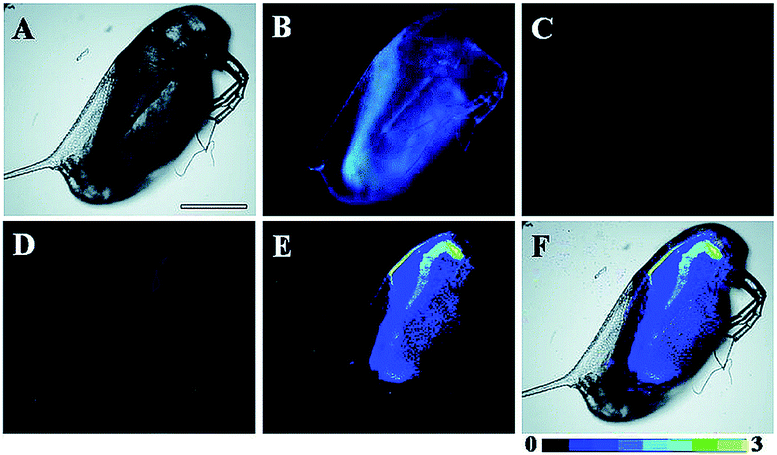 | ||
| Fig. 24 Bright-field (A), steady-state (B) and time-gated ((C) Tb3+ luminescence; (D) rhodamine luminescence) luminescence images of the 47 loaded D. magna after being treated with 0.5 mM NOC-13 for 10 min. (E) is the ratiometric (ratio = Ired/Igreen) luminescence image of the D. magna. (F) is the merged image of brightfield and ratiometric images. Scale bar: 200 μm. Adapted with permission from ref. 48d. Copyright 2017 Royal Society of Chemistry. | ||
5.3. Imaging of RSS in lysosomes
RSS are also important for lysosomal microenvironment adjustment and cellular signal conduction.53a,94 Recently, some attention has been paid to the design and application of novel probes for lysosome-targeted detection of RSS. However, this type of probe is much rarer than mitochondrion-targeted ones (Fig. 25). | ||
| Fig. 25 Fluorescent probes for imaging RSS in lysosomes. | ||
Zhou and Zhang et al. reported a coumarin based probe (52) for lysosome-targeted H2S imaging.9552 was facilely prepared by connecting two coumarin fluorophores with disulfide bonds. Before the reaction with H2S, the emission of 52 remained weak. However, after coming into contact with H2S, 15-fold fluorescence enhancement was observed due to the specific cleavage of disulfide bonds. Interestingly, co-localization experiments demonstrated the lysosome-targeting capability of 52. Fluorescence imaging of endogenous and exogenous H2S in HeLa cells, D. melanogaster and C. elegans was successfully realized with this probe.
5.4. Challenges for lysosome-targeted probes
Significant progress has been made in the development of lysosome-targeted fluorescent probes in recent years. However, there are still several limitations for related probes. For example, the targeting mechanisms of most developed probes are the trapping of protonated lipophilic amines, which cannot be used to distinguish between endosomes, autophagosomes, autolysosomes and other acidic compartments. Moreover, these ligands are toxic to living cells because they can alkalize lysosome microenvironments and hinder the smooth progress of diverse lysosome activities, which make them unsuitable for long-term tracing. Therefore, lysosome selective probes without the alkalization effect are required. As discussed above, genetically encoded probes need to be frequently washed to diminish the background during imaging, and this can also influence the lysosomal microenvironments. Therefore, designing bio-orthogonal probes with strong anti-interference capability, excellent selectivity and high sensitivity is still hard. Moreover, some probes may be quenched in the acidic lysosomal microenvironments, which poses extra challenges for designing related probes. Most of the current developed probes emit in the visible region, indicating that they cannot be used for deep tissue imaging due to their poor penetration capacity.6. Endoplasmic reticulum targeted probes
The ER is the main place for protein fabrication, modification, folding and transportation. It also takes charge of the synthesis of other molecules, like lipids and carbohydrates, as well as Ca2+ storage.30b,96 During synthesis of diverse proteins, RONSS are indispensable for controlling their functional three-dimensional structures and chemical groups.50b,97 Therefore, maintaining suitable oxidative microenvironments is essential for ER functions. Once the redox homeostasis in the ER is out of control, it can hamper the formation of proper protein structures and cause diverse disorders as well as ER stress.30a,96b,c Development of ER-targeted probes and tracing localized RONSS has been recognized as a new strategy for investigating the molecular mechanisms of different diseases.6.1. Imaging of ROS in the endoplasmic reticulum
ROS are critical for protein synthesis because they are involved in the formation of the disulfide bond, which is essential for protein folding. However, overexpression of ROS in the ER may cause oxidative stress, which can lead to protein crosslinking and further result in morbigenous effects on living cells. Developing novel fluorescent probes capable of tracing the ROS flux in the ER is significant for revealing the potential physiological and pathological functions of ROS during different bio-events, which should also help to improve the understanding of different major diseases (Fig. 26). | ||
| Fig. 26 Fluorescent probes for imaging of ROS and RNS in the ER. | ||
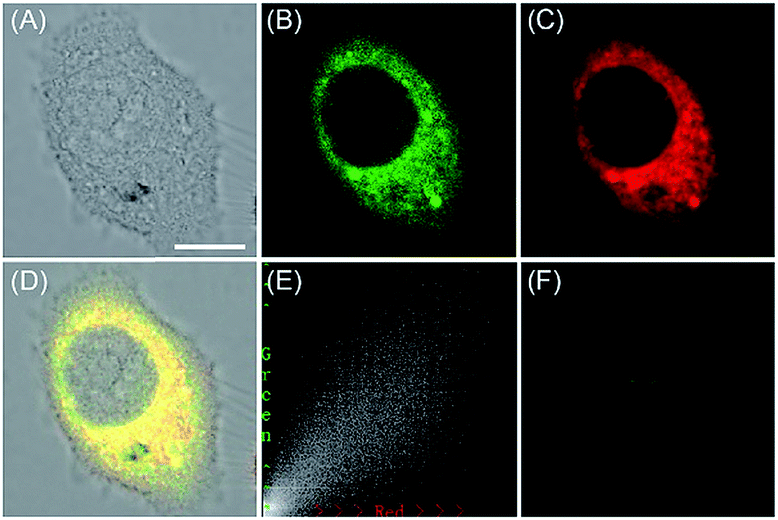 | ||
| Fig. 27 The colocalization imaging of 53 and ER-tracker Red in Tm-treated HepG2 cells. (A) The bright-field image. (B) The fluorescence image of 53 (10 μM, Ex = 405 nm, collected 420–480 nm). (C) The fluorescence image of ER-Tracker Red (0.5 μM, Ex = 543 nm, collected 580–630 nm). (D) The merged image of (A)–(C). (E) Intensity correlation plot of stain 53 and ER-Tracker Red. (F) The fluorescence image of ER-BZT in Tm-induced HepG2 cells pretreated with Tiron (5.0 mM) for 20 min. Scale bar: 10 μm. Adapted with permission from ref. 30a. Copyright 2016 Royal Society of Chemistry. | ||
ER-selective fluorescent probes based on other groups have also been developed. Yoon, Bouffard and Kim reported N-heterocyclic carbene borane-based HOCl probe 56 with ER-targeting capability.98 In the presence of HOCl, the specific oxidation of the B-H bond occurred, yielding the emissive naphthoimidazolium derivative and a 470-fold ratiometric signal response. Confocal imaging experiments revealed that probe 56 overlapped well with commercialized ER-Tracker Red (Fig. 28), and HOCl imaging in RAW264.7 cells as well as rat hippocampal slices was realized.
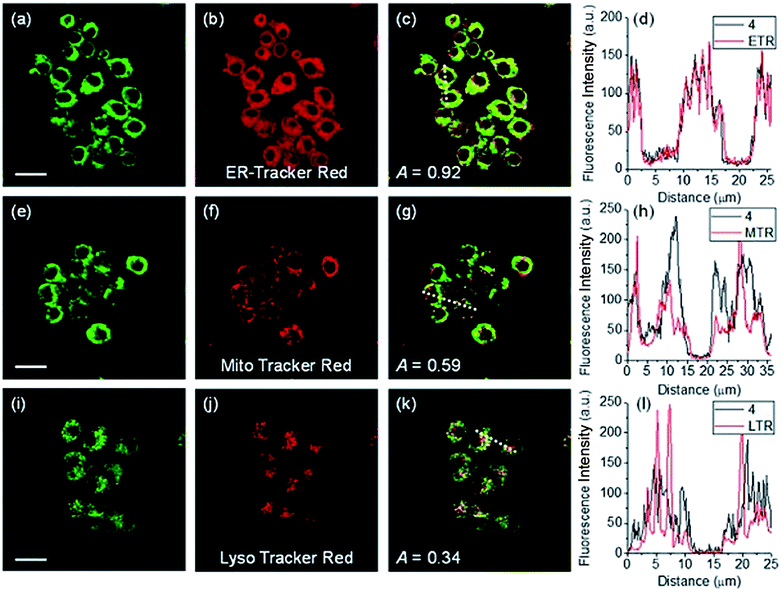 | ||
| Fig. 28 TPM (a, e, and i), OPM (b, f, and j), and merged (c, g, and k) images of RAW264.7 cells colabeled with 56 (10 μM) and organelle trackers (1.0 μM). Line profile of fluorescence intensity (d, h, and l) obtained from the corresponding cells images. Scale bars = 20 μm. Adapted with permission from ref. 98. Copyright 2016 Royal Society of Chemistry. | ||
6.2. Imaging of RNS in the endoplasmic reticulum
Although fluorescent probes for tracing RNS in living cells have attracted significant interest during the past few decades, probes capable of imaging the RNS fluctuations in subcellular structures are still rare, especially in the ER. However, ER nitrative stress caused by RNS overproduction also has deadly effects on living cells. Therefore, ER-targeted RNS probes are urgently needed due to their potential in early warning and diagnosis. Several groups have devoted efforts and made progress in related areas in the past 3 years. | ||
| Fig. 29 (A) Preparation of normal tissues and tunicamycin-treated tissues from a mouse model. (B) Two-photon fluorescence images of tissues during ER stress: (a) normal tissues, (b) tunicamycin-treated tissues, and (c) relative pixel intensities for images (a) and (b). Scale bar is 100 μm. Adapted with permission from ref. 96a. Copyright 2016 Royal Society of Chemistry. | ||
6.3. Imaging of RSS in the endoplasmic reticulum
ER-targeted RSS probes are beneficial for mapping the fluctuation of RSS during diverse processes, and this can offer direct evidence for the cellular functions of RSS. Related investigations have gradually received attention, and several probes have been devised and successfully used for RSS imaging in the ER. But it should be recognized that this is still in its infancy, there is plenty of room for better performance fluorescent probes.Recently, Das, Thomas and co-workers devised probe 62 (MB-Sn) for ER H2Sn imaging.97c The fluorescence of the probe is blocked initially. Upon reacting with H2Sn, the cleavage of a thioester and subsequent nucleophilic attack occurs, releasing a fluorophore. The LOD of 62 was as low as 26.01 nM. Benefiting from the excellent photostability and ER-targeting capability, 62 was employed for imaging H2Sn in the ER of living cells with the assistance of super-resolution structured illumination microscopy, which showed promise for mapping the intracellular distribution of H2Sn and further exploring their biological functions. Very recently, Zhou's group reported a probe, 63 (ER-H2S2), and successfully traced H2S2 in the ER of living cells as well as zebrafish.100
6.4. Challenges for endoplasmic reticulum targeted probes
Over the past three years, a series of ER-targeted fluorescent probes have been developed for selectively staining bioactive species in different research models, such as diabetes, inflammation and depression. However, the targeting mechanisms of ER-selective probes are still unknown, which severely limited our pursuit of better performing fluorescent tools and further understanding the chemical–biological functions of different species. Conjugating responsive groups with ER-targeted dyes seems to be feasible, but the subtle alteration of molecular charges and bulk usually hampers the localization capability. The protein tagging technique has also been employed to resolve the localization of fluorescent probes, but the complexity as well as efficiency remain to be further resolved. Moreover, almost all the reported probes emit visible light, and the in vivo applications of these probes are limited. Many bioactive species still cannot be visualized in the ER due to the lack of applicable probes. Probes for the quantification of the basal concentrations of diverse species in the ER are also rare (Fig. 30). | ||
| Fig. 30 Fluorescent probes for imaging RSS in the ER (60–63) and ROS in the Golgi apparatus (64 and 65). | ||
7. Golgi apparatus-targeted probes
To reveal the intrinsic molecular mechanisms of hepatic IRI, our group developed an L-cysteine containing TP probe, 64 (CCA), for reversible imaging of the O2˙− fluctuation in the Golgi apparatus.31a64 was prepared by connecting caffeic acid with L-cysteine through an amide bond. When reacting with O2˙−, the fluorescence intensity of 64 increased linearly to the change of O2˙− concentration in the range of 0–5 μM, and the LOD of this probe was estimated to be 18 nM. Benefiting from the abundance of cysteine residue recognition receptors in the Golgi apparatus, probe 64 could target this organelle effectively. With this probe, the significant increase of O2˙− levels in the Golgi apparatus of mice liver during IRI was disclosed by using TP microscopy. More significantly, the tumor necrosis factor (TNF-α) as the positive O2˙− mediator was demonstrated (Fig. 31). Therefore, 64 not only revealed the molecular mechanism of hepatic IRI, but also provided a promising treatment strategy, which further proved the significance of organelle-targeted fluorescent probes for disease diagnosis and drug discovery.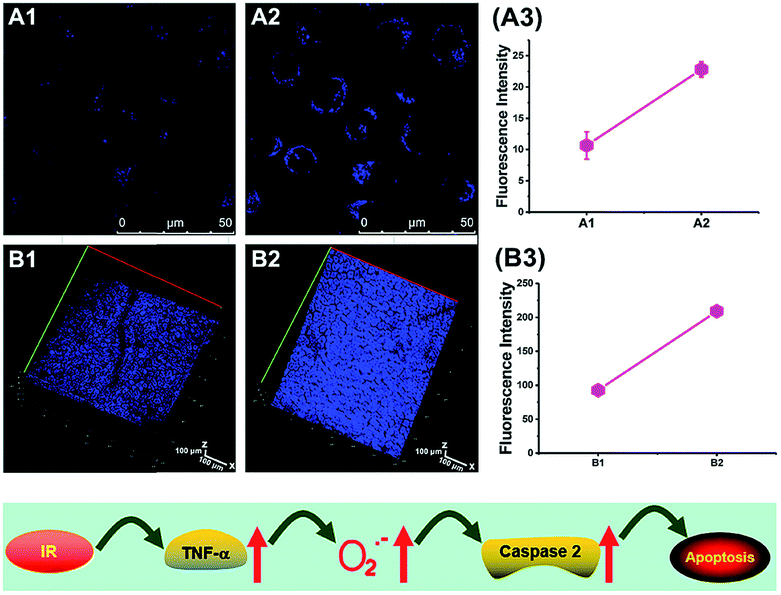 | ||
| Fig. 31 Top: Increased O2˙− levels in IR cells and mice. (A) The normal (A1) and IR hepatic (A2) cells loaded with 10 μM 64 for TP fluorescence imaging, and (A3) the average fluorescence intensity output of (A). (B) The in vivo 3D images of normal (B1) and IR mice (B2) injected with 10 μM 64 for TP fluorescence imaging, and (B3) the average fluorescence intensity output of (B). Images were acquired at an excitation wavelength of 800 nm and emission wavelengths corresponding to the blue channel of 430–530 nm. Bottom: Model for the signalling role of O2˙− in IR cells and mice. Adapted with permission from ref. 31a. Copyright 2019 Royal Society of Chemistry. | ||
Das, Thomas and Smythe et al. reported BODIPY derivative probe 65 (SF-1) for HOCl imaging.97b Interestingly, the low charge/intermediate amphiphilicity made 65 selectively accumulate in the Golgi apparatus and lysosomes, and “turn-on” fluorescence is observed only upon addition of HOCl into the probe solution, with a LOD of 4.3 nM. Endogenous HOCl in the Golgi apparatus and lysosomes of macrophage cells under LPS stimulation was visualized with 65 with the assistance of structured illumination microscopy, with a lateral resolution of 120 nm.
7.1. Challenges for Golgi apparatus-targeted probes
Golgi apparatus-targeted RONSS fluorescent probes have not received enough interest in the past decade, mostly due to the lack of efficient targeting ligands. Although several probes have been reported for imaging metal ions in the Golgi apparatus, the targeting mechanisms should be explored for further accelerating the development of related probes. L-Cysteine has been proved to be a promising targeting ligand, but the universality remains to be further checked. Though enzyme-activated probes hold promise for localization selective imaging, the scope has not been extended to Golgi apparatus-targeted RONSS probes.8. Probes for multi-parameter imaging
Apart from the above-discussed probes that can only image one species in different organelles, probes for sequential or simultaneous tracing of two or more targets have been recognized to be another group of important tools for bioanalysis, owing to their potential in clarifying the inter-conversion and “crosstalk” between different species as well as different organelles.8c,101 Such interesting tasks can be realized by using fluorescent probes with two or more responsive groups or employing several probes without spectral overlap. Nanoprobes containing several recognition molecules are also alternative candidates.8.1. Simultaneous imaging of bioactive species in an organelle
RONSS are highly reactive, interactive and interconvertible molecules. Sequential or simultaneous imaging of two or more species can reveal their internal relationships in specific organelles and further advance our understanding of diverse biological events.30b,101c,102 Probes for sequential or simultaneous imaging can be applied for tracing diverse critical biological events. For example, probes capable of simultaneous imaging are helpful for monitoring the real-time distribution of different species, and they can even be explored to identify the “crosstalk” between different subcellular structures. On the other hand, probes for sequential imaging are helpful for revealing the upstream and downstream relationships between different species. Recently, several probes have been developed for organelle-targeted multiple analyte imaging, and they can be divided into molecular probes and nanoprobes (Fig. 32). | ||
| Fig. 32 Molecular probes for simultaneous imaging of bioactive species in one organelle. | ||
The inter-relationship of mitochondrial O2˙− and pH is unclear for cellular signal transduction and homeostasis. Novel probes for precise evaluation of the subcellular pH and ROS fluctuation in the mitochondria are beneficial for revealing their interactions. A TP probe, 67 (CFT), was reported by our group.102d67 is prepared by covalent linkage of caffeoyl, fluorescein and TPP. This probe could make distinguishable optical signal changes upon coming into contact with different analytes. Benefiting from the reversible response and specificity, instantaneous, dynamic and reversible monitoring of the concentration fluctuations of H+ and O2˙− in mitochondria of 4T1 cells was achieved with the assistance of TP microscopy. Importantly, with this probe, Drp1 deficiency was revealed to be a promising signal pathway to induce mitochondrial hyperfusion and subsequent cell apoptosis.
Unlike the complicated design of multi-responsive probes, simultaneous employment of different probes with distinguishable spectral properties to detect bioactive species in specific organelles is more feasible and easier to perform. Using probe 68-1 (M-H2O2, H2O2 probe, LOD = 18 nM) and 68-2 (M–Zn, Zn2+ probe, LOD = 11 nM), simultaneous imaging of H2O2 and Zn2+ fluctuation in the mitochondria of living cells was realized by our group.102bIn vitro and in vivo tests revealed the synergistic concentration enhancement of H2O2 and Zn2+ in mitochondria during apoptosis and inflammation (Fig. 33), and this offered a new idea for investigating the correlations between different bioactive species in diverse organelles of living cells.
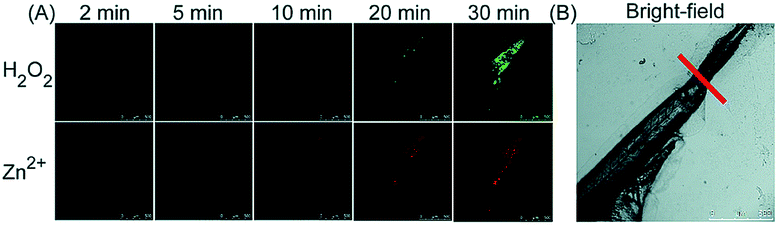 | ||
| Fig. 33 The simultaneous fluorescence imaging of H2O2 and Zn2+ in zebrafish upon local injury of the tail fin. (A) The fluorescence images of 68-1 (50 mM, green channel) and 68-2 (50 mM, red channel). (B) The bright-field image (the red line indicates a cut injury). Adapted with permission from ref. 102b. Copyright 2016 Royal Society of Chemistry. | ||
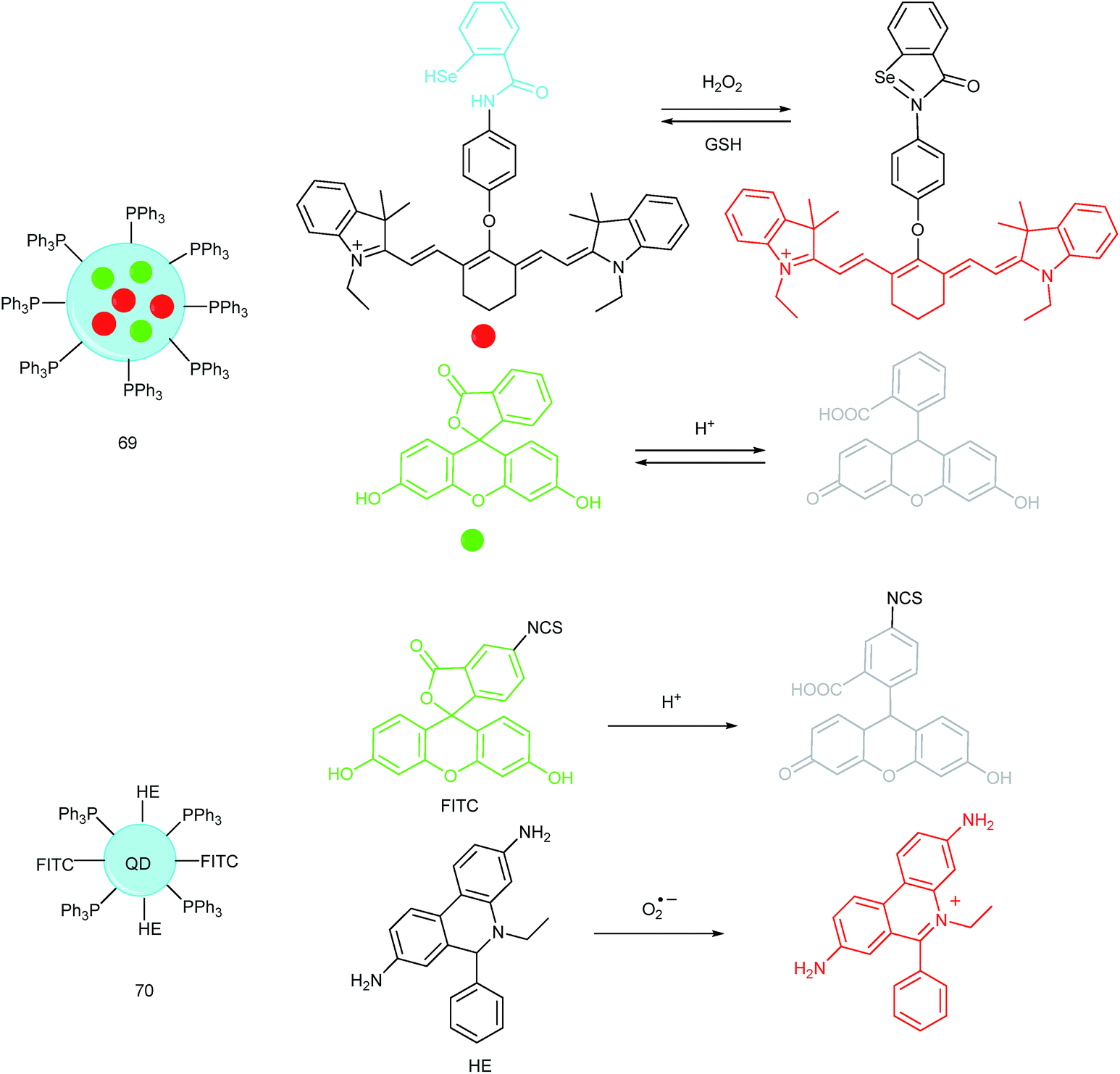 | ||
| Fig. 34 Nanoprobes for simultaneous imaging of bioactive species in mitochondria. | ||
A QD based ratiometric probe (70) was developed by Tian's group.104 Silica encapsulated CdSe/ZnS QDs were employed as the inner reference. The pH responsive probe, fluorescein isothiocyanate (FITC) and O2˙− selective probe, hydroethidine (HE) were grafted onto the surface of the nanocomposites, respectively. The mitochondrion-targeted ratiometric probe was obtained after further TPP modification. With this probe, they revealed that O2˙− could enter into the mitochondria and elevate the O2˙− amount as well as pH, while mitochondrial acidification could cause the O2˙− burst. This probe offered an admirable candidate for investigating the relationships between mitochondrial O2˙− and pH.
8.2. Simultaneous imaging of bioactive species in different organelles
Apart from using multi-analyte responsive molecular or nanoprobes for imaging bioactive species in organelles, simultaneous visualization of bioactive species in different organelles is another important task, since it can facilitate the investigation of bioactive species' fluctuations in different subcellular structures. Visualization of bioactive species in different organelles may offer more opportunities for tracing the “cross-talk” between different organelles and further advance our understanding of diverse physiological and pathological processes (Fig. 35).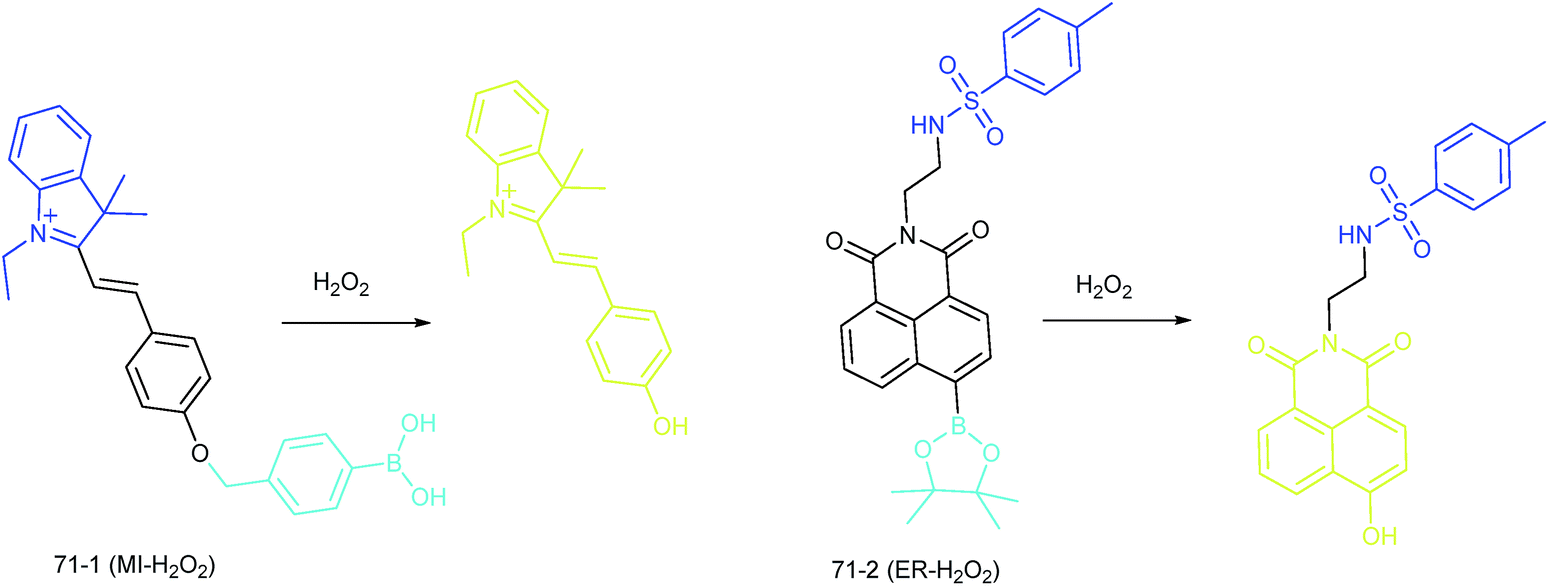 | ||
| Fig. 35 Structures and the responses of probes 71-1 and 71-2. | ||
Our group demonstrated the simultaneous visualization of H2O2 fluctuations in the mitochondria and ER with two molecular probes (71-1, MI-H2O2, mitochondrion-targeted H2O2 probe, LOD = 80 nM and 71-2, ER-H2O2, ER targeted H2O2 probe, LOD = 120 nM).30b71-1 and 71-2 could accumulate in mitochondria and lysosomes, respectively. Under different stimulations, the H2O2 concentration in the two subcellular structures could be easily tracked owing to the sensitivity and specificity of the probes, which offered the possibility of real-time discriminating the subcellular H2O2 fluctuation (Fig. 36). Interestingly, delayed H2O2 rise effects between the ER and mitochondria during different apoptosis pathways were observed for the first time; this work offered a new method for investigating the “organelle crosstalk” during different processes.
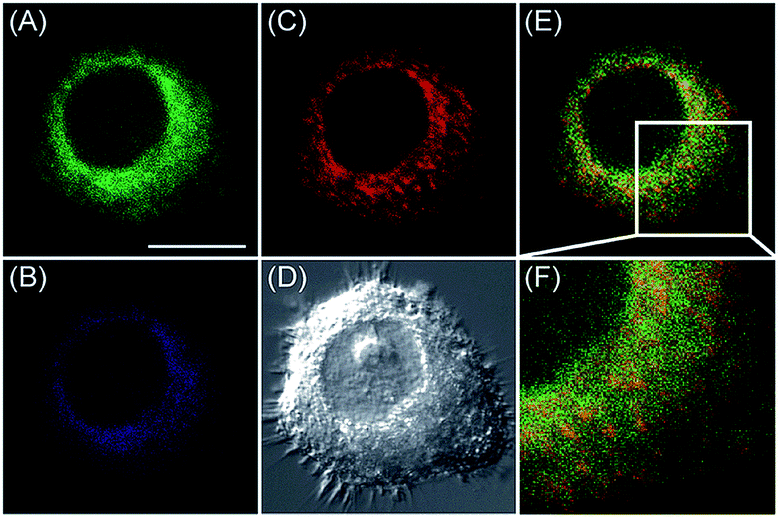 | ||
| Fig. 36 CLSM images of HepG2 cells stained simultaneously with 71-1 and 71-2 (10 mM). 71-2 was excited at 405 nm, and collected at 500–620 nm for the green channel (image A) and at 430–470 nm for the blue channel (image B). 71-1 was excited at 543 nm, and collected at 550–600 nm for the red channel (image C). (D) The bright-field image. (E) The overlay of (A) and (C). (F) The enlarged image from the square marked in image (E). Adapted with permission from ref. 30b. Copyright 2016 Royal Society of Chemistry. | ||
8.3. Challenges for multi-parameter imaging
Simultaneous or sequential imaging of the fluctuations of bioactive species in one or different organelles holds great promise for clarifying their upstream and downstream relationships, which will further deepen our understanding of the biological functions of these species and facilitate related bioanalysis. However, construction of molecular probes with two or more responsive moieties is complicated. On the other hand, organelle-targeted nanoprobes usually suffer from inferior targeting efficiency because nanostructures are less permeable during traveling through the complicated intracellular barriers, which makes them usually require longer incubation time during cell imaging. Employment of different molecular tools to image diverse analytes is feasible, and it can easily be extended to investigate communication between different subcellular compartments and open new avenues for bioanalysis. However, probes that have distinct emission bands and can work under single wavelength excitation have not been demonstrated yet.9. Conclusion and outlook
Organelle-targeted RONSS fluorescent probes have aroused increasing interest over the past decade, owing to their potential for better monitoring diverse physiological and pathological activities, and elucidating the molecular mechanisms of different biological events. Based on various fluorophores, recognition groups and targeting ligands, a great number of subcellular localized probes have been elaborately prepared and successfully employed for different applications, including monitoring cancer, diabetes, depression, PDT, drug induced oxidative/nitrative stress, IRI, inflammation and so on. These probes have significantly facilitated our understanding of life science and have undoubtedly become powerful fluorescent tools for bioanalysis. By highlighting the latest and most representative probes, this review summarized current strategies for designing organelle-targeted RONSS probes. The targeting, detection and response mechanisms are carefully discussed, and they will offer valuable references to investigators interested in this area and facilitate the pursuit of better organelle-targeted fluorescent probes. Although the significant superiority of organelle-targeted fluorescent probes over their traditional counterparts has been demonstrated, several obstacles still exist for their practical application. The current challenges as well as the potential future directions are listed below.9.1. Solubility
During living cell imaging, water solubility is a critical parameter for fluorescent probes. The large aromatic structures in fluorescent probes may cause fluorophore aggregation through π–π stacking, which can not only reduce cellular permeability and subcellular localization, but also quench the fluorescence (aggregation caused quenching effect, ACQ) of fluorescent probes. Organic solvents like DMSO are often used to enhance the solubility of organelle-targeted probes, and this can place extra burden on the cell/tissue samples, and interfere with the subcellular microenvironments. Therefore, development of organelle-targeted probes that have better solubility and can avoid ACQ is highly desired in future work. For this purpose, devising efficient modification strategies and developing water-soluble aggregation induced emission nanoprobes are ideal tactics.9.2. Localization capability
Precisely driving probes into organelles of interest is the prerequisite for subcellular imaging. However, the current available probes suffer from several limitations, including poor targeting efficiency, potential toxic effects and so on. For instance, the widely used mitochondrion targeting group TPP can reduce MMP owing to its cationic feature, and such kind of probe may leak from mitochondria when the MMP is lost. On the other hand, lysosome targeting group morpholine modified probes have an alkalization effect. The high concentration of morpholine decorating the probes can elevate the pH value of lysosomes, which also has certain toxic effects on the analysed samples. Moreover, highly efficient targeting strategies toward many other important organelles, including the Golgi apparatus, nucleus and lipid droplets, are still rare, which severely limits the determination of critical biological events in these subcellular structures. Genetically encoded probes also suffer from several limitations as discussed above. Therefore, exploration of reliable targeting mechanisms and novel targeting ligands toward diverse subcellular organelles is still urgently required. This aspect relies on the expansion of biological research to identify the intrinsic characteristics and metabolic pathways of diverse organelles. It also relies greatly on chemical synthesis to develop and optimize the structure of targeting ligands. Therefore, collaboration of chemists and biologists is essential for developing highly selective, efficient and non-toxic organelle-targeted ligands, which would further advance chemo-biological investigations.9.3. Detection performance
After reaching the organelles of interest, probes need to specifically and effectively interact with the detected targets for in situ imaging. However, it is well known that bioactive species suffer from ultralow concentration, short life span and environmental sensitivity, while most developed probes failed in tracing the bioactive species in the resting state due to the high LOD, and poor specificity and sensitivity. Therefore, new organelle-targeted probes with higher specificity and sensitivity are highly required. Exploration of new reaction mechanisms, and combination with theoretical calculation as well as high throughput probe screening systems should be promising solutions. In addition, subcellular localized probes with signal amplification have not been investigated, although they should also be study-worthy schemes in future work.9.4. Optical performance
Signal-noise ratio is another critical parameter during subcellular analysis. Many biomolecules, like reduced nicotinamide adenine dinucleotide and some proteins, are emissive under laser irradiation, and can interfere with the resolution and reality of detection information. But the emission wavelengths of most developed organelle-targeted fluorescent probes locate in the visible region, making them unsuitable for background-free/in vivo fluorescent imaging. Therefore, probes with high quantum yields, excellent photostability, long lifetime and strong brightness are attractive. Probes with two or more photon excitation, NIR-I and even NIR-II emissions are also useful for live cell imaging. Development of new fluorophores with higher performances may pave the road to deep penetration, high resolution fluorescence imaging in the near future.9.5. Multifunctionality
Nowadays, theranostic probes have been extensively explored owing to their simplicity and multifunctionality. As reaction-based probes are capable of capturing/adjusting diverse bioactive species, they are promising for controlling diverse disorders caused by RONSS overexpression. However, the therapeutic effects of these probes have rarely been explored. Furthermore, probes with cascade targeting, multiparameter detection and multimodal imaging functions are still rare. Related investigations will tremendously accelerate personalized diagnosis and therapy. Recently, photo-caged probes have drawn great interest for bio-imaging, and they enable us to monitor biomolecules of spatiotemporal interest. However, to the best of our knowledge, such function has not been introduced in organelle-targeted probes.9.6. Research models
Bioactive species participate in almost all the bioactivities and are closely related to diverse major diseases. Research models are indispensable for shedding light on the molecular mechanisms of different biological phenomena. From this review, it can be clearly found that the developed models are still limited. Thus, other models with different important biological activities, including cell immunity, drug resistance, cell communication, cancer metastasis and so on, remain to be further developed and investigated. Besides, communication between bioactive species from different organelles has been demonstrated to be another promising research field, and it should also be explored to further advance our understanding of different biological events.In summary, considerable research efforts have been devoted to the development of organelle-targeted fluorescent probes during the past decade, with the focus on the preparation of powerful fluorescent tools to enhance the resolution and sensitivity during cell imaging and better explain the molecular mechanisms of diverse biological events. Although research in this field is still in its infancy and many challenges remain to be resolved, it is expected that more organelle-targeted fluorescent probes with high performance will emerge and further advance our understanding in the near future through the conjoint efforts from researchers worldwide.
Finally, a summary of the physiochemical properties of organelle-targeted RONSS probes is provided in the Appendix. However, it is difficult to compare the selectivity/anti-interference capability of probes, even those based on the same detection mechanism, by studying the summary table. We encourage the reader to examine the primary papers and further assess the selectivity during choosing probes for bioanalysis (Table 2).
| Analyte | Targeting sitea | Probe | λ Ex/λEmb (nm) | Probe concentration vs. dynamic range | Response time | LOD | Φ | Commentsd | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Nu = nucleus; Mito = mitochondria; Lyso = lysosomes; ER = endoplasmic reticulum; Gol = Golgi apparatus. b λ Ex is the excitation wavelength of the probe in the presence of an analyte. λEm is the maximum emission wavelength/emission range of the probe in the presence of an analyte. c Φ is the quantum yield of the probe in the presence of an analyte. d TP probe = two-photon excitation probe; RT probe = ratiometric probe. | ||||||||||
| ROS | O2˙− | Mito | 8 (MF-DBZH) | 483, 800/512 | 20 μM/3–12 μM | — | 9.5 nM | — | TP probe for in vivo imaging | 35 |
| 9 (HKSOX-1m) | 509/534 | 10 μM/0–13 μM | <10 min | 23 nM | — | Imaging O2˙− in the resting state, imaging O2˙− in quantitative microplate assays | 34 | |||
| 10 | 494/520 | 10 μM/0.65–25 μM | <10 min | — | — | Off–on probe | 24a | |||
| ER | 53 (ER-BZT) | 365, 700/450 | 10 μM/0.1–15 μM | In seconds | 60 nM | 0.19 | TP probe for in vivo imaging | 30a | ||
| Gol | 64 (CCA) | 370, 800/495 | 10 μM/0–5 μM | — | 18 nM | 0.21 | TP probe for in vivo imaging | 31a | ||
| H2O2 | Nu | 1 (NucPE1) | 505/530 | — | — | — | 0.626 | Off–on probe | 17 | |
| 2 (pep-NP1) | 380/F555/403 | 5 μM/1–10 μM | — | 0.17 μM | — | RT probe | 19 | |||
| 3 (SPG2) | 495/513 | — | — | — | 0.57 | Targets diverse organelles | 65 | |||
| Mito | 11 (Cy-B) | 685/709 | 10 μM/0–18 μM | — | 13 nM | — | NIR probe for in vivo imaging | 36a | ||
| 12 (Mito-NIRHP) | 670/704 | 10 μM/0–50 μM | ∼20 min | 26 nM | 0.636 | NIR probe for in vivo imaging | 36c | |||
| Lyso | 38 (Lyso-HP) | 450, 780/550 | 10 μM/1–20 μM | <1 min | 1.21 μM | — | TP probe for imaging in tissues | 91 | ||
| 39 (LyNC) | 450/537 | 5 μM/0–220 μM | <1 h | 0.22 μM | 0.17 | Imaging in nematodes | 37 | |||
| 40 (HP-L1) | 520/584 | 1 μM/0–80 μM | ∼8 min | 0.23 μM | — | Logic probe, reduced background | 28 | |||
| ER | 54 (α-Naph) | 395/F540/465 | 10 μM/0–10 μM | — | 37 nM | — | RT probe for quantitative measurement of H2O2 in the ER | 97a | ||
| HOCl | Mito | 13 (PMN-TPP) | 410/F522/640 | 5 μM/0–10 μM | In seconds | 0.43 μM | — | RT probe for in vivo imaging | 38c | |
| 14 (RMClO-2) | Ex412/540/Em570/476 | — | <5 s | 0.84 nM | — | RT probe for quantifying cancer mitochondrial basal HOCl | 39a | |||
| 15 (RSTPP) | 553/580 | 10 μM/2–35 μM | <1 min | 9 nM | 0.34 | Off–on probe | 39b | |||
| Lyso | 41-1 LYSOTP | 375, 750/500 | 5 μM/50 nM-10 μM | In seconds | 19.6 nM | 0.35 | TP probe for in vivo imaging | 40b | ||
| 42 (Lyso-NDMTC) | 410,800/547 | 5 μM/1–20 μM | ∼1 min | 7.6 pM | — | TP probe for imaging HOCl at the pM level | 88 | |||
| 43 (Lyso-HA) | 420/F586/450 | 10 μM/0–50 μM | In seconds | 0.11 μM | — | RT probe with a large Stokes shift | 92 | |||
| ER | 55 (ER-ClO) | 425/F480/554 | 10 μM/0–120 μM | ∼30 s | 0.59 μM | — | RT probe | 96c | ||
| 56 | 326, 720/F450/361 | — | <1 min | 3.6 μM | 0.29 | TP and RT probe | 98 | |||
| Gol & lyso | 65 (SF-1) | 550/586 | — | <15 min | 4.3 nM | 0.116 | For structured illumination microscopy imaging | 97b | ||
| 1O2 | Mito | 18 (Si-DMA) | 640/678 | — | — | — | 0.17 | NIR probe | 41b | |
| 19 (MNAH) | 440, 820/536 | — | — | — | — | TP probe for deep tissue imaging | 41a | |||
| ˙OH | Mito | 20 (MPT-Cy2) | 450/595 | 10 μM/0–10 μM | — | 1.16 μM | — | In vivo imaging | 42 | |
| HOBr | Mito | 16 (RhSN-mito) | 624/663 | 6 μM/0–100 nM | — | 20 pM | 0.68 | Large Stokes shift, high quantum yield | 43a | |
| 17 (HAP) | 405/F437/528 | 5 μM/15–135 nM | ∼30 s | 1.8 ± 0.2 nM | 0.113 | RT probe | 43b | |||
| hROS | Lyso | 44 (PSiR) | 620/680 | 2 μM/0–2 μM HOCl, 0–1.5 μM ONOO−, 0–20 μM ˙OH | In seconds | 10.6 nM, 7.9 nM, 0.14 μM | 0.12 | NIR probe for in vivo imaging | 89 | |
| RNS | ONOO− | Mito | 24 (PNCy3Cy5) | 530/F560/660 | 2 μM/0–700 nM | <30 s | 0.65 nM | — | RT probe for semiquantitatively image | 45 |
| 25 (MITO-CC) | 420/F473/651 | 2 μM/0–7.5 μM | <20 s | 11.3 nM | 0.546 | RT and TP probe for in vivo imaging | 78 | |||
| 26 (Rhod-ONOO−) | 500, 800/558 | — | ∼30 min | 43 nM | — | TP probe | 47 | |||
| ER | 59 (ABAH-LW) | 370/F481/405 | — | In seconds | 21.4 nM | — | RT probe | 96b | ||
| NO | Nu | 4 (HTDAF-2DA) | 488/512 | — | — | — | — | Targets diverse organelles, fast cell permeability | 66 | |
| 5 (TMR-NO-SNAP) | 533/564 | — | In seconds | 0.5 μM | 0.59 | Targets diverse organelles | 67 | |||
| Mito | 21 (Mito-RhNO) | 555/585 | 1 μM/1–10 μM | — | 4.0 nM | — | Off–on probe | 48c | ||
| 22 | 570/616 | 2 μM/0–120 μM | — | 12 nM | 0.25 | Dual-color NO imaging | 48a | |||
| 23 | 490/518 | 4 μM/0–12.8 μM | In seconds | 4.8 nM | — | Imaging NO in the resting state | 49 | |||
| Lyso | 45 | 440, 840/530 | 11 μM/1–1.3 μM | — | 5 nM | 0.3 | TP probe | 93b | ||
| 46 | 645/680 | 4 μM/0–20 μM | In seconds | 0.12 nM | — | NIR probe for in vivo imaging | 48b | |||
| 47 (TRP-NO) | 330/F565/540 | 15 μM/0–160 μM | In seconds | 1.8 μM | — | RT probe for quantitative time-gated luminescence detection | 48d | |||
| ER | 57 (ER-Nap-NO) | 440/538 | 10 μM/0.01–20 μM | <6 min | 3.3 nM | 0.214 | TP probe for in vivo imaging | 96a | ||
| HNO | Mito | 27 (MitoHNO) | 690/727 | 5 μM/0–10 μM | ∼12 min | 60 nM | 0.23 | NIR probe | 81a | |
| Lyso | 48 (Lyso-JN) | 690/700 | 2 μM/0–10 μM | — | 0.06 μM | 0.37 | NIR probe for background-free imaging | 50a | ||
| ER | 58 (ER-HNO) | 530/586 | 10 μM/2–15 μM | <20 min | 2 μM | 0.105 | For structured illumination microscopy imaging | 50b | ||
| RSS | GSH | Nu | 7 (HaloRT Ligand) | Ex405, Em480/Ex488, Em565 | 10 μM/1.25–15 mM | Within minutes | K d = 23.7 mM | — | Targets diverse organelles, reversible GSH imaging | 70 |
| Mito | 28 (MitoGP) | 600/810 | 10 μM/0–10 μM | — | 26 nM | 0.187 | NIR probe with high targeting efficiency | 52 | ||
| 29 | 550/599 | 10 μM/1–15 μM | <30 min | 109 nM | 0.238 | Off–on probe | 83 | |||
| 30 (MitoRT) | Ex405, Em488/Ex488, Em567 | 6 μM/1–10 mM | 26.5 s for 90% | K d = 3.7 mM | 0.2, >0.97 | Reversible GSH imaging | 51 | |||
| Lyso | 50 (RhB-1) | 520/582 | — | 9 s | 190 nM | — | Off–on probe | 53a | ||
| Cys | Mito | 31 (NFL1) | 630/735 | 10 μM/0–3 μM | <10 min | 14.5 nM | 0.05 | NIR probe for in vivo imaging | 53b | |
| H2S | Nu | 6 (CouN3-BC) | 405/450 | — | — | — | — | Targets diverse organelles | 68 | |
| Mito | 32 | 394/532 | 10 μM/0–100 μM | ∼40 min | 2.46 μM | 0.11 | Off–on probe | 85 | ||
| 33 (TAB-2) | 365,760/500–550 | 10 μM/0–35 μM | — | 47 nM | 0.501 | TP probe | 56 | |||
| 34 (HSN2-BG) | 435/515 | — | — | — | — | Targets diverse organelles | 12b | |||
| Lyso | 51 (SulpHensor) | 530/555 | 5 μM/0–10 μM | ∼60 min | 0.5 μM | 0.6 | Logic probe for background-free imaging | 94a | ||
| 52 | 345/453 | 10 μM/0–300 μM | <30 min | 7.9 × 10−7 M | — | Off–on probe | 95 | |||
| ER | 61 (Na-H2S-ER) | 440, 760/545 | — | <30 min | 7.77 × 10−6 M | — | TP probe for in vivo imaging | 99 | ||
| H2Sn | Mito | 35 (Mito-ss) | 635/730 | 10 μM/0–10 μM | <30 s | 25 nM | — | NIR probe for in vivo imaging | 58 | |
| ER | 62 (MB-Sn) | 530/584 | 10 μM/0–10 μM | <10 min | 26.01 nM | 0.125 | For super-resolution structured illumination microscopy | 97c | ||
| 63 (ER-H2S2) | 435/540 | 10 μM/0.1–100 μM | 12 min | 26 nM | — | Imaging H2S2 in zebrafish | 100 | |||
| SO2 | Mito | 36 (Mito-Ratio-SO2) | 405/F480/650 | 5 μM/0.3–50 μM | <3 min | 0.09 mM | — | RT probe | 59a | |
| 37 (DSPT) | 390/F540/590 | 2.5 μM/0–1.5 equiv. | 1.5 min | 69 nM | — | RT probe | 87 | |||
10. Appendix
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21535004, 91753111, 21874086, and 21775094) and the Key Research and Development Program of Shandong Province (2018YFJH0502).Notes and references
- (a) K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed; (b) H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed; (c) J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed; (d) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC; (e) V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC; (f) L.-Y. Niu, Y.-Z. Chen, H.-R. Zheng, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Soc. Rev., 2015, 44, 6143–6160 RSC; (g) D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC; (h) M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2013, 114, 4496–4539 CrossRef PubMed.
- (a) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed; (b) W. Chyan, D. Y. Zhang, S. J. Lippard and R. J. Radford, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 143–148 CrossRef CAS PubMed; (c) H. W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X. B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140–7180 RSC; (d) X. Qian and Z. Xu, Chem. Soc. Rev., 2015, 44, 4487–4493 RSC; (e) A. C. Sedgwick, L. Wu, H. H. Han, S. D. Bull, X. P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC; (f) Y. Tang, D. Lee, J. Wang, G. Li, J. Yu, W. Lin and J. Yoon, Chem. Soc. Rev., 2015, 44, 5003–5015 RSC; (g) Roopa, N. Kumar, V. Bhalla and M. Kumar, Chem. Commun., 2015, 51, 15614–15628 RSC.
- (a) W. Xu, Z. Zeng, J.-H. Jiang, Y.-T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed; (b) H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed; (c) X. Jiao, Y. Li, J. Niu, X. Xie, X. Wang and B. Tang, Anal. Chem., 2018, 90, 533–555 CrossRef CAS PubMed; (d) Z. Xu and L. Xu, Chem. Commun., 2016, 52, 1094–1119 RSC; (e) R. Zhang, B. Song and J. Yuan, TrAC, Trends Anal. Chem., 2018, 99, 1–33 CrossRef CAS.
- (a) J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed; (b) A. Fernandez and M. Vendrell, Chem. Soc. Rev., 2016, 45, 1182–1196 RSC; (c) M. H. Lee, A. Sharma, M. J. Chang, J. Lee, S. Son, J. L. Sessler, C. Kang and J. S. Kim, Chem. Soc. Rev., 2018, 47, 28–52 RSC; (d) K. Qiu, Y. Chen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2019, 378, 66–86 CrossRef CAS.
- (a) T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC; (b) M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- (a) Q. Wan, S. Chen, W. Shi, L. Li and H. Ma, Angew. Chem., Int. Ed., 2014, 53, 10916–10920 CrossRef CAS PubMed; (b) K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS; (c) M. H. Lee, N. Park, C. Yi, J. H. Han, J. H. Hong, K. P. Kim, D. H. Kang, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 14136–14142 CrossRef CAS PubMed; (d) Y. Liu, J. Zhou, L. Wang, X. Hu, X. Liu, M. Liu, Z. Cao, D. Shangguan and W. Tan, J. Am. Chem. Soc., 2016, 138, 12368–12374 CrossRef CAS PubMed.
- (a) N. Jiang, J. Fan, F. Xu, X. Peng, H. Mu, J. Wang and X. Xiong, Angew. Chem., Int. Ed., 2015, 54, 2510–2514 CrossRef CAS PubMed; (b) H. Xiao, C. Wu, P. Li and B. Tang, Anal. Chem., 2018, 90, 6081–6088 CrossRef CAS PubMed; (c) H. Xiao, P. Li, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 1588–1593 RSC; (d) H. Xiao, C. Wu, P. Li, W. Gao, W. Zhang, W. Zhang, L. Tong and B. Tang, Chem. Sci., 2017, 8, 7025–7030 RSC.
- (a) P. Chabosseau, E. Tuncay, G. Meur, E. A. Bellomo, A. Hessels, S. Hughes, P. R. V. Johnson, M. Bugliani, P. Marchetti, B. Turan, A. R. Lyon, M. Merkx and G. A. Rutter, ACS Chem. Biol., 2014, 9, 2111–2120 CrossRef CAS PubMed; (b) A. M. Hessels, P. Chabosseau, M. H. Bakker, W. Engelen, G. A. Rutter, K. M. Taylor and M. Merkx, ACS Chem. Biol., 2015, 10, 2126–2134 CrossRef CAS PubMed; (c) Y. Han, C. Ding, J. Zhou and Y. Tian, Anal. Chem., 2015, 87, 5333–5339 CrossRef CAS PubMed; (d) K. P. Divya, S. Sreejith, P. Ashokkumar, K. Yuzhan, Q. Peng, S. K. Maji, Y. Tong, H. Yu, Y. Zhao, P. Ramamurthy and A. Ajayaghosh, Chem. Sci., 2014, 5, 3469–3474 RSC; (e) Y. Chen, Y. Bai, Z. Han, W. He and Z. Guo, Chem. Soc. Rev., 2015, 44, 4517–4546 RSC; (f) Y. Matsui, Y. Funato, H. Imamura, H. Miki, S. Mizukami and K. Kikuchi, Chem. Sci., 2017, 8, 8255–8264 RSC; (g) G. Zhang, J. J. Gruskos, M. S. Afzal and D. Buccella, Chem. Sci., 2015, 6, 6841–6846 RSC; (h) A. Rodriguez-Garcia, J. Rojo-Ruiz, P. Navas-Navarro, F. J. Aulestia, S. Gallego-Sandin, J. Garcia-Sancho and M. T. Alonso, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2584–2589 CrossRef CAS PubMed.
- (a) Z. Yang, Y. He, J.-H. Lee, N. Park, M. Suh, W. S. Chae, J. Cao, X. Peng, H. Jung, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 9181–9185 CrossRef CAS PubMed; (b) D. Li, X. Tian, A. Wang, L. Guan, J. Zheng, F. Li, S. Li, H. Zhou, J. Wu and Y. Tian, Chem. Sci., 2016, 7, 2257–2263 RSC; (c) M. K. Kuimova, S. W. Botchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69–73 CrossRef CAS PubMed.
- (a) L. M. Hyman and K. J. Franz, Coord. Chem. Rev., 2012, 256, 2333–2356 CrossRef CAS PubMed; (b) A. J. Shuhendler, K. Pu, L. Cui, J. P. Uetrecht and J. Rao, Nat. Biotechnol., 2014, 32, 373–380 CrossRef CAS PubMed; (c) H.-S. Wang, TrAC, Trends Anal. Chem., 2016, 85, 181–202 CrossRef CAS.
- (a) X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC; (b) X. Jiang, L. Wang, S. L. Carroll, J. Chen, M. C. Wang and J. Wang, Antioxid. Redox Signaling, 2018, 29, 518–540 CrossRef CAS PubMed.
- (a) X. Yang, Y. Zhou, X. Zhang, S. Yang, Y. Chen, J. Guo, X. Li, Z. Qing and R. Yang, Chem. Commun., 2016, 52, 10289–10292 RSC; (b) L. A. Montoya and M. D. Pluth, Anal. Chem., 2016, 88, 5769–5774 CrossRef CAS PubMed; (c) H. D. Li, Q. C. Yao, J. L. Fan, N. Jiang, J. Y. Wang, J. Xia and X. J. Peng, Chem. Commun., 2015, 51, 16225–16228 RSC; (d) V. S. Lin, A. R. Lippert and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7131–7135 CrossRef CAS PubMed; (e) B. Tang, L. Yin, X. Wang, Z. Chen, L. Tong and K. Xu, Chem. Commun., 2009, 5293–5295 RSC; (f) Q. Gao, W. Zhang, B. Song, R. Zhang, W. Guo and J. Yuan, Anal. Chem., 2017, 89, 4517–4524 CrossRef CAS PubMed; (g) F. Yang, H. Gao, S. S. Li, R. B. An, X. Y. Sun, B. Kang, J. J. Xu and H. Y. Chen, Chem. Sci., 2018, 9, 5556–5563 RSC.
- (a) K.-Y. Tan, C.-Y. Li, Y.-F. Li, J. Fei, B. Yang, Y.-J. Fu and F. Li, Anal. Chem., 2017, 89, 1749–1756 CrossRef CAS PubMed; (b) L. Wang, L. Yuan, X. Zeng, J. Peng, Y. Ni, J. C. Er, W. Xu, B. K. Agrawalla, D. Su, B. Kim and Y.-T. Chang, Angew. Chem., Int. Ed., 2016, 55, 1773–1776 CrossRef CAS PubMed; (c) J. Deng, K. Wang, M. Wang, P. Yu and L. Mao, J. Am. Chem. Soc., 2017, 139, 5877–5882 CrossRef CAS PubMed.
- (a) Y. Tang, X. Kong, A. Xu, B. Dong and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 3356–3359 CrossRef CAS PubMed; (b) X. Xie, F. Tang, X. Shangguan, S. Che, J. Niu, Y. Xiao, X. Wang and B. Tang, Chem. Commun., 2017, 53, 6520–6523 RSC; (c) T. F. Brewer and C. J. Chang, J. Am. Chem. Soc., 2015, 137, 10886–10889 CrossRef CAS PubMed; (d) A. Roth, H. Li, C. Anorma and J. Chan, J. Am. Chem. Soc., 2015, 137, 10890–10893 CrossRef CAS PubMed.
- (a) V. S. Harrison, C. E. Carney, K. W. MacRenaris, E. A. Waters and T. J. Meade, J. Am. Chem. Soc., 2015, 137, 9108–9116 CrossRef CAS PubMed; (b) H. Li, G. Parigi, C. Luchinat and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 6224–6233 CrossRef CAS PubMed.
- (a) A. Nakamura, K. Takigawa, Y. Kurishita, K. Kuwata, M. Ishida, Y. Shimoda, I. Hamachi and S. Tsukiji, Chem. Commun., 2014, 50, 6149–6152 RSC; (b) Y. Cheng, C. Sun, X. Ou, B. Liu, X. Lou and F. Xia, Chem. Sci., 2017, 8, 4571–4578 RSC.
- B. C. Dickinson, Y. Tang, Z. Chang and C. J. Chang, Chem. Biol., 2011, 18, 943–948 CrossRef CAS PubMed.
- G. Lukinavicius, C. Blaukopf, E. Pershagen, A. Schena, L. Reymond, E. Derivery, M. Gonzalez-Gaitan, E. D'Este, S. W. Hell, D. W. Gerlich and K. Johnsson, Nat. Commun., 2015, 6, 8497 CrossRef CAS PubMed.
- Y. Wen, K. Liu, H. Yang, Y. Li, H. Lan, Y. Liu, X. Zhang and T. Yi, Anal. Chem., 2014, 86, 9970–9976 CrossRef CAS PubMed.
- (a) K. Henze and W. Martin, Nature, 2003, 426, 127–128 CrossRef CAS PubMed; (b) M. T. Lin and M. F. Beal, Nature, 2006, 443, 787–795 CrossRef CAS PubMed; (c) K. Krumova, L. E. Greene and G. Cosa, J. Am. Chem. Soc., 2013, 135, 17135–17143 CrossRef CAS PubMed; (d) E. K. Lei and S. O. Kelley, J. Am. Chem. Soc., 2017, 139, 9455–9458 CrossRef CAS PubMed; (e) C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62–65 CrossRef CAS PubMed.
- (a) B. C. Dickinson and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 9638–9639 CrossRef CAS PubMed; (b) J. Qian and B. Z. Tang, Chem, 2017, 3, 56–91 CrossRef CAS.
- (a) L. V. Johnson, M. L. Walsh and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 990–994 CrossRef CAS PubMed; (b) K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 CrossRef CAS PubMed; (c) L. B. Chen, Annu. Rev. Cell Biol., 1988, 4, 155–181 CrossRef CAS PubMed.
- (a) A. R. Sarkar, C. H. Heo, H. W. Lee, K. H. Park, Y. H. Suh and H. M. Kim, Anal. Chem., 2014, 86, 5638–5641 CrossRef CAS PubMed; (b) Y. Wen, K. Liu, H. Yang, Y. Liu, L. Chen, Z. Liu, C. Huang and T. Yi, Anal. Chem., 2015, 87, 10579–10584 CrossRef CAS PubMed; (c) Z. Xue, R. Zhu, S. Wang, J. Li, J. Han, J. Liu and S. Han, Anal. Chem., 2018, 90, 2954–2962 CrossRef CAS PubMed; (d) Y. Chen, C. Zhu, J. Cen, Y. Bai, W. He and Z. Guo, Chem. Sci., 2015, 6, 3187–3194 RSC; (e) T. B. Ren, Q. L. Zhang, D. Su, X. X. Zhang, L. Yuan and X. B. Zhang, Chem. Sci., 2018, 9, 5461–5466 RSC.
- (a) F. Si, Y. Liu, K. Yan and W. Zhong, Chem. Commun., 2015, 51, 7931–7934 RSC; (b) H. Wang, Z. Gao, X. Liu, P. Agarwal, S. Zhao, D. W. Conroy, G. Ji, J. Yu, C. P. Jaroniec, Z. Liu, X. Lu, X. Li and X. He, Nat. Commun., 2018, 9, 562–577 CrossRef PubMed; (c) L. D. Field, J. B. Delehanty, Y. Chen and I. L. Medintz, Acc. Chem. Res., 2015, 48, 1380–1390 CrossRef CAS PubMed; (d) S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 CrossRef CAS PubMed.
- (a) C. Wang, T. Zhao, Y. Li, G. Huang, M. A. White and J. Gao, Adv. Drug Delivery Rev., 2017, 113, 87–96 CrossRef CAS PubMed; (b) H. Lan, F. Xue, Z. Liu, L. Chen, C. Huang and T. Yi, Adv. Opt. Mater., 2016, 4, 1367–1372 CrossRef CAS; (c) Z. Wu and X. Tang, Anal. Chem., 2015, 87, 8613–8617 CrossRef CAS PubMed.
- (a) X. Zhang, B. Wang, C. Wang, L. Chen and Y. Xiao, Anal. Chem., 2015, 87, 8292–8300 CrossRef CAS PubMed; (b) Z. Wu, D. Liang and X. Tang, Anal. Chem., 2016, 88, 9213–9218 CrossRef CAS PubMed; (c) W. Zhu, X. Chai, B. Wang, Y. Zou, T. Wang, Q. Meng and Q. Wu, Chem. Commun., 2015, 51, 9608–9611 RSC; (d) Y. Cai, C. Gui, K. Samedov, H. Su, X. Gu, S. Li, W. Luo, H. H. Y. Sung, J. W. Y. Lam, R. T. K. Kwok, I. D. Williams, A. Qin and B. Z. Tang, Chem. Sci., 2017, 8, 7593–7603 RSC; (e) R. Duncan, J. Kopecek and J. B. Lloyd, Biochem. Soc. Trans., 1984, 12, 913–915 CrossRef CAS PubMed.
- (a) G. Niu, P. Zhang, W. Liu, M. Wang, H. Zhang, J. Wu, L. Zhang and P. Wang, Anal. Chem., 2017, 89, 1922–1929 CrossRef CAS PubMed; (b) R. A. Firestone, J. M. Pisano and R. J. Bonney, J. Med. Chem., 1979, 22, 1130–1133 CrossRef CAS PubMed.
- J. Liu, J. Ren, X. Bao, W. Gao, C. Wu and Y. Zhao, Anal. Chem., 2016, 88, 5865–5870 CrossRef CAS PubMed.
- (a) J. M. Meinig, L. Fu and B. R. Peterson, Angew. Chem., Int. Ed., 2015, 54, 9696–9699 CrossRef CAS PubMed; (b) M. Terasaki, J. Song, J. R. Wong, M. J. Weiss and L. B. Chen, Cell, 1984, 38, 101–108 CrossRef CAS PubMed.
- (a) H. Xiao, X. Liu, C. Wu, Y. Wu, P. Li, X. Guo and B. Tang, Biosens. Bioelectron., 2017, 91, 449–455 CrossRef CAS PubMed; (b) H. Xiao, P. Li, X. Hu, X. Shi, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 6153–6159 RSC; (c) P. Li, X. Shi, H. Xiao, Q. Ding, X. Bai, C. Wu, W. Zhang and B. Tang, Analyst, 2018, 144, 191–196 RSC.
- (a) W. Zhang, J. Zhang, P. Li, J. Liu, D. Su and B. Tang, Chem. Sci., 2018, 10, 879–883 RSC; (b) R. S. Li, P. F. Gao, H. Z. Zhang, L. L. Zheng, C. M. Li, J. Wang, Y. F. Li, F. Liu, N. Li and C. Z. Huang, Chem. Sci., 2017, 8, 6829–6835 RSC; (c) P. Li, X. Guo, X. Bai, X. Wang, Q. Ding, W. Zhang, W. Zhang and B. Tang, Anal. Chem., 2019, 91, 3382–3388 CrossRef CAS PubMed; (d) T. Hirayama, M. Inden, H. Tsuboi, M. Niwa, Y. Uchida, Y. Naka, I. Hozumi and H. Nagasawa, Chem. Sci., 2019, 10, 1514–1521 RSC.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- J. J. Hu, N.-K. Wong, S. Ye, X. Chen, M.-Y. Lu, A. Q. Zhao, Y. Guo, A. C.-H. Ma, A. Y.-H. Leung, J. Shen and D. Yang, J. Am. Chem. Soc., 2015, 137, 6837–6843 CrossRef CAS PubMed.
- P. Li, W. Zhang, K. Li, X. Liu, H. Xiao, W. Zhang and B. Tang, Anal. Chem., 2013, 85, 9877–9881 CrossRef CAS PubMed.
- (a) F. Xu, H. Li, Q. Yao, J. Fan, J. Wang and X. Peng, J. Mater. Chem. B, 2016, 4, 7363–7367 RSC; (b) G. Masanta, C. H. Heo, C. S. Lim, S. K. Bae, B. R. Cho and H. M. Kim, Chem. Commun., 2012, 48, 3518–3520 RSC; (c) X. Xie, X. Yang, T. Wu, Y. Li, M. Li, Q. Tan, X. Wang and B. Tang, Anal. Chem., 2016, 88, 8019–8025 CrossRef CAS PubMed.
- S. I. Reja, M. Gupta, N. Gupta, V. Bhalla, P. Ohri, G. Kaur and M. Kumar, Chem. Commun., 2017, 53, 3701–3704 RSC.
- (a) Q. Xu, C. H. Heo, J. A. Kim, H. S. Lee, Y. Hu, D. Kim, K. M. K. Swamy, G. Kim, S.-J. Nam, H. M. Kim and J. Yoon, Anal. Chem., 2016, 88, 6615–6620 CrossRef CAS PubMed; (b) G. Cheng, J. Fan, W. Sun, K. Sui, X. Jin, J. Wang and X. Peng, Analyst, 2013, 138, 6091–6096 RSC; (c) H. Xiao, J. Li, J. Zhao, G. Yin, Y. Quan, J. Wang and R. Wang, J. Mater. Chem. B, 2015, 3, 1633–1638 RSC.
- (a) J.-T. Hou, K. Li, J. Yang, K.-K. Yu, Y.-X. Liao, Y.-Z. Ran, Y.-H. Liu, X.-D. Zhou and X.-Q. Yu, Chem. Commun., 2015, 51, 6781–6784 RSC; (b) J. Zhou, L. Li, W. Shi, X. Gao, X. Li and H. Ma, Chem. Sci., 2015, 6, 4884–4888 RSC.
- (a) B. Zhang, X. Yang, R. Zhang, Y. Liu, X. Ren, M. Xian, Y. Ye and Y. Zhao, Anal. Chem., 2017, 89, 10384–10390 CrossRef CAS PubMed; (b) L. Yuan, L. Wang, B. K. Agrawalla, S.-J. Park, H. Zhu, B. Sivaraman, J. Peng, Q.-H. Xu and Y.-T. Chang, J. Am. Chem. Soc., 2015, 137, 5930–5938 CrossRef CAS PubMed; (c) F. Tian, Y. Jia, Y. Zhang, W. Song, G. Zhao, Z. Qu, C. Li, Y. Chen and P. Li, Biosens. Bioelectron., 2016, 86, 68–74 CrossRef CAS PubMed; (d) Z. Qu, J. Ding, M. Zhao and P. Li, J. Photochem. Photobiol., A, 2015, 299, 1–8 CrossRef CAS.
- (a) H.-W. Liu, S. Xu, P. Wang, X.-X. Hu, J. Zhang, L. Yuan, X.-B. Zhang and W. Tan, Chem. Commun., 2016, 52, 12330–12333 RSC; (b) S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS PubMed.
- F. Liu, J. Du, D. Song, M. Xu and G. Sun, Chem. Commun., 2016, 52, 4636–4639 RSC.
- (a) X. Liu, A. Zheng, D. Luan, X. Wang, F. Kong, L. Tong, K. Xu and B. Tang, Anal. Chem., 2017, 89, 1787–1792 CrossRef CAS PubMed; (b) H. Huang and Y. Tian, Chem. Commun., 2018, 54, 12198–12201 RSC.
- H. Li, X. Li, X. Wu, W. Shi and H. Ma, Anal. Chem., 2017, 89, 5519–5525 CrossRef CAS PubMed.
- X. Jia, Q. Chen, Y. Yang, Y. Tang, R. Wang, Y. Xu, W. Zhu and X. Qian, J. Am. Chem. Soc., 2016, 138, 10778–10781 CrossRef CAS PubMed.
- A. C. Sedgwick, H.-H. Han, J. E. Gardiner, S. D. Bull, X.-P. He and T. D. James, Chem. Commun., 2017, 53, 12822–12825 RSC.
- D. Cheng, W. Xu, L. Yuan and X. Zhang, Anal. Chem., 2017, 89, 7693–7700 CrossRef CAS PubMed.
- (a) Y.-Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS PubMed; (b) Y. Huo, J. Miao, L. Han, Y. Li, Z. Li, Y. Shi and W. Guo, Chem. Sci., 2017, 8, 6857–6864 RSC; (c) H. Yu, X. Zhang, Y. Xiao, W. Zou, L. Wang and L. Jin, Anal. Chem., 2013, 85, 7076–7084 CrossRef CAS PubMed; (d) Z. Dai, L. Tian, B. Song, X. Liu and J. Yuan, Chem. Sci., 2017, 8, 1969–1976 RSC.
- J. Miao, Y. Huo, X. Lv, Z. Li, H. Cao, H. Shi, Y. Shi and W. Guo, Biomaterials, 2016, 78, 11–19 CrossRef CAS PubMed.
- (a) X. Jing, F. Yu and L. Chen, Chem. Commun., 2014, 50, 14253–14256 RSC; (b) F. Ali, S. Sreedharan, A. H. Ashoka, H. K. Saeed, C. G. W. Smythe, J. A. Thomas and A. Das, Anal. Chem., 2017, 89, 12087–12093 CrossRef CAS PubMed.
- J. Chen, X. Jiang, C. Zhang, K. R. MacKenzie, F. Stossi, T. Palzkill, M. C. Wang and J. Wang, ACS Sens., 2017, 2, 1257–1261 CrossRef CAS PubMed.
- S.-Y. Lim, K.-H. Hong, D. I. Kim, H. Kwon and H. J. Kim, J. Am. Chem. Soc., 2014, 136, 7018–7025 CrossRef CAS PubMed.
- (a) R. Huang, B.-B. Wang, X.-M. Si-Tu, T. Gao, F.-F. Wang, H. He, X.-Y. Fan, F.-L. Jiang and Y. Liu, Chem. Commun., 2016, 52, 11579–11582 RSC; (b) C. Han, H. Yang, M. Chen, Q. Su, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2015, 7, 27968–27975 CrossRef CAS PubMed; (c) C.-Y. Kim, H.-J. Kang, S.-J. Chung, H.-K. Kim, S.-Y. Na and H.-J. Kim, Anal. Chem., 2016, 88, 7178–7182 CrossRef CAS PubMed.
- X. Zhang, H. Tan, Y. Yan, Y. Hang, F. Yu, X. Qu and J. Hua, J. Mater. Chem. B, 2017, 5, 2172–2180 RSC.
- L. Zhou, D. Lu, Q. Wang, S. Liu, Q. Lin and H. Sun, Biosens. Bioelectron., 2017, 91, 699–705 CrossRef CAS PubMed.
- J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed.
- X. Feng, T. Zhang, J.-T. Liu, J.-Y. Miao and B.-X. Zhao, Chem. Commun., 2016, 52, 3131–3134 RSC.
- M. Gao, F. Yu, H. Chen and L. Chen, Anal. Chem., 2015, 87, 3631–3638 CrossRef CAS PubMed.
- (a) W. Xu, C. L. Teoh, J. Peng, D. Su, L. Yuan and Y.-T. Chang, Biomaterials, 2015, 56, 1–9 CrossRef CAS PubMed; (b) Y. Liu, K. Li, M.-Y. Wu, Y.-H. Liu, Y.-M. Xie and X.-Q. Yu, Chem. Commun., 2015, 51, 10236–10239 RSC.
- W. Pan, Y. Li, M. Wang, H. Yang, N. Li and B. Tang, Chem. Commun., 2016, 52, 4569–4572 RSC.
- M. Luan, N. Li, W. Pan, L. Yang, Z. Yu and B. Tang, Chem. Commun., 2017, 53, 356–359 RSC.
- W. Zhang, P. Li, F. Yang, X. Hu, C. Sun, W. Zhang, D. Chen and B. Tang, J. Am. Chem. Soc., 2013, 135, 14956–14959 CrossRef CAS PubMed.
- X. Li, S. Zhang, J. Cao, N. Xie, T. Liu, B. Yang, Q. He and Y. Hu, Chem. Commun., 2013, 49, 8656–8658 RSC.
- (a) J. Seo, S. Kim and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 11154–11155 CrossRef CAS PubMed; (b) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- D. Srikun, A. E. Albers, C. I. Nam, A. T. Iavarone and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 4455–4465 CrossRef CAS PubMed.
- J. Wang, Y. Zhao, C. Wang, Q. Zhu, Z. Du, A. Hu and Y. Yang, PLoS One, 2015, 10, e0123986 CrossRef PubMed.
- C. Wang, X. B. Song, Z. Han, X. Y. Li, Y. P. Xu and Y. Xiao, ACS Chem. Biol., 2016, 11, 2033–2040 CrossRef CAS PubMed.
- J. Chen, M. Zhao, X. Jiang, A. Sizovs, M. C. Wang, C. R. Provost, J. Huang and J. Wang, Analyst, 2016, 141, 1209–1213 RSC.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Correa Jr, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- X. Jiang, C. Zhang, J. Chen, S. Choi, Y. Zhou, M. Zhao, X. Song, X. Chen, M. Maletic-Savatic, T. Palzkill, D. Moore, M. C. Wang and J. Wang, Antioxid. Redox Signaling, 2019, 30, 1900–1910 CrossRef CAS PubMed.
- (a) L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed; (b) Z. Yu, W. Pan, N. Li and B. Tang, Chem. Sci., 2016, 7, 4237–4244 RSC.
- A. T. Hoye, J. E. Davoren, P. Wipf, M. P. Fink and V. E. Kagan, Acc. Chem. Res., 2008, 41, 87–97 CrossRef CAS PubMed.
- (a) J. Qiao, Z. Liu, Y. Tian, M. Wu and Z. Niu, Chem. Commun., 2015, 51, 3641–3644 RSC; (b) F. Du, Y. Min, F. Zeng, C. Yu and S. Wu, Small, 2014, 10, 964–972 CrossRef CAS.
- Q. Hu, C. Qin, L. Huang, H. Wang, Q. Liu and L. Zeng, Dyes Pigm., 2018, 149, 253–260 CrossRef CAS.
- (a) J.-T. Hou, M.-Y. Wu, K. Li, J. Yang, K.-K. Yu, Y.-M. Xie and X.-Q. Yu, Chem. Commun., 2014, 50, 8640–8643 RSC; (b) K. Li, J.-T. Hou, J. Yang and X.-Q. Yu, Chem. Commun., 2017, 53, 5539–5541 RSC.
- K. Xu, D. Luan, X. Wang, B. Hu, X. Liu, F. Kong and B. Tang, Angew. Chem., Int. Ed., 2016, 55, 12751–12754 CrossRef CAS PubMed.
- (a) Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225–14229 CrossRef CAS PubMed; (b) W. Lv, Z. Zhang, K. Y. Zhang, H. Yang, S. Liu, A. Xu, S. Guo, Q. Zhao and W. Huang, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 CrossRef CAS PubMed; (c) C. Gui, E. Zhao, R. T. K. Kwok, A. C. S. Leung, J. W. Y. Lam, M. Jiang, H. Deng, Y. Cai, W. Zhang, H. Su and B. Z. Tang, Chem. Sci., 2017, 8, 1822–1830 RSC; (d) H.-W. Liu, X.-X. Hu, K. Li, Y. Liu, Q. Rong, L. Zhu, L. Yuan, F.-L. Qu, X.-B. Zhang and W. Tan, Chem. Sci., 2017, 8, 7689–7695 RSC; (e) S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kogler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519 CrossRef CAS.
- D. Cheng, Y. Pan, L. Wang, Z. Zeng, L. Yuan, X. Zhang and Y.-T. Chang, J. Am. Chem. Soc., 2017, 139, 285–292 CrossRef CAS PubMed.
- B. C. Zhu, M. Zhang, L. Wu, Z. Y. Zhao, C. Y. Liu, Z. K. Wang, Q. X. Duan, Y. W. Wang and P. Jia, Sens. Actuators, B, 2018, 257, 436–441 CrossRef CAS.
- K. Sunwoo, K. N. Bobba, J.-Y. Lim, T. Park, A. Podder, J. S. Heo, S. G. Lee, S. Bhuniya and J. S. Kim, Chem. Commun., 2017, 53, 1723–1726 RSC.
- (a) X. Gong, X.-F. Yang, Y. Zhong, Y. Chen and Z. Li, Dyes Pigm., 2016, 131, 24–32 CrossRef CAS; (b) M. Ren, B. Deng, K. Zhou, J.-Y. Wang, X. Kong and W. Lin, J. Mater. Chem. B, 2017, 5, 1954–1961 RSC.
- D. P. Li, Z.-Y. Wang, X.-J. Cao, J. Cui, X. Wang, H.-Z. Cui, J.-Y. Miao and B.-X. Zhao, Chem. Commun., 2016, 52, 2760–2763 RSC.
- J. Zhang, X. Bao, J. Zhou, F. Peng, H. Ren, X. Dong and W. Zhao, Biosens. Bioelectron., 2016, 85, 164–170 CrossRef CAS.
- (a) W. Niu, L. Guo, Y. Li, S. Shuang, C. Dong and M. S. Wong, Anal. Chem., 2016, 88, 1908–1914 CrossRef CAS PubMed; (b) K. Yin, F. Yu, W. Zhang and L. Chen, Biosens. Bioelectron., 2015, 74, 156–164 CrossRef CAS.
- Y. L. Pak, J. Li, K. C. Ko, G. Kim, J. Y. Lee and J. Yoon, Anal. Chem., 2016, 88, 5476–5481 CrossRef CAS PubMed.
- L. Tang, P. He, X. Yan, J. Sun, K. Zhong, S. Hou and Y. Bian, Sens. Actuators, B, 2017, 247, 421–427 CrossRef CAS.
- W.-L. Wu, H.-L. Ma, M.-F. Huang, J.-Y. Miao and B.-X. Zhao, Sens. Actuators, B, 2017, 241, 239–244 CrossRef CAS.
- B. Zhu, P. Li, W. Shu, X. Wang, C. Liu, Y. Wang, Z. Wang, Y. Wang and B. Tang, Anal. Chem., 2016, 88, 12532–12538 CrossRef CAS PubMed.
- H. Zhang, J. Liu, C. Liu, P. Yu, M. Sun, X. Yan, J.-P. Guo and W. Guo, Biomaterials, 2017, 133, 60–69 CrossRef CAS PubMed.
- (a) W. Gao, W. Cao, H. Zhang, P. Li, K. Xu and B. Tang, Chem. Commun., 2014, 50, 8117–8120 RSC; (b) W. Gao, L. Ji, L. Li, G. Cui, K. Xu, P. Li and B. Tang, Biomaterials, 2012, 33, 3710–3718 CrossRef CAS.
- M. Ren, B. Deng, J.-Y. Wang, X. Kong, Z.-R. Liu, K. Zhou, L. He and W. Lin, Biosens. Bioelectron., 2016, 79, 237–243 CrossRef CAS.
- M. Ren, B. Deng, K. Zhou, X. Kong, J.-Y. Wang, G. Xu and W. Lin, J. Mater. Chem. B, 2016, 4, 4739–4745 RSC.
- (a) B. Wang, S. Yu, X. Chai, T. Li, Q. Wu and T. Wang, Chem.–Eur. J., 2016, 22, 5649–5656 CrossRef CAS PubMed; (b) H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486–17489 CrossRef CAS PubMed.
- (a) S. Yang, Y. Qi, C. Liu, Y. Wang, Y. Zhao, L. Wang, J. Li, W. Tan and R. Yang, Anal. Chem., 2014, 86, 7508–7515 CrossRef CAS PubMed; (b) J. Fan, Z. Han, Y. Kang and X. Peng, Sci. Rep., 2016, 6, 19562–19569 CrossRef CAS PubMed.
- X. J. Zou, Y. C. Ma, L. E. Guo, W. X. Liu, M. J. Liu, C. G. Zou, Y. Zhou and J. F. Zhang, Chem. Commun., 2014, 50, 13833–13836 RSC.
- (a) S. J. Li, D. Y. Zhou, Y. Li, H. W. Liu, P. Wu, J. Ou-Yang, W. L. Jiang and C. Y. Li, ACS Sens., 2018, 3, 2311–2319 CrossRef CAS PubMed; (b) L. Wu, Y. Wang, M. Weber, L. Liu, A. C. Sedgwick, S. D. Bull, C. Huang and T. D. James, Chem. Commun., 2018, 54, 9953–9956 RSC; (c) J.-T. Hou, H. S. Kim, C. Duan, M. S. Ji, S. Wang, L. Zeng, W. X. Ren and J. S. Kim, Chem. Commun., 2019, 55, 2533–2536 RSC.
- (a) C. Gao, Y. Tian, R. Zhang, J. Jing and X. Zhang, Anal. Chem., 2017, 89, 12945–12950 CrossRef CAS PubMed; (b) F. Ali, S. Aute, S. Sreedharan, H. A. Anila, H. K. Saeed, C. G. Smythe, J. A. Thomas and A. Das, Chem. Commun., 2018, 54, 1849–1852 RSC; (c) A. Hoskere, S. Sreedharan, F. Ali, C. G. Smythe, J. A. Thomas and A. Das, Chem. Commun., 2018, 54, 3735–3738 RSC.
- Y. L. Pak, S. J. Park, G. Song, Y. Yim, H. Kang, H. M. Kim, J. Bouffard and J. Yoon, Anal. Chem., 2018, 90, 12937–12943 CrossRef CAS PubMed.
- Y. Tang, A. Xu, Y. Ma, G. Xu, S. Gao and W. Lin, Sci. Rep., 2017, 7, 12944 CrossRef.
- H. Zhou, J. Tang, L. Sun, J. Zhang, B. Chen, J. Kan, W. Zhang, J. Zhang and J. Zhou, Sens. Actuators, B, 2019, 278, 64–72 CrossRef CAS.
- (a) L. Yang, Y. Chen, Z. Yu, W. Pan, H. Wang, N. Li and B. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 27512–27521 CrossRef CAS; (b) Z. Liu, H. Pei, L. Zhang and Y. Tian, ACS Nano, 2018, 12357–12368 CrossRef CAS PubMed; (c) L. Yang, N. Li, W. Pan, Z. Yu and B. Tang, Anal. Chem., 2015, 87, 3678–3684 CrossRef CAS PubMed; (d) K. Dou, G. Chen, F. Yu, Z. Sun, G. Li, X. Zhao, L. Chen and J. You, J. Mater. Chem. B, 2017, 5, 8389–8398 RSC.
- (a) Y. Zhou, X. Zhang, S. Yang, Y. Li, Z. Qing, J. Zheng, J. Li and R. Yang, Anal. Chem., 2017, 89, 4587–4594 CrossRef CAS PubMed; (b) H. Xiao, P. Li, S. Zhang, W. Zhang, W. Zhang and B. Tang, Chem. Commun., 2016, 52, 12741–12744 RSC; (c) F. Yu, M. Gao, M. Li and L. Chen, Biomaterials, 2015, 63, 93–101 CrossRef CAS; (d) W. Zhang, X. Wang, P. Li, H. B. Xiao, W. Zhang, H. Wang and B. Tang, Anal. Chem., 2017, 89, 6840–6845 CrossRef CAS.
- W. Pan, H. Wang, L. Yang, Z. Yu, N. Li and B. Tang, Anal. Chem., 2016, 88, 6743–6748 CrossRef CAS.
- H. Huang, F. Dong and Y. Tian, Anal. Chem., 2016, 88, 12294–12302 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |