
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMain-chain scission of individual macromolecules induced by solvent swelling†
Daniel
Messmer
 *a,
Oscar
Bertran
b,
Reinhard
Kissner
c,
Carlos
Alemán
d and
A. Dieter
Schlüter
*a
*a,
Oscar
Bertran
b,
Reinhard
Kissner
c,
Carlos
Alemán
d and
A. Dieter
Schlüter
*a
aPolymer Chemistry, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: daniel.messmer@mat.ethz.ch; ads@mat.ethz.ch
bDepartment of Physics, EETAC, Universitat Politècnica de Catalunya, c/ Esteve Terrades, 7, 08860, Castelldefels, Spain
cLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 3, 8093 Zürich, Switzerland
dDepartament d’Enginyeria Química (EEBE), Barcelona Research Center for Multiscale Science and Engineering, Universitat Politècnica de Catalunya, C/ Eduard Maristany, 10-14, Ed. I2, 08019, Barcelona, Spain
First published on 8th May 2019
Abstract
We present a comprehensive investigation of main-chain scission processes affecting peripherally charged and neutral members of a class of dendronized polymers (DPs) studied in our laboratory. In these thick, sterically highly congested macromolecules, scission occurs by exposure to solvents, in some cases at room temperature, in others requiring modest heating. Our investigations rely on gel permeation chromatography and atomic force microscopy and are supported by molecular dynamics simulations as well as by electron paramagnetic resonance spectroscopy. Strikingly, DP main-chain scission depends strongly on two factors: first the solvent, which must be highly polar to induce scission of the DPs, and second the dendritic generation g. In DPs of generations 1 ≤ g ≤ 8, scission occurs readily only for g = 5, no matter whether the polymer is charged or neutral. Much more forcing conditions are required to induce degradation in DPs of g ≠ 5. We propose solvent swelling as the cause for the main-chain scission in these individual polymer molecules, explaining in particular the strong dependence on g: g < 5 DPs resemble classical polymers and are accessible to the strongly interacting, polar solvents, whereas g > 5 DPs are essentially closed off to solvent due to their more closely colloidal character. g = 5 DPs mark the transition between these two regimes, bearing strongly sterically congested side chains which are still solvent accessible to some degree. Our results suggest that, even in the absence of structural elements which favour scission such as cross-links, solvent swelling may be a generally applicable mechanochemical trigger. This may be relevant not only for DPs, but also for other types of sterically strongly congested macromolecules.
Introduction
Reactions leading to changes in the molar mass of polymers significantly affect the mechanical properties of polymeric materials1,2 and are therefore an important factor in polymer ageing and processing. These reactions fall into three broad categories: depolymerization above the ceiling temperature Tc, random or site-specific scission of individual backbone bonds and (post-polymerization) cross-linking. The triggers and mechanisms involved in these reactions elements which favour scission and depend on the particular polymer structures and environmental conditions. For instance, many polymers undergo random scission under oxidative stress3 or (usually chain-centered) scission under mechanical stress, and distinct mechanophores can be introduced into polymer chains to cause targeted chain rupture.4 When radical mechanisms are involved, random chain scission and cross-linking often compete, as is the case e.g. in the photooxidation of polyolefins.5,6 Reactions leading to changes in polymer molar mass are desirable e.g. for drug delivery7,8 (chain scission), in lithography9,10 (chain scission and/or cross-linking), or for recycling purposes (depolymerization).11,12 Furthermore, intriguing mechanoresponsivity, e.g. mechanochromicity13–18 or mechanoluminescence,19–23 results from the introduction of backbone mechanophores.We have previously observed a process of main-chain scission in dendronized polymers (DPs), which are linear polymers bearing a dendritic side chain on each repeating unit.24 This unexpected degradation reaction occurred in the course of the divergent synthesis of DPs of dendritic generation g = 6. The synthesis of this class of DPs (PGgXn; g denotes the dendritic generation, n the main-chain degree of polymerization Pn, and X the dendrons' terminal groups, see Fig. 1) relies on a graft-from protocol in which the dendritic side chains are constructed one g at a time.25 Specifically, this protocol comprises first the deprotection of terminal NHBoc groups in PGgNHBocn with trifluoroacetic acid to afford the polyelectrolyte PGgNHn3TFA. In a second step, the liberated amines are reacted with the N-hydroxysuccinimide ester 1 (see Fig. 1) under basic conditions to afford the next higher g DP, PG(g + 1)NHBocn. This simple two-step sequence proceeds with very high functional group conversion and has provided facile access to DPs of up to g = 5.25 However, the path to higher g DPs using this procedure was blocked by degradation: PG5NHBocn (n ≫ 100) undergoes main-chain scission upon deprotection, mainly yielding oligomers 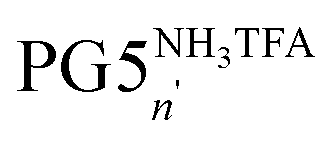 (n′ ≈ 50).24 Chain scission was thought to be the result of coulombic repulsion between partially unscreened terminal ammonium cations. These repulsive forces were thought to mechanochemically activate the backbone C–C bonds – the points of balance between the forces exerted by neighboring dendrons – toward spontaneous scission at ambient temperature.
(n′ ≈ 50).24 Chain scission was thought to be the result of coulombic repulsion between partially unscreened terminal ammonium cations. These repulsive forces were thought to mechanochemically activate the backbone C–C bonds – the points of balance between the forces exerted by neighboring dendrons – toward spontaneous scission at ambient temperature.
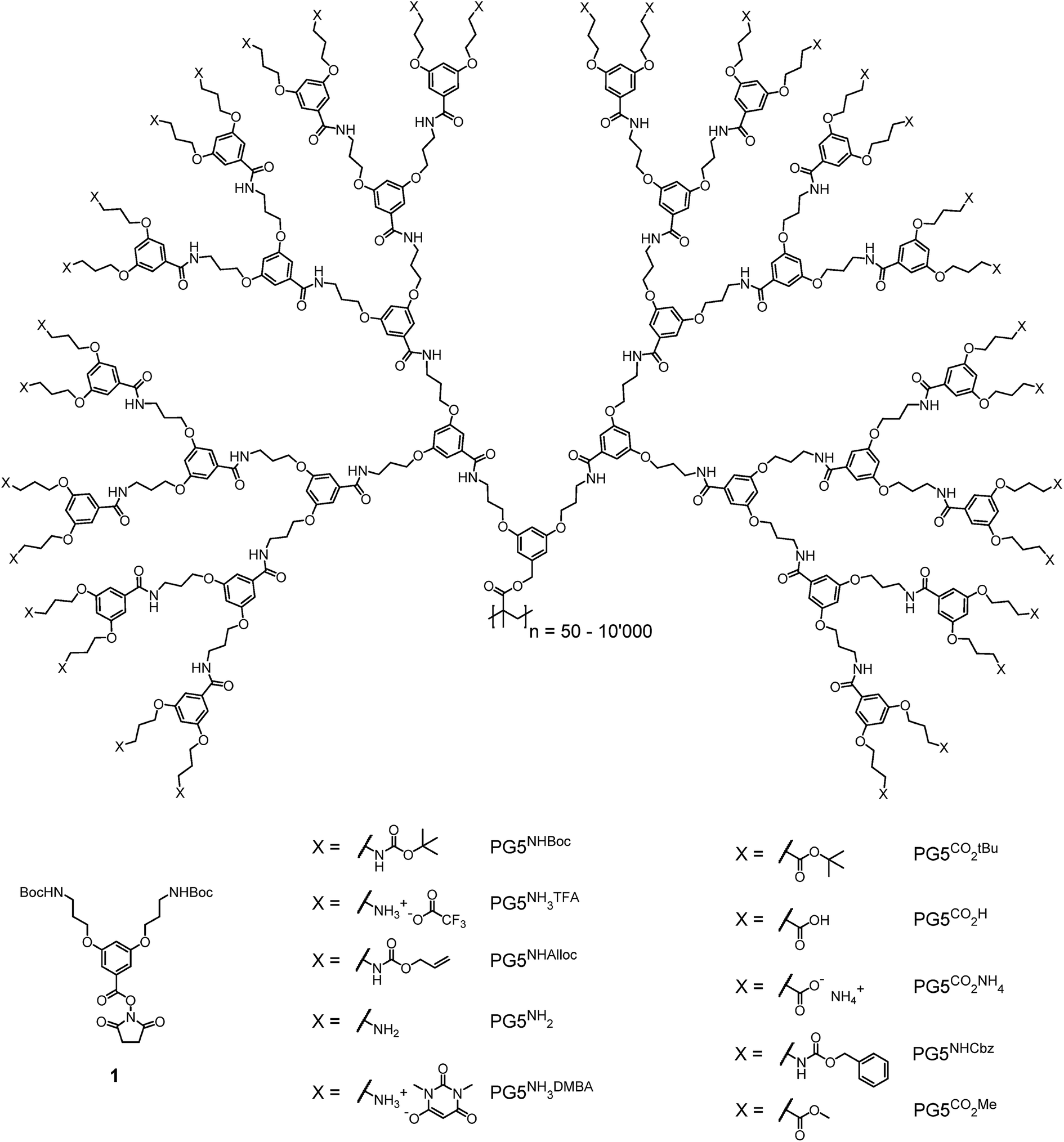 | ||
| Fig. 1 Chemical structures of dendronization agent 1 and the g = 5 DPs discussed in this publication. | ||
The synthesis of g > 5 DPs has long been of significant interest: as postulated by de Gennes and Hervet,26 for any dendritic structure there is a value of g, gmax, above which steric congestion prevents the existence of defect-free dendrons. For DPs of the type PGgNHBocn, this regime was predicted to be around gmax ≈ 6–7.27–29 However, because of the unexpected chain scission, the aforementioned two-step synthetic protocol could not be employed in the synthesis of PG6NHBocn. Recently, an alternative deprotection/dendronization protocol relying on the NHAlloc protecting group was successful in circumventing main-chain scission at g = 5 by proceeding via the charge-neutral intermediate PG5NHn2. The resulting DPs of g = 6–8 are of very high structural perfection and for the first time permitted a discussion of the effects of reaching and surpassing gmax.30
This successful alternative route seemed to vindicate the hypothesis of charge-induced main-chain scission, but a growing body of evidence pointed toward a more fundamental cause behind backbone cleavage than just coulombic repulsion. Here, we will first present more in-depth investigations of charge-induced main-chain scission in DPs of g = 5, utilizing the protonation of terminal amines. Then, we will turn to main-chain scission reactions observed for neutral DPs, mainly of g = 5, though our investigations encompass entire homologous series of g = 1–8. This study relies on results from gel permeation chromatography (GPC), atomic force microscopy (AFM), and electron paramagnetic resonance (EPR) spectroscopy. We discuss how these results form a consistent picture which suggests that the main-chain scission reactions are generally caused by swelling of the dendritic periphery. This is supported by a molecular dynamics (MD) simulation of the DP swelling process. To our knowledge, this represents the first instance of a sterically induced degradation process of single polymer chains in solution, which may be a general feature of strongly sterically congested macromolecules. Furthermore, these results provide further evidence that DPs of g > gmax are essentially “molecular colloidal particles” rather than classical linear polymers, interacting with their environments only via their very periphery.31
Results
The phenomenon of main chain scission was investigated on charged and on neutral DPs in a variety of ways. g = 5 DPs were the focus of investigations concerning charged species, extending on previous studies.24 Additionally, we present investigations for a variety of factors potentially affecting the degradation of charge-neutral DPs in the entire range of g = 1–8. For both categories of experiments, details and additional data can be found in the ESI† (Sections 3 and 4, respectively).Main-chain scission of peripherally charged dendronized polymers
The recent synthesis of PG6NHBoc500 from PG5NHAlloc500 proceeded via the neutral intermediate PG5NH5002, obtained from the N-deprotection of PG5NHAlloc500 in the presence of a base, e.g. triethylamine.30 If no excess base is added, deprotected amines are immediately protonated (eventually to the positively charged polyelectrolyte PG5NHn3DMBA) by the allyl scavenger N,N′-dimethylbarbituric acid (DMBA), which is quite acidic (pKa ≈ 4.7 (ref. 32)). Thus, there were two options to prepare peripherally positively charged g = 5 DPs: either to protonate the neutral intermediate PG5NH5002e.g. with excess trifluoroacetic acid (TFA) to furnish the polyelectrolyte PG5NHn3TFA (Scheme 1a), or to omit the base during deprotection to afford PG5NHn3DMBA directly (Scheme 1b). To facilitate analysis, PG5NHn3TFA and PG5NHn3DMBA were converted into their neutral g = 6 homologs PG6NHBocn by reaction with the g = 1 dendronization reagent 1 prior to AFM imaging (Fig. 2a–c) and GPC analysis (Fig. 2d). AFM imaging revealed drastic reductions in main-chain lengths for intermediate protonation by either TFA (Fig. 2b) or DMBA (Fig. 2c). These findings were confirmed by GPC analysis (Fig. 2d).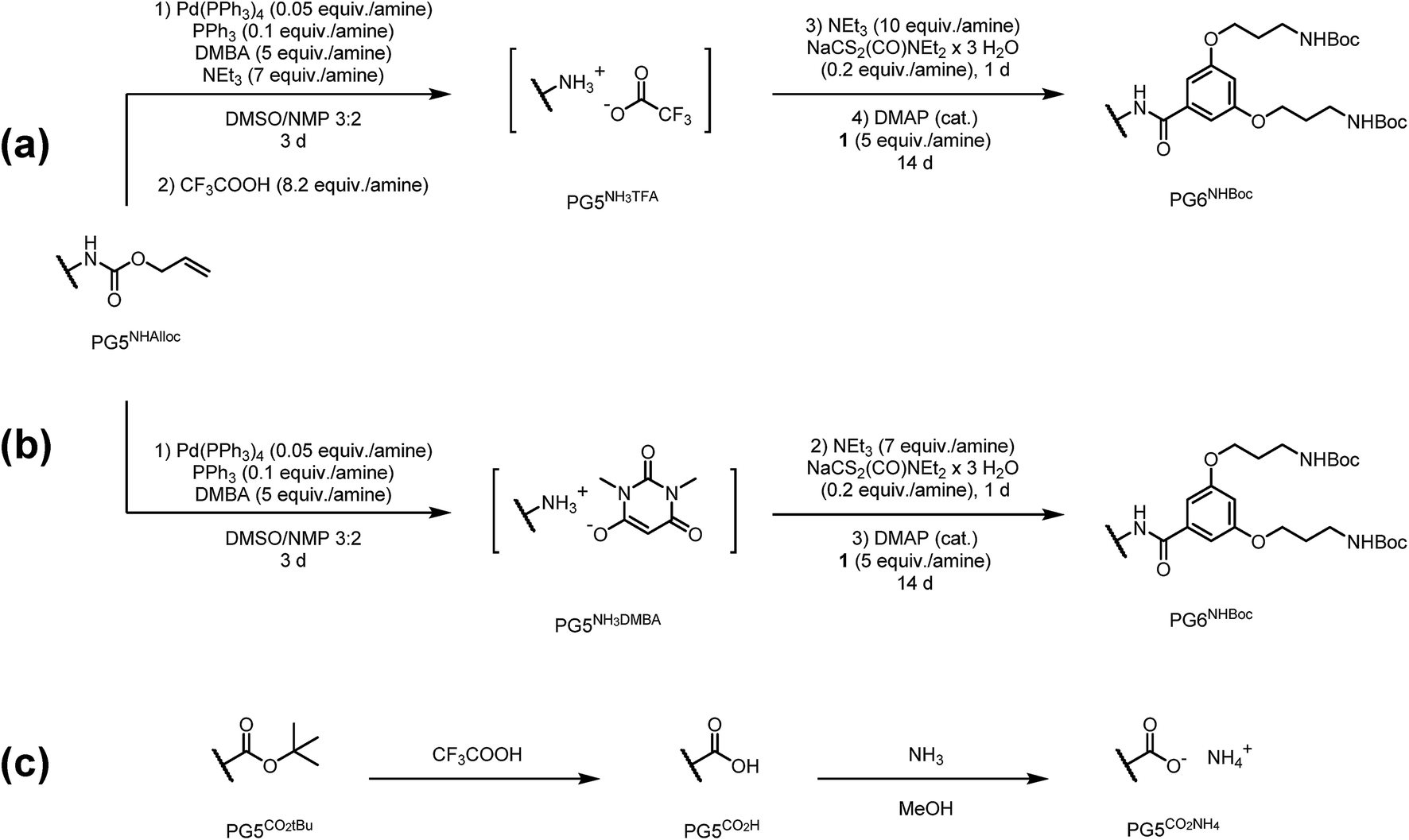 | ||
Scheme 1 Reactions involving charged g = 5 DPs; the transformations of only one terminal group are shown for clarity. (a) Intermittent protonation of PG5NH5002 with TFA, followed by dendronization under basic conditions (compare ref. 30); (b) base-free deprotection of PG5NHAlloc500, resulting in the polyelectrolyte 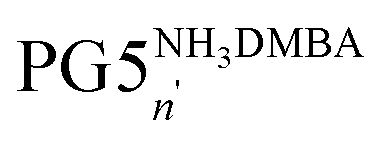 , again followed by dendronization; (c) deprotection of PG5CO5002tBu, followed by deprotonation of the resulting polyacid PG5COn2H. , again followed by dendronization; (c) deprotection of PG5CO5002tBu, followed by deprotonation of the resulting polyacid PG5COn2H. | ||
 | ||
Fig. 2 Main-chain scission of charged g = 5 dendronized polymers: (a–d) DPs bearing derivatives of amines and (e–h) DPs bearing derivatives of carboxylic acids. Tapping mode AFM height images (tapping mode, mica) of (a) PG5NHAlloc500, (b) PG6NHBocn prepared via PG5NHn3TFA (Scheme 1a), PG6NHBocn prepared via PG5NHn3DMBA (Scheme 1b); (d) corresponding GPC retention curves (DMF, 0.1% LiBr). AFM height images (tapping mode, mica) of (e) PG5CO5002tBu, (f) PG5COn2H and (g)  ; (h) corresponding DLS curves (MeOH, 0.1 mg mL−1). PG5COn2H: Dh ∼ 19. 5 nm; ; (h) corresponding DLS curves (MeOH, 0.1 mg mL−1). PG5COn2H: Dh ∼ 19. 5 nm;  : Dh ∼ 16 nm. : Dh ∼ 16 nm. | ||
Aside from the above, fully protonated intermediates, partially charged DPs were also investigated in order to determine a lower threshold of charged peripheral groups necessary to induce backbone scission. To that end, g = 5 DPs carrying varying ratios of orthogonally (Boc- and Alloc-) protected amines were prepared. NHAlloc groups were selectively deprotected under weakly acidic conditions (same conditions as in Scheme 1b). No significant main-chain scission was found even for the DP bearing the largest proportion (80%) of charged peripheral groups (see ESI, Fig. S4† for details).
Next, the main-chain scission of a negatively charged DP was investigated. The terminal tert-butyl esters in PG5CO5002tBu were deprotected using neat TFA, liberating PG5COn2H with terminal carboxylic acid moieties. Subsequent deprotonation with ammonia afforded the corresponding polyelectrolyte 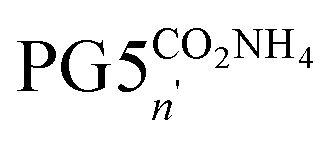 (Scheme 1c). AFM height images (Fig. 2e–g) indicated significant chain scission already upon ester deprotection (Fig. 2f), and not only upon deprotonation to carboxylate (Fig. 2g). Short, dot-like objects were observed in both cases, although the features of PG5COn2H appear larger and less uniform. Dynamic light scattering (DLS) measurements in methanol (Fig. 2h) showed reduced particle sizes upon deprotection, qualitatively confirming main chain scission, though DLS indicated little change in particle size upon deprotonation of PG5COn2H to
(Scheme 1c). AFM height images (Fig. 2e–g) indicated significant chain scission already upon ester deprotection (Fig. 2f), and not only upon deprotonation to carboxylate (Fig. 2g). Short, dot-like objects were observed in both cases, although the features of PG5COn2H appear larger and less uniform. Dynamic light scattering (DLS) measurements in methanol (Fig. 2h) showed reduced particle sizes upon deprotection, qualitatively confirming main chain scission, though DLS indicated little change in particle size upon deprotonation of PG5COn2H to  .
.
Main-chain scission of non-charged DPs
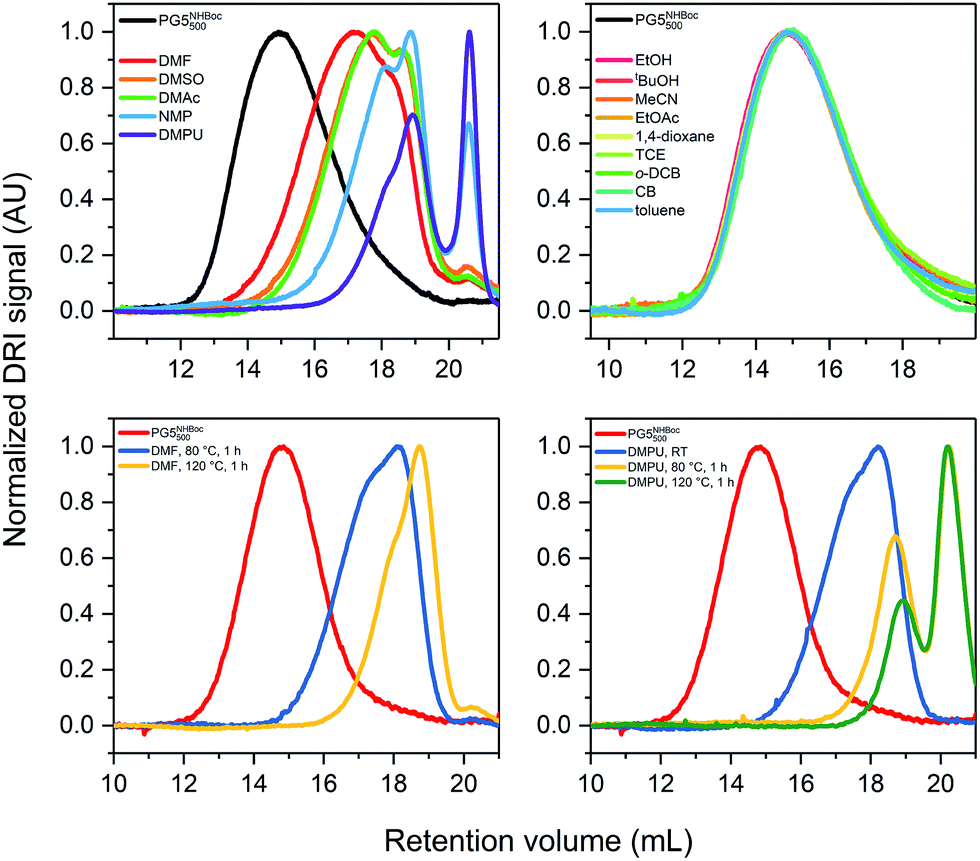 | ||
| Fig. 3 GPC retention curves demonstrating the effects of solvent and temperature. Solutions of PG5NHBoc500 (1% w/V) were prepared at RT, then heated to the indicated temperature for 1 h. (a) Results obtained for polar-aprotic solvents in which scission was found to occur at 80 °C; (b) results obtained for other solvent categories (alcohols, intermediate polarity aprotic solvents, chlorinated solvents, aromatics) in which no scission was observed for T ≤ 80 °C. Further heating of already degraded samples in (c) DMF and (d) DMPU up to 120 °C. | ||
To probe differences in the extent of scission in dependence of temperature, DPs which had already undergone some degradation were heated further. Solutions of ca. 1% w/V PG5NHBoc500 were prepared at RT and samples were analyzed by GPC after full dissolution. Further samples were analyzed after successive heating to 80 °C, then to 120 °C for 1 h each. For all polar-aprotic solvents, higher ultimate temperatures led to shorter scission products, as shown here for the extreme cases DMF (Fig. 3c) and DMPU (Fig. 3d): among the tested polar-aprotic solvents, DMF leads to the lowest extent of scission at any given temperature, while scission in DMPU already sets in at RT.§ For more data, see ESI (Fig. S9†).
To find the lower temperature threshold for main-chain scission in DMF (the synthetically most relevant solvent for DPs25,30,33), a concentrated solution of PG5NHBoc500 was prepared (ca. 20% w/w in DMF) and gradually heated while performing rheological tests. This suggested an onset of scission between 60 and 65 °C (see ESI, Fig. S11† and the accompanying text).
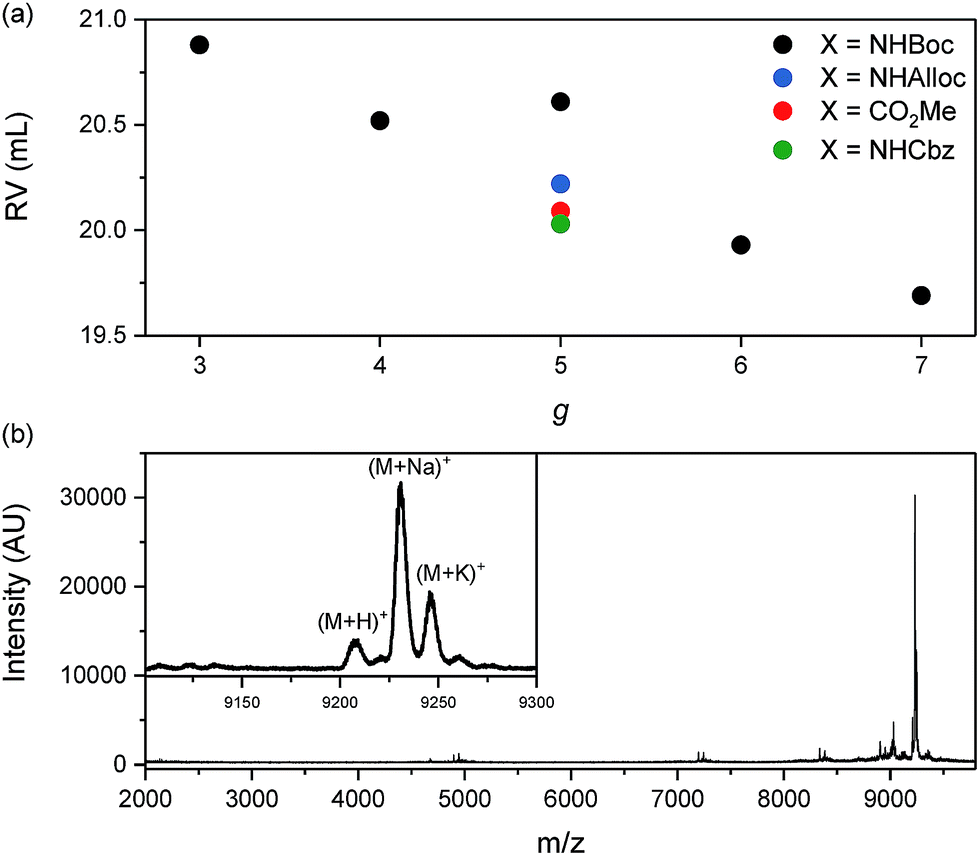
The influence of g on the scission process was studied by heating solutions of DPs (1% w/V) in DMF or DMPU to 80 °C for 1 h. These experiments involved a wide variety of DPs, including PGgNHBoc10000 of g = 1–4, PG5NHBoc∼5000, an entire homologous series of PGgNHBoc500 with high structural perfection from g = 1–8,30 as well as additional samples of PGgNHBoc500 with both low33 and high structural perfection of g = 6–8. The differences in structural perfection between the latter two sample populations have been characterized using the well-established defect labeling method34,35 and are discussed extensively elsewhere.30,33Fig. 4a shows the collected results of this large number of scission experiments, expressed by the shifts in GPC peak retention volumes (ΔRV) of the products relative to the respective starting polymers (see ESI for GPC curves, Fig. S12–S15†). ΔRV values were used as a measure to judge whether scission occurs rather than more readily transferrable quantities such as relative changes in molar mass due to experimental constraints which are discussed in the ESI (Section 4.4†). With one exception (one batch of PG6NHBoc500; see ESI, Fig. S16,† for discussion), only the DPs of g = 5 showed significant degradation, as indicated by large positive values of ΔRV.
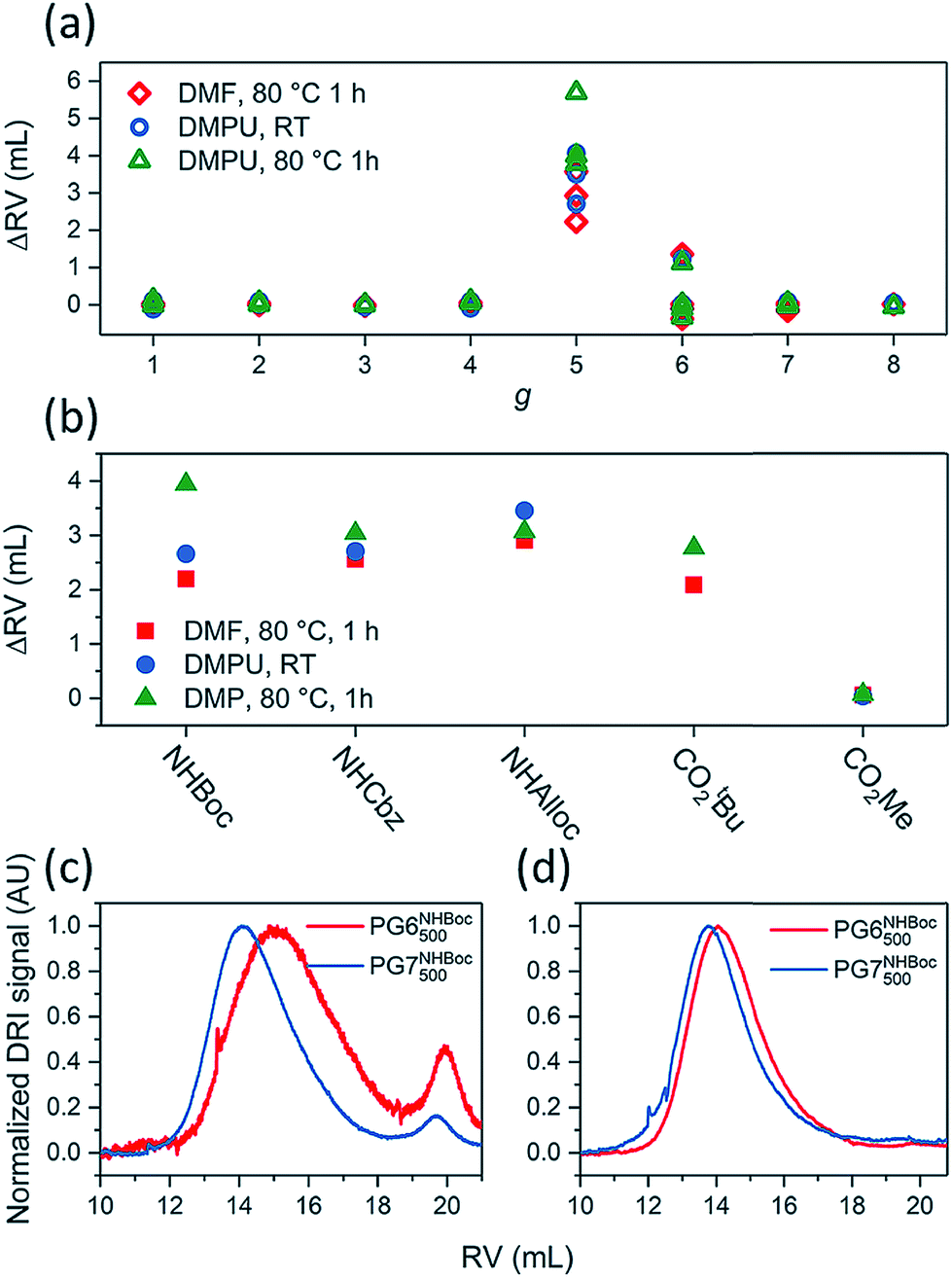 | ||
Fig. 4 (a) Influence of dendritic generation g as evidenced by shifts in GPC peak retention volumes ΔRV; DP solutions (∼1% w/V in DMF or DMPU) were heated to 80 °C for 1 h. Three groups of DPs were employed: a series of g = 1–8 and Pn = 500 described in ref. 30, a series including g = 5–8 and Pn = 500 prepared as described in ref. 33, and a series of and Pn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 from g = 1–5. (b) Influence of peripheral substitution: solutions of PG5NHBoc500, PG5NHAlloc500, PG5NHCbz500, PG5CO5002tBu and PG5CO5002Me (∼1% w/V in DMF or DMPU) were heated to 80 °C for 1 h. GPC chromatograms and molar mass data corresponding to Fig. 4a and b can be found in the ESI, Fig. S12–S19.† Influence of dendritic structural perfection: solutions of PG6NHBoc500 and PG7NHBoc500 (∼1% w/V in DMPU) were heated to 120 °C for 2 h, using DPs of low structural perfection ((c) see ref. 30) and high structural perfection ((d) see ref. 33). See ESI, Fig. S20† for the comparison with starting materials. 000 from g = 1–5. (b) Influence of peripheral substitution: solutions of PG5NHBoc500, PG5NHAlloc500, PG5NHCbz500, PG5CO5002tBu and PG5CO5002Me (∼1% w/V in DMF or DMPU) were heated to 80 °C for 1 h. GPC chromatograms and molar mass data corresponding to Fig. 4a and b can be found in the ESI, Fig. S12–S19.† Influence of dendritic structural perfection: solutions of PG6NHBoc500 and PG7NHBoc500 (∼1% w/V in DMPU) were heated to 120 °C for 2 h, using DPs of low structural perfection ((c) see ref. 30) and high structural perfection ((d) see ref. 33). See ESI, Fig. S20† for the comparison with starting materials. | ||
g = 5 DPs bearing terminal groups other than NHBoc (PG5NHCbz500, PG5NHAlloc500, PG5NHAlloc10000, PG5CO5002tBu, PG5CO5002Me) were also found to suffer scission. Upon heating 1% w/V solutions in DMF or DMPU to 80 °C for 1 h, all of these g = 5 DPs except PG5CO5002Me underwent significant main-chain degradation (Fig. 4b; see ESI† for GPC curves). PG5CO5002Me only degraded notably when exposed to higher final temperatures (DMF or DMPU at 120 °C, see ESI, Fig. S18d†). As judged by comparison of the relative concentrations of the secondary products appearing at high RV (see GPC curves in the ESI, Fig. S18†), the extent of scission followed the rough order PG5NHBoc500 > PG5NHCbz500 > PG5NHAlloc500 for the carbamates and PG5CO5002tBu ≫ PG5CO5002Me for the two esters.
ΔRV of the main polymer peak in GPC is a fairly crude measure of main-chain scission: closer inspection of chromatograms revealed additional features when harsh degradation conditions (DMPU, ≥80 °C) were applied to DPs of g = 3–7. These experiments revealed additional species with RV ≈ 19–21 mL (compare Fig. 6 and the accompanying text). However, DPs of different structural perfection did not behave the same: when heated to 120 °C in DMPU, samples of PG6NHBoc500 and PG7NHBoc500 with low structural perfection33 produced significant amounts of low-molar mass scission products (Fig. 4c), whereas PG6NHBoc500 and PG7NHBoc500 with high structural perfection (Fig. 4d)30 did not show signs of degradation even under these harsh conditions. In these experiments, ΔRV was usually negligible. Even harsher conditions (≫ 2 h at 120 °C in DMPU) were required to afford substantial shifts in peak RV (see ESI, Fig. S33†).
Some other factors were of little or indeterminate impact on main-chain scission: the variation of concentration (Fig. 5a), externally applied shear forces (Fig. 5b), and prolonged heating (≫ 1 h; Fig. 5c) produced little change. The impact of chain length is unclear: both samples shown in Fig. 5a (PG5NHBoc500 and PG5NHBoc∼5000) suffered significant degradation, their molar masses decreasing by ∼70% relative to the initial DPs (see ESI, Table S1†). It should be noted that these two DP samples were prepared by different methods (PG5NHBoc500 by RAFT polymerization,36 PG5NHBoc∼5000 by free radical polymerization25).
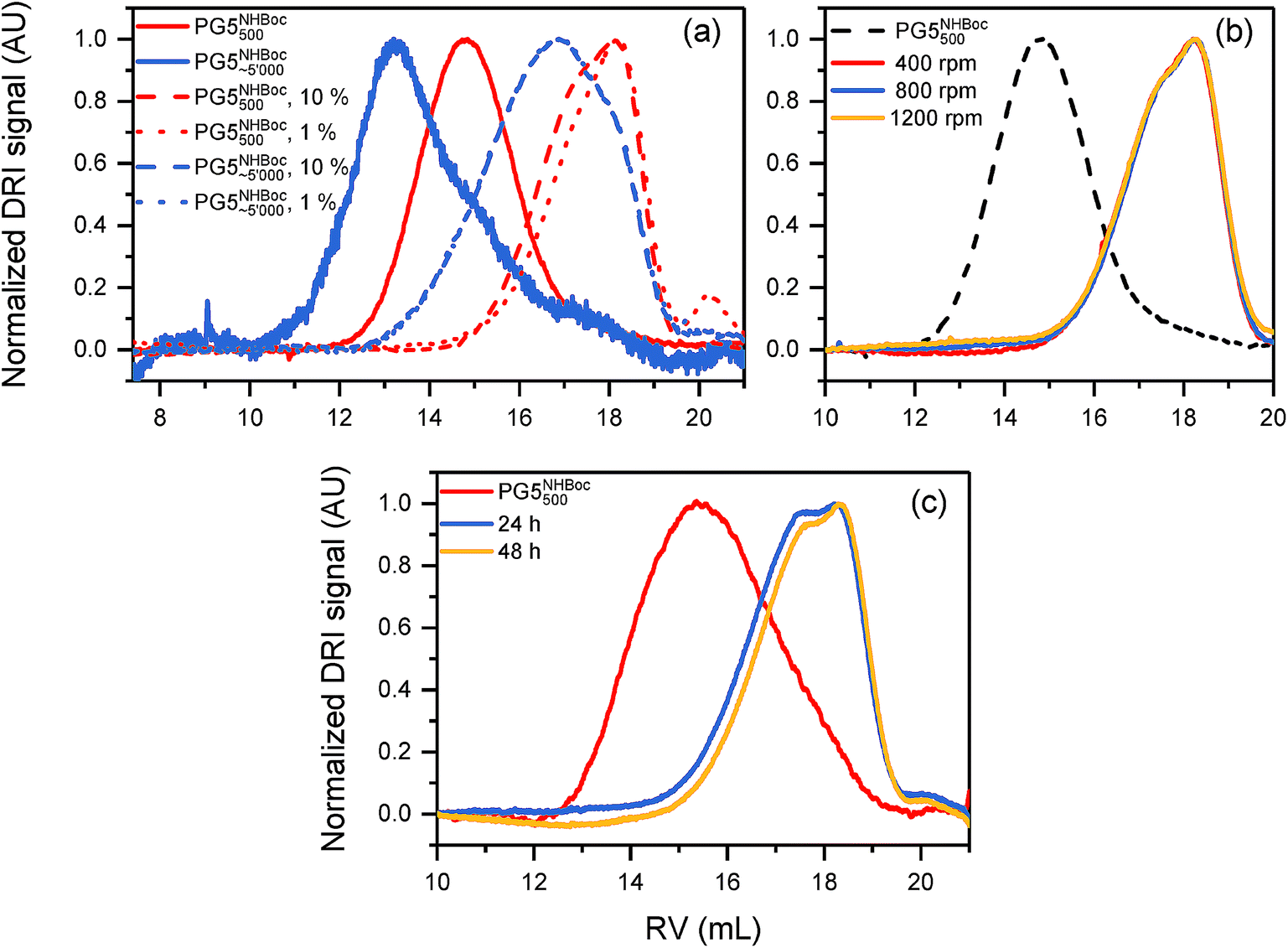 | ||
| Fig. 5 Parameters of little impact on main-chain scission: (a) DP concentration: GPC retention curves (DMF, 0.1% LiBr) of polymers obtained after heating PG5NHBoc500 and PG5NHBoc∼5000 to 80 °C for 1 h, both at concentrations of ca. 10% w/w and 1% w/V in DMF. (b) Shear rate: GPC retention curves of DPs (5% w/V in DMPU, 24 h at RT) exposed to different shear rates (orbital shaker). (c) Time: GPC curves of PG5NHBoc500 (1% w/V in DMF) maintained at 80 °C for a total of 48 h, showing little shift in peak RV after initial scission. | ||
Next, we set out to determine the identities of the low molar mass side products generated in “hot solvent” mediated degradation (e.g. the peaks appearing at RV ≈ 20.5 mL in Fig. 2a in the degradation of PG5NHBoc500). To that end, samples containing ∼1% w/V of DP in DMPU were heated to 120 °C for 1–2 h and the products were analyzed by GPC and MALDI-TOF-MS. Previous experiments had indicated some degradation in DMPU at 80 °C (see ESI, Fig. S13 and S15†) for the investigated samples. In GPC, the low molar mass peak RV was found to decrease with g, ranging from RV ≈ 20.9 mL for the degradation products of PG3NHBoc500 to RV ≈ 19.7 mL for those of PG7NHBoc500 (Fig. 6a; see ESI, Fig. S21† for corresponding chromatograms). MALDI-TOF-MS provided evidence for the corresponding macromonomers for all DPs of g < 6 (see e.g.Fig. 6b, see ESI Fig. S22–S31† for all mass spectra). For PG6NHBoc500 and PG7NHBoc500, no interpretable peaks were found (see ESI, Fig. S22, S30 & S31†).
 | ||
| Fig. 6 (a) GPC peak retention volumes of secondary, low molar mass DP scission products as a function of g; see ESI, Fig. S21† for corresponding chromatograms. (b) MALDI-TOF mass spectrum (positive mode, DCTB matrix) of the degradation products of PG5CO5002Me; the main peak (m/z = 9230.9) likely corresponds to the sodium adduct of the macromonomer, (M + Na)+, calculated m/z = 9229.92. | ||
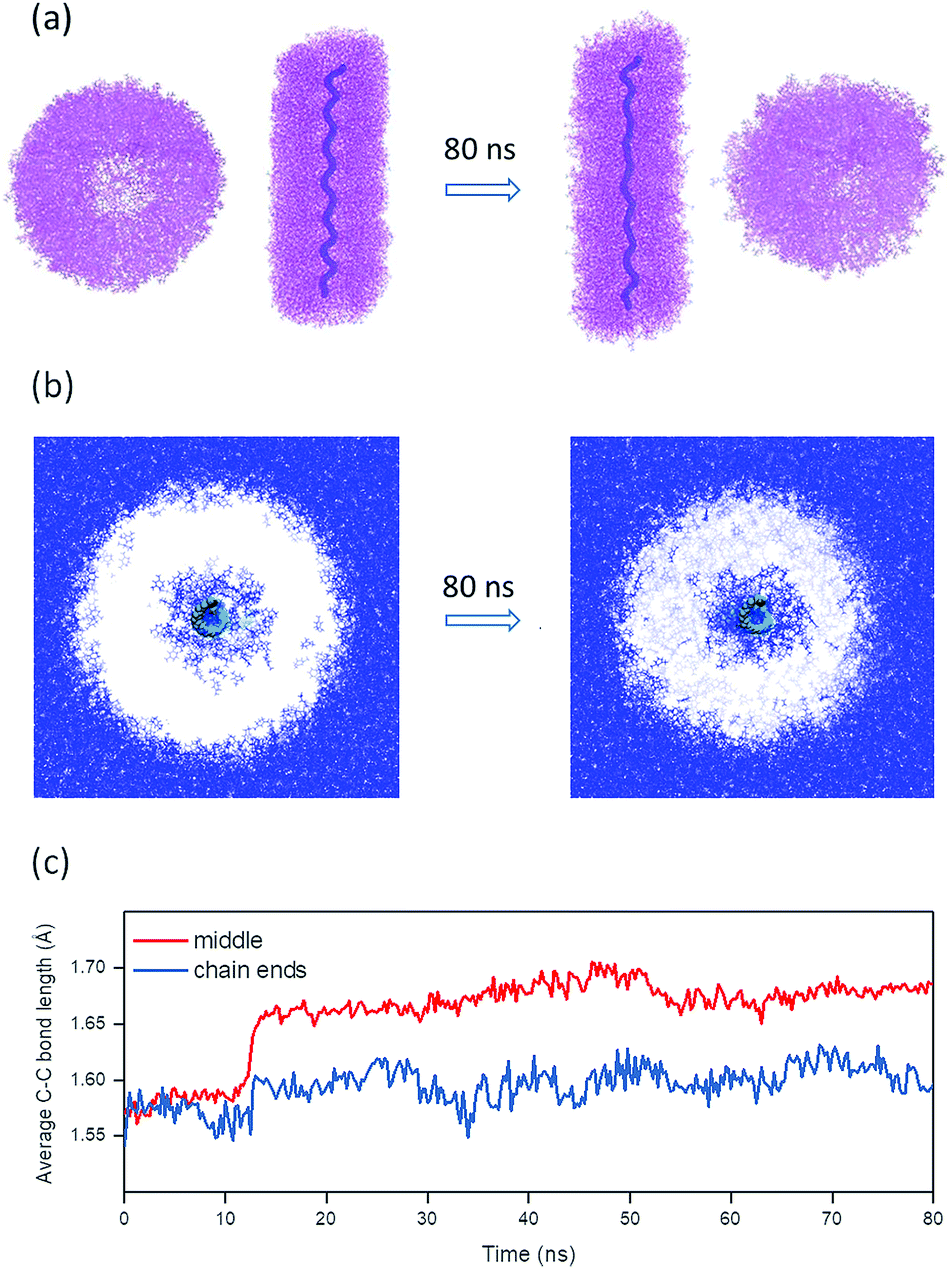 | ||
| Fig. 7 Molecular dynamics simulation of PG5NHBoc100 swelling. (a) Axial and cross-sectional projections obtained from MD simulations after solvent equilibration in DMPU38 and after 80 ns of productive simulation; solvent molecules are omitted for clarity, the DP backbone is represented by the thick purple line. (b) Cross-sectional profile through the DP, showing the distribution of DMPU (blue; backbone indicated in green, DP branches omitted for clarity). (c) Time evolution of average C–C bond lengths in the middle of the chain (30 bonds) and at the two ends (15 bonds each). | ||
The evolution of the backbone is summarized in Table 1 (also see ESI, Fig. S37†). The following backbone parameters significantly increased over the course of the simulation trajectory: end-to end distance, average dihedral angle, average bond length, and the average bond angle. The changes are particularly drastic in the middle of the chain: the center of the DP segment (average over 30 bonds) experienced far greater changes than the two chain ends (average over 15 bonds from either chain end, Fig. 7c).
| Parameter | Beginning of simulation | End of simulationb | Change |
|---|---|---|---|
| a Time-average during t = 0–10 ns. b Time-average during t = 70–80 ns. c Starting value at t = 0 ns, directly after equilibration. | |||
| End-to-end distance | 208.9 nma | 214.7 nm | +2.8% |
| Average backbone dihedral angle (entire chain) | 160°c | 170.8° | +6.8% |
| Average backbone bond angle (entire chain) | 113.1°c | 116.6 ± 1.3° | +1.8% |
| Average backbone bond length (entire chain) | 1.574 Åc | 1.603 ± 0.004 Å | +3.1% |
| Average backbone bond length (30 bonds in the middle) | 1.58 Åa | 1.68 ± 0.01 Å | +6.3% |
| Average backbone bond length (15 + 15 bonds at chain ends) | 1.57 Åa | 1.61 ± 0.01 Å | +2.5% |
EPR spectroscopy
EPR spectra of scission products were obtained by heating solutions of PG5NHBoc500 (∼5% w/V in DMPU) in the thermostat of the spectrometer, followed by vitrification of the reaction mixture and measurement at 200 K. The neat DP solutions revealed a weak, broad signal with a Landé g-factor of gJ ≈ 2.01 (see ESI, Fig. S39a†). In order to capture the most likely transient radicals involved in DP degradation, oxygen was excluded and a radical trapping agent (5,5-dimethyl-1-pyrroline N-oxide, [DMPO, 2], see Fig. 8a) was employed in subsequent experiments (Fig. 8), which revealed a much more intense EPR signal (Fig. 8a). The signal was fitted by a simple deconvolution approach based on the typical 4-line pattern of DMPO radical adducts (Fig. 8b, see ESI, Fig. S41†) and likely stems from at least four distinct radical species. Prolonged heating suggested that these species are not thermally stable (see ESI, Fig. S40b†).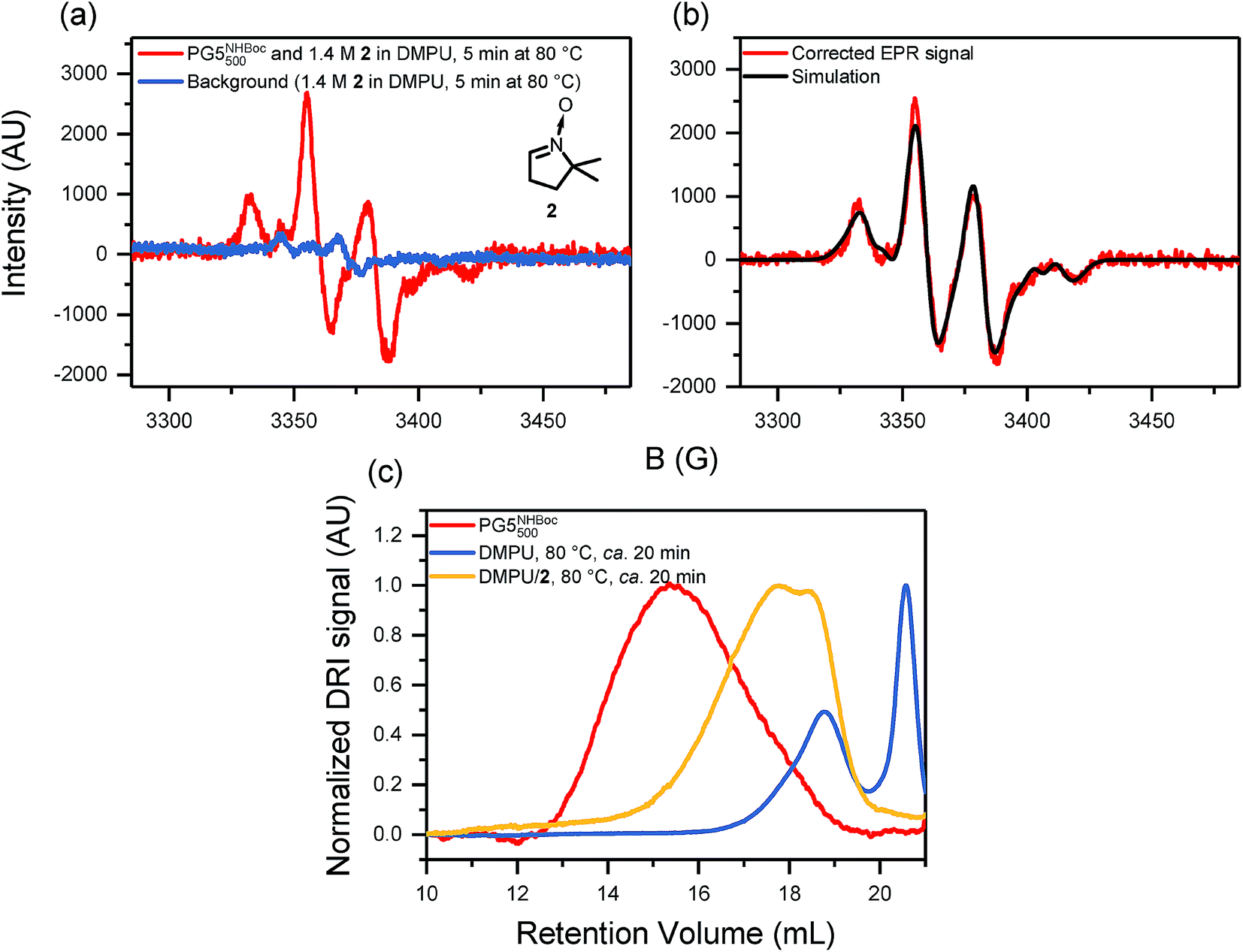 | ||
| Fig. 8 (a) EPR spectra (200 K, X-band, 1 scan) after “hot solvent” treatment of PG5NHBoc500 (ca. 5% w/V in 1.4 M DMPO/DMPU, 80 °C for 5 min). The inset shows the structure of the radical trapping agent DMPO (2). (b) Deconvolution of the background- and baseline-corrected EPR signal, consisting of 4 component radicals, see Fig. S41.† (c) GPC retention curves (DMF, 0.1% LiBr) of DPs isolated from EPR experiments after heating without and with a radical scavenger. Both samples were kept at 80 °C in DMPU for approx. 20 min. | ||
The degradation products obtained from EPR experiments were also investigated by GPC (Fig. 8c), revealing that the radical trap DMPO significantly affected the extent of degradation: in the presence of DMPO, longer polymeric fragments remained when compared to standard scission conditions (i.e. in the presence of DMPU alone, see Fig. 8c). Furthermore, unlike in all other cases of g = 5 DP scission, neither GPC (Fig. 8c) nor MALDI-TOF-MS (see ESI, Fig. S40†) provided any indication for macromonomer formation.
Discussion
Main-chain scission in charged DPs
The divergent synthesis of PGgNHBocn usually relies on the preparation of a polyelectrolytic intermediate (most commonly PGgNHn3TFA). After synthetic attempts to produce PG6NHBocn resulted in short, oligomeric products instead of the desired long polymers, it became clear that the polyelectrolytes of g = 5, such as PG5NHn3TFA (n ≫ 50) are unstable.24 The initial hypothesis that peripheral charges are responsible for this instability motivated alterations in the synthetic strategy, eventually leading to the use of the Alloc instead of the Boc protecting group.30 This change not only permitted the successful circumvention of main-chain scission, but also provided direct access to PG5NH5002. This charge-neutral intermediate offered a new tool to explore main-chain scission further, as it permitted for the controlled generation of polyelectrolytes e.g. by full protonation of PG5NH5002, which should result in backbone degradation according the initial working hypothesis. The treatment of PG5NH5002 with excess TFA (Scheme 1a, Fig. 2a, b and d) indeed resulted in main-chain scission. Furthermore, the Alloc-based protecting group chemistry made it possible to generate polyelectrolytes by protonation with weaker acids, the most straightforward option being DMBA, the allyl scavenger involved in the deprotection reaction. The protonation by this weak acid (pKa ≈ 4.7,32 producing PG5NHn3DMBA, Scheme 1b) likewise led to main-chain scission (Fig. 2a, c and d). These results seemed to vindicate the initial hypothesis that peripheral charges are responsible for DP degradation at g = 5.Conjecturing that main-chain scission might not be limited to positively charged DPs, we attempted similar experiments with negatively charged, carboxylate-bearing DPs generated from the ester-protected DP PG5CO5002tBu. Though carboxylates certainly differ from ammonium cations in many aspects affecting charge–charge interactions (charge delocalization, counterion pairing, solvation etc.), still the generation of negative charges in the periphery of a DP such as PG5CO5002H was expected to result in scission, considering the ease with which the process occurs in PGgNHn3TFA. We aimed to first prepare PG5CO5002H by removal of the tert-butyl group with TFA, and to then deprotonate the polyacid using ammonia, resulting in a polyelectrolyte, PG5COn2NH4 (Scheme 1c).
To our surprise and counter to the working hypothesis, we found that the ester deprotection already resulted in a substantial reduction in main-chain length, as evidenced by AFM (Fig. 2e and f) and by DLS (Fig. 2h): PG5CO5002H was not obtained, even though peripheral carboxylic acids should be charge-neutral in the given medium (neat TFA). The effect of the subsequent treatment with ammonia to generate a negatively charged polyelectrolyte is uncertain: DLS measurements show little change in average particle size (Fig. 2h; PG5COn2H: Dh ≈ 19.5 nm; 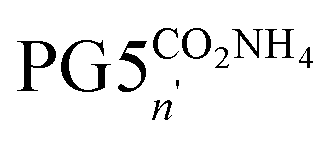 : Dh ≈ 16 nm). The reduction in particle size evident in AFM height images (Fig. 2f and g) might originate from differences in aggregation behavior between PG5COn2H and PG5COn2NH4.
: Dh ≈ 16 nm). The reduction in particle size evident in AFM height images (Fig. 2f and g) might originate from differences in aggregation behavior between PG5COn2H and PG5COn2NH4.
The above results cast doubts on the initial working hypothesis, which were reinforced by other observations involving charged DPs: for structurally deficient DPs of g > 5 prepared previously, polyelectrolytic intermediates of the type PGgNHn3TFA were found to be stable, failing the expectation that the g > 5 DPs should be even more prone to “charge-induced” degradation than PG5NHn3TFA, due to their higher numbers of peripheral charges. The surprising stability had been rationalized by structural defects resulting in reduced peripheral charge densities,33 but for the recently prepared, more structurally perfect DPs of g > 5,30 this argument cannot be applied: these DPs contain comparatively few structural defects, and the peripheral charge densities in the corresponding polyelectrolytes are higher than in PG5NHn3TFA. However, the preparation of g > 5 polyelectrolytes, e.g. PG6NH5003TFA and PG7NH5003TFA, proceeded without any backbone degradation.30 PG6NH5003TFA does degrade in TFA at elevated temperatures, however the backbone of PG7NH5003TFA remains intact even under such harsh conditions (see ESI, Fig. S3†).
Our initial working hypothesis that peripheral charges are responsible for the degradation of g = 5 polyelectrolytes was therefore inadequate: the results discussed above suggest that peripheral charges affect the backbone stability of DPs to a degree, but that there is a more fundamental cause behind main-chain scission.
Main-chain scission in non-charged DPs
Further insights into main-chain scission were gained by closer examination of a serendipitous discovery: during synthesis and handling of PGgNHBocn, DPs are usually not subjected to temperatures > 45 °C. In an attempt to prepare highly concentrated solutions of PG5NHBoc∼5000 (∼20% w/w in DMF), the DP-solvent mixture was heated to ca. 60 °C to accelerate dissolution. Alarmingly, the resulting solution was of very low viscosity, which GPC and AFM analysis revealed to be due to a drastic decrease in chain length (see ESI, Fig. S7†).Following this readily reproduced finding, a number of factors of potential relevance to this process of “hot solvent” mediated main-chain scission were investigated as shown in the Results section and in the accompanying ESI (Section 4†). The nature of the solvent (Fig. 3a and b), the ultimate temperature (Fig. 3c and d), the dendritic generation number g (Fig. 4a), the nature of the peripheral groups (Fig. 4b) and the dendritic structural perfection (Fig. 4c and d) were found to impact the outcome of scission experiments significantly.
Among these factors, particularly solvent/temperature and g are of crucial importance: “hot solvent” mediated main-chain scission has only been observed for polar-aprotic solvents (Fig. 3a). The temperature above which scission occurs varies by solvent, ranging from room temperature for DMPU to ≥60 °C for DMF (see ESI, Fig. S11†). In all polar-aprotic solvents, the extent of scission increases with the ultimately reached temperature, and already degraded samples undergo further backbone scission when subjected to even higher temperatures (Fig. 4c and d). No scission at all was found to occur in other solvent classes, including alcohols, moderately polar aprotic, chlorinated, and aromatic solvents (Fig. 3b). One key difference between all of these solvent classes and the polar-aprotics is that the latter are chemically quite similar to the dendritic branchwork. Previous investigations employing a solvatochromic probe suggested that the polar-aprotic solvents are particularly good swelling agents for DPs of the type PGgNHBocn.38
The g dependence of the “hot solvent” mediated process (Fig. 4a) closely matches that of the degradation in polyelectrolytic DPs: in both cases, only DPs of g = 5 readily undergo main-chain scission. While degradation has been induced in neutral DPs of g = 3–7 (see Fig. 6) and in polyelectrolytes of g = 4–6 (see ESI† and ref. 24), however very harsh conditions are necessary to cause the degradation of g ≠ 5 DPs.
Swelling as a common, underlying cause of main-chain scission
The solvent- and g-dependence of scission and the similarities between the “hot solvent” and “charge induced” main-chain scission processes led us to formulate a new hypothesis: the fundamental cause behind main-chain scission in dendronized polymers is solvent swelling.Main-chain scission only occurs in the presence of good swelling agents,¶ such as TFA|| or the polar-aprotic solvents akin to DMF and DMPU. Backbone degradation also only occurs in an intermediate range of steric congestion (Fig. 4a): DPs of g < 5 are more readily accessible to solvent than their higher g homologs, but solvent swelling does not result in significant strain on the polymer backbone, as there is sufficient space to accommodate the swelling agent within the DP branchwork. g = 5 DPs are still open to some solvent swelling, but the combined steric demand of the bulky dendritic side chains and the added swelling agent apparently exerts sufficient tension to break the DP backbone. The side chains of g > 5 DPs are even bulkier, however effects of essentially dense intramolecular packing become apparent for these polymers, which are near to gmax ≈ 6–7:29 no or only very little swelling occurs, even with good swelling agents such as DMPU or TFA. Rather, all solvent interactions take place in the periphery, much like in a dense colloid. Consequently, solvation of these very high g DPs is not accompanied by internal rearrangements of the dendritic matter, and no scission occurs, as little additional intramolecular tension results from dispersing such molecular objects in a solvent.
The above hypothesis is corroborated by a number of other factors which were studied for the “hot solvent” mediated variety of swelling-induced main-chain scission: first, the polymer concentration (Fig. 5a) and (moderate) shear (Fig. 5b) were not found to alter the outcome of degradation experiments. This suggests that forces acting on the DP molecules (either transmitted through entanglements in a gel or applied externally by stirring or shaking) do not significantly affect main-chain scission. In this, the essentially mechanochemical activation of DP backbones is different from other swelling-induced degradation reactions of polymeric materials. Rather than in individual molecules, such phenomena have been observed previously in cross-linked gels13,19 and in dense polymer brushes attached to surfaces.39–41 In contrast to DPs, these topologies feature points outside a scissile polymer segment – i.e., cross-links and attachment points – against which the forces caused by the swelling of the overall structure can brace. In DPs, the forces ultimately leading to backbone degradation appear to originate from within the solvent-swollen polymer segment itself, and it is not necessary to apply external force for a sufficiently swollen DP segment to undergo scission.
Second, there is some influence of peripheral substitution: generally, DPs with bulkier peripheral groups undergo more severe degradation under identical external conditions. For instance, PG5CO5002tBu behaves very similarly to the “workhorse” polymer of this study, PG5NHBoc500, and consequently undergoes main-chain scission e.g. at room temperature in DMPU. PG5CO5002Me, bearing sterically less demanding terminal groups, requires more forcing conditions, and only degrades when heated to >80 °C (Fig. 4b, also see corresponding chromatograms and Fig. S18d in the ESI†).
Third, it was found that structural perfection impacts scission: recently prepared, structurally virtually perfect DPs of g = 6 or g = 7 (ref. 30) do not undergo main-chain scission, whereas their cousins prepared by older synthetic routes33,42 do experience some degradation (Fig. 4c and d). Crucially, while the former are essentially densely packed, the latter are structurally deficient, and consequently their structures are looser and more accessible to swelling agents. The differences in structural perfection were determined using defect labeling34,35 and are owed to the different modes of synthesis of these two series of DPs as discussed elsewhere.30,33 Structural perfection and the accessibility of such high g DPs to solvent relate closely to the question whether such high g DPs can be considered molecular colloidal particles.30
Solvent swelling as the cause for scission is further corroborated by an MD simulation of a DP of g = 5 (PG5NHBoc100) in DMPU as shown in Fig. 7. The penetration of the solvent into the DP structure is accompanied by significant internal rearrangements. Swelling is associated with a contraction of the overall DP diameter (Fig. 7b, also see ESI, Fig. S36†) and an elongation of the DP (Fig. 7a). The overall extension of the simulated DP (Fig. 7a and Table 1) is caused by significant changes not only in backbone conformation (average C–C–C–C dihedral angles increase), but also by changes in fundamental bonding parameters, namely C–C–C bond angles and C–C bond lengths. The changes are most drastic in middle of this short DP chain (Fig. 7c): under confinement to locally cylindrical geometry, C–C bonds experience an average extension of 6.3% over the course of swelling. Meanwhile, the bonds at the chain ends are stretched less (+2.5% on average), as the dendrons they bear are not subject to the geometric restrictions present in the middle of the chain.
An analysis of the different energy terms shows a ∼20% increase of the stretching and bending contributions by the end of the productive simulation trajectory (t = 80 ns). Although this is partially compensated for by the favourable DMPU⋯DP interaction arising from solvent penetration, the increase in the bonding contributions clearly reflects substantial strain on the backbone due to swelling. At the end-point of simulation, the force applied to individual strained bonds in the middle segment amounts to 2.7 ± 0.3 nN, with peaks reaching up to 2.9 ± 0.2 nN (see ESI, Fig. S38†). The mechanochemical literature reports that tensions of ∼2–3 nN are sufficient for bond rupture in macromolecules,43–47 suggesting that no external stress (originating e.g. in shear fields48,49) in addition to swelling is necessary to induce DP main-chain scission. It should however be noted that the harmonic potentials used in MD simulations50 cannot appropriately capture the electronic changes necessarily involved in bond breaking.
As the above paragraphs mainly dealt with neutral species, some additional comments regarding polyelectrolytic DPs are required. Prior MD simulations of PG5NH753TFA (Fig. 2b) in aqueous solution37 suggested that coulombic repulsion between peripheral ammonium cations leads to a porous, water-swollen DP structure. Similarly, the degradation of PG5NHn3DMBA might be due to swelling of the DP structure by DMSO and NMP (which are good swelling agents) promoted by the unfolding of the DP structure due to charge–charge repulsion. Similar reasoning can be applied to cases where TFA induces scission (Fig. 2c, ref. 24), and additionally TFA is a fairly strong acid (pKa ≈ 0.5) – strong enough to partially protonate free amides, potentially generating charges in the interior of the DP structure.** Even if no additional protonation occurs, TFA appears to be an excellent swelling agent: PG5NHn3TFA degrades to shorter fragments†† than PG5NHn3DMBA (Fig. 2d), and the deprotection of PG5CO5002tBu results in main-chain scission even though in that case peripheral charges are not present. Solvent swelling must play a significant role in all these degradation processes, as the non-swellable polyelectrolytic DPs of g > 5 do not undergo backbone scission. Overall however, the picture for charged DPs is still somewhat murky, and further investigation of the relevant factors is necessary.
Mechanistic insights into main-chain scission
The investigation of main-chain scission in charge-neutral DPs has not only provided deeper insights into the causes of main-chain scission, but has also permitted mechanistic conclusions. Swelling-induced main-chain scission in general should follow the course already proposed for the specific case of “charge-induced” degradation, which was thought to proceed by backbone scission, followed by some depolymerization of the active chain end and finally either by reactive termination or loss of driving force for the depolymerization reaction.24 For the degradation of PG5NHn3TFA, this was previously demonstrated by the presence of macromonomer in the scission product mixtures, detected by MALDI-TOF-MS and by 1H-NMR spectroscopy.24 For the case of “hot solvent” mediated main-chain scission of charge-neutral DPs, macromonomer was similarly found to be produced: in all scission experiments of g = 5 DPs, a secondary peak was observed in GPC, at much higher retention volumes than the oligomeric product fraction. Similar peaks were also observed in the “hot solvent” treatment of PGgNHBocn with g = 3, 4, 6, and 7 (Fig. S21†). The retention volume of the second peak was found to decrease consistently with increasing g (Fig. 6a), suggesting well-defined, small molecule scission products the masses of which are related to that of the DP repeating unit. This supposition was readily confirmed by MALDI-TOF-MS: for all DPs of g ≤ 5, the principal ion either corresponded to an adduct of the macromonomer of the respective DP or else was very close in mass (e.g.Fig. 4c; see ESI for all mass spectra and a detailed evaluation, Fig. S22–S31, Table S3†). For the case of PG5NHBocn, scission products were also investigated by 1H-NMR spectroscopy, and again signals in the olefinic spectral region suggested the presence of macromonomer (see ESI, Fig. S32†). For PG6NHBoc500 and PG7NHBoc500, MALDI-TOF-MS showed no clear peaks. This is likely due to a combination of poor detection efficiency for the heavy ions (expected M > 20 kDa) and poor ionization efficiency, perhaps in part promoted by interdigitation of the few generated macromonomers with the remaining polymer.EPR spectroscopy provided even deeper insights into the process of main-chain scission. Swelling-induced main-chain scission is essentially a mechanochemical process.4,51,52 For DP swelling, the forces exerted onto the polymer backbone are generated internally rather than applied externally, as is possible e.g. by bulk crushing or milling,53–55 by sonochemical activation56–58 or by surface adhesion forces.44,45,59 Under external application of rupturing forces, polymer backbones usually undergo chain-centered homolytic scission57,60,61 (unless e.g. an off-center mechanophore is incorporated),4 and the resulting radicals can be detected and characterized by EPR spectroscopy.53–55 By vitrification of an ongoing DP degradation reaction (PG5NHBoc500 in DMPU at 80 °C), we found a low concentration of transient radicals by EPR spectroscopy (see ESI, Fig. S39a†). A significantly improved signal was obtained when the degradation proceeded in the presence of a radical trapping reagent (DMPO, 2, see Fig. 8a). The deconvolution of the EPR signal indicates that at least four different radical adducts are generated (Fig. 8b, see ESI, Fig. S41 & Table S4,† for details). Due to the very broad lines, the present data unfortunately does not permit for the identification and structural assignment of concrete radical species.
While this directly confirms our long-held suspicion that a radical process is involved in DP main-chain scission, backbone bond heterolysis (though likely subject to a much higher activation barrier than homolysis) should not be discounted out of hand, particularly as DP degradation proceeds exclusively in very polar media (polar-aprotic solvents, TFA). In the presence of a radical trapping agent, though, no depolymerization occurs, as evidenced by the lack of a secondary peak at RV ≈ 20.5 mL in the GPC retention curves of the scission products obtained from EPR studies (Fig. 8c). Likewise, MALDI-TOF-MS of the scission products (see ESI, Fig. S40†) did not indicate the formation of macromonomer. An alternative polar pathway therefore seems unlikely, as the active chain ends generated in bond heterolysis should be able to depropagate and produce macromonomer even in the presence of 2. The presence of a shoulder when depropagation of chain ends is prevented by reaction with 2 suggests that chain scission proceeds in a chain-centered manner, similar to e.g. sonication-induced polymer degradation.56,57 Shoulders observed in other cases (e.g.Fig. 2d and 5c) may likewise correspond to different degrees of chain-centered fragmentation, though the occurrence of depropagation complicates the interpretation of those fragment populations.
The results discussed above form the following overall mechanistic picture of DP scission: the swelling of the dendritic branchwork of a suitable DP results in an increased steric demand of the already bulky side chains. The repulsive forces between neighbouring dendrons are transmitted to and concentrated in the backbone via the branched side chain structures.52 As the backbone represents the point of balance between neighbouring dendrons, the main chain rather than some side chain ruptures. The strained backbone bonds undergo homolysis, and the resulting radicals depropagate, expelling the corresponding macromonomer.‡‡ Depropagation proceeds until the radical chain end is quenched chemically (e.g. by dioxygen) or until the driving force disappears, e.g. through spatial rearrangement of the dendrons at the newly formed chain end.
Summary
Since the material presented above covers a fairly large range of interrelated results, we would like to summarize the most salient points. Initial experiments with charged DPs (Fig. 2a–d) supported our previous working hypothesis, which had proposed coulombic repulsion between peripheral charges as the determining factor in DP main-chain scission. However, the stability of g > 5 polyelectrolytes30 and degradation in the acid-mediated deprotection of PG5COn2tBu (forming a nominally charge-neutral product, Fig. 2e, f and h) contradicted this hypothesis, and suggested that the main cause of main-chain scission must be sought elsewhere.The discovery and subsequent investigation of scission in charge neutral DPs – without any peripheral chemical reactions occurring – provided key evidence in the search for an alternative explanation (Fig. 3–6). In striking similarity to DP scission involving charged species, it was found that DPs of g = 5 suffer degradation in the presence of polar-aprotic solvents, mostly at elevated temperatures. Very limited backbone scission could be provoked in DPs of g ≠ 5, but only at much higher temperatures. The strong dependence of DP degradation on g (Fig. 4a) and the solvent (Fig. 3a and b) suggested that swelling of the dendritic branches is the root cause behind main-chain scission. The impact (or lack thereof) of a number of other factors (temperature, dendritic structural perfection, peripheral substitution, shear, concentration; Fig. 4b–d and 5) confirmed this notion, and an MD simulation of a g = 5 DP in a good swelling agent demonstrated that solvent uptake is connected to substantial strain on the DP backbone (Fig. 7, Table 1). Using EPR spectroscopy and a radical trapping agent, it was shown that degradation proceeds initially via homolytic C–C bond scission along the backbone, followed by partial depolymerization (Fig. 8). This is consistent with an essentially mechanochemical process, induced by internally generated tension on the polymer backbone, exerted by the inherently very bulky and additionally solvent-swollen dendritic side chains.
Swelling as the root cause for scission not only explains the observations made for neutral DPs, but can also readily accommodate the degradation of charged DPs: TFA in particular is likely an excellent swelling agent for DPs, in addition to protonating peripheral amines e.g. in the deprotection of PG5NHBocn. Scission also occurs upon protonation of terminal amines by weak electrolytes such as DMBA because coulombic repulsion among peripheral groups opens the DP structure to additional solvent swelling.
Conclusions & outlook
Solvent-swelling induced main-chain scission is essentially caused by chain-internal mechanical stress and thereby stands apart from classical chain degradation reactions: it is not caused by external mechanical forces, nor by chemical reactants attacking the backbone, nor is it related to an intrinsic temperature instability of the backbone. DP chain scission also differs from scission phenomena observed previously in polymer networks13,19 and brushes,39–41 where scission is topologically restricted to segments braced between cross-links or against attachment points. Rather, DP backbone scission is the consequence of endowing an individual linear polymer with densely packed, compact side groups. This dense functionalization has been enabled by divergent dendritic synthesis which uses highly efficient amide bond formation chemistry.25,30 Similar solvent instabilities might well be observed in other very densely functionalized macromolecules, such as hypergraft62 or bottle-brush59 polymers.The observation that DP main chain scission occurs most readily at g = 5 – just below the onset of dense packing at the projected maximum dendritic generation gmax ≈ 6 (ref. 29) – reinforces the notion that DPs of such high g are essentially molecular colloids: DPs of g > 5 are densely packed and only interact with solvent peripherally. DPs of g = 5 still offer the possibility of interaction that is more than superficial, but for solvents which strongly interact with the DP branchwork, swelling results in substantial stresses on the polymer backbone and therefore leads to main-chain scission. In contrast, g < 5 DPs more closely resemble standard linear polymers, in that they can by and large accommodate solvent swelling without backbone degradation.
The parameter space for the investigation of DP degradation is large. The work presented here mainly used the example of PG5NHBoc500. In-depth investigations regarding charged DPs and the purposeful variation of structural parameters such as repeating unit spacing, dendron tether length, and peripheral substitution may deliver further insights into DP main-chain scission. Particularly the functionalization of DPs with groups providing LCST/UCST behavior will be of great interest.63 The knowledge of degradation tendencies may also prove synthetically useful in the preparation of g > 6, high Pn DPs in the quest towards individual macromolecules of GDa molar mass.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to express our gratitude to Prof. Nicholas Spencer (ETH Zürich) for access to AFM instrumentation, Dr Thomas Schweizer (ETH Zürich) for conducting and evaluating rheological measurements, and the personnel at MoBiAs (particularly Louis Bertschi, ETH Zürich) for MALDI-TOF-MS. CA's and OB's research is funded by MINECO (MAT2015-69367-R), AGAUR (2017 SGR 359) and ICREA Foundation (“ICREA Academia” to CA). DM and ADS thank ETH Zürich for financial support.Notes and references
- B. M. Fanconi, Chain Scission and Mechanical Failure of Polyethylene, J. Appl. Phys., 1983, 54(10), 5577–5582 CrossRef CAS.
- G. A. V. Timóteo, G. J. M. Fechine and M. S. Rabello, Stress Cracking and Photodegradation Behavior of Polycarbonate. The Combination of Two Major Causes of Polymer Failure, Polym. Eng. Sci., 2008, 48(10), 2003–2010 CrossRef.
- J. Verdu, Structural Changes Caused by Oxidation, in Oxidative Ageing of Polymers, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013, pp. 163–201 Search PubMed.
- J. Li, C. Nagamani and J. S. Moore, Polymer Mechanochemistry: From Destructive to Productive, Acc. Chem. Res., 2015, 48(8), 2181–2190 CrossRef CAS PubMed.
- G. Geuskens, F. Debie, M. S. Kabamba and G. Nedelkos, New Aspects of the Photooxidation of Polyolefins, Polym. Photochem., 1984, 5(1–6), 313–331 CrossRef CAS.
- F. Gugumus, Mechanisms of Photooxidation of Polyolefins, Macromol. Mater. Eng., 1990, 176, 27–42 Search PubMed.
- B. L. Banik, P. Fattahi and J. L. Brown, Polymeric Nanoparticles: The Future of Nanomedicine, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8(2), 271–299 Search PubMed.
- K. M. El-Say and H. S. El-Sawy, Polymeric Nanoparticles: Promising Platform for Drug Delivery, Int. J. Pharm., 2017, 528, 675–691 CrossRef CAS PubMed.
- K. Nakamura, Photopolymers: Photoresist Materials, Processes, and Applications, CRC Press, 2014 Search PubMed.
- Y. Chen, Nanofabrication by Electron Beam Lithography and Its Applications: A Review, Microelectron. Eng., 2015, 135, 57–72 CrossRef CAS.
- D. S. Achilias, Chemical Recycling of Poly(Methyl Methacrylate) by Pyrolysis. Potential Use of the Liquid Fraction as a Raw Material for the Reproduction of the Polymer, Eur. Polym. J., 2007, 43(6), 2564–2575 CrossRef CAS.
- D. S. Achilias, I. Kanellopoulou, P. Megalokonomos, E. Antonakou and A. A. Lappas, Chemical Recycling of Polystyrene by Pyrolysis: Potential Use of the Liquid Product for the Reproduction of Polymer, Macromol. Mater. Eng., 2007, 292, 923–934 CrossRef CAS.
- C. K. Lee, C. E. Diesendruck, E. Lu, A. N. Pickett, P. A. May, J. S. Moore and P. V. Braun, Solvent Swelling Activation of a Mechanophore in a Polymer Network, Macromolecules, 2014, 47, 2690–2694 CrossRef CAS.
- A. R. Sulkanen, J. Sung, M. J. Robb, J. S. Moore, N. R. Sottos and G. Liu, Spatially Selective and Density-Controlled Activation of Interfacial Mechanophores, J. Am. Chem. Soc., 2019, 141, 4080–4085 CrossRef CAS PubMed.
- M. J. Robb, T. Ann Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, Regioisomer-Specific Mechanochromism of Naphthopyran in Polymeric Materials Scheme 1. Transformation of Naphthopyran into a Colored Merocyanine Species Is Accomplished Using Mechanical Force A, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- T. Ann Kim, M. J. Robb, J. S. Moore, S. R. White and N. R. Sottos, Mechanical Reactivity of Two Different Spiropyran Mechanophores in Polydimethylsiloxane, Macromolecules, 2018, 51, 9177–9183 CrossRef.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, Mechanochromic Dynamic Covalent Elastomers: Quantitative Stress Evaluation and Autonomous Recovery, ACS Macro Lett., 2015, 4, 1307–1311 CrossRef CAS.
- L. Wang, W. Zhou, Q. Tang, H. Yang, Q. Zhou, X. Zhang, L. Wang, W. Zhou, Q. Tang and H. Yang, et al., Rhodamine-Functionalized Mechanochromic and Mechanofluorescent Hydrogels with Enhanced Mechanoresponsive Sensitivity, Polymers, 2018, 10(9), 994 CrossRef PubMed.
- J. M. Clough, J. Van Der Gucht and R. P. Sijbesma, Mechanoluminescent Imaging of Osmotic Stress-Induced Damage in a Glassy Polymer Network, Macromolecules, 2017, 50, 2043–2053 CrossRef CAS PubMed.
- Y. Yuan, W. Yuan and Y. Chen, Recent Advances in Mechanoluminescent Polymers, Sci. China Mater., 2016, 59(6), 507–520 CrossRef CAS.
- W. Yuan, Y. Yuan, F. Yang, M. Wu and Y. Chen, Improving Mechanoluminescent Sensitivity of 1,2-Dioxetane-Containing Thermoplastic Polyurethanes by Controlling Energy Transfer across Polymer Chains, Macromolecules, 2018, 51, 9019–9025 CrossRef CAS.
- G. A. Filonenko, J. A. M. Lugger, C. Liu, E. P. A. van Heeswijk, M. M. R. M. Hendrix, M. Weber, C. Müller, E. J. M. Hensen, R. P. Sijbesma and E. A. Pidko, Tracking Local Mechanical Impact in Heterogeneous Polymers with Direct Optical Imaging, Angew. Chem., Int. Ed., 2018, 57(50), 16385–16390 CrossRef CAS PubMed.
- Y. Yuan, W. Chen, Z. Ma, Y. Deng, Y. Chen, Y. Chen and W. Hu, Enhanced Optomechanical Properties of Mechanochemiluminescent Poly(Methyl Acrylate) Composites with Granulated Fluorescent Conjugated Microporous Polymer Fillers, Chem. Sci., 2019, 10, 2206–2211 RSC.
- H. Yu, A. D. Schlüter and B. Zhang, Main-Chain Scission of a Charged Fifth-Generation Dendronized Polymer, Helv. Chim. Acta, 2012, 95(12), 2399–2410 CrossRef CAS.
- B. Zhang, R. Wepf, K. Fischer, M. Schmidt, S. Besse, P. Lindner, B. T. King, R. Sigel, P. Schurtenberger and Y. Talmon, et al., The Largest Synthetic Structure with Molecular Precision: Towards a Molecular Object, Angew. Chem., Int. Ed., 2011, 50(3), 737–740 CrossRef CAS PubMed.
- P. G. de Gennes and H. Hervet, Statistics of “Starburst” Polymers, J. Phys., Lett., 1983, 44(3), 351–360 CrossRef CAS.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard, Starburst Dendrimers: Molecular-Level Control of Size, Shape, Surface Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter, Angew. Chem., Int. Ed., 1990, 29(2), 138–175 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- B. Zhang, R. Wepf, M. Kröger, A. Halperin and A. D. Schlüter, Height and Width of Adsorbed Dendronized Polymers: Electron and Atomic Force Microscopy of Homologous Series, Macromolecules, 2011, 44(17), 6785–6792 CrossRef CAS.
- D. Messmer, M. Kröger and A. D. Schlüter, Pushing Synthesis towards the Maximum Generation Range of Dendritic Macromolecules, Macromolecules, 2018, 51(14), 5420–5429 CrossRef CAS.
- A. D. Schlüter, A. Halperin, M. Kröger, D. Vlassopoulos, G. Wegner and B. Zhang, Dendronized Polymers: Molecular Objects between Conventional Linear Polymers and Colloidal Particles, ACS Macro Lett., 2014, 3(10), 991–998 CrossRef.
- N. I. Korotkikh, A. H. Cowley, J. A. Moore, N. V. Glinyanaya, I. S. Panov, G. F. Rayenko, T. M. Pekhtereva and O. P. Shvaika, Reaction of 1-Tert-Butyl-3,4-Diphenyl-1,2,4-Triazol-5-Ylidenes with a Malonic Ester, Org. Biomol. Chem., 2008, 6(1), 195–199 RSC.
- H. Yu, A. D. Schlüter and B. Zhang, Synthesis of High Generation Dendronized Polymers and Quantification of Their Structure Perfection, Macromolecules, 2014, 47(13), 4127–4135 CrossRef CAS.
- L. Shu, I. Gössl, J. P. Rabe and A. D. Schlüter, Quantitative Aspects of the Dendronization of Dendronized Linear Polystyrenes, Macromol. Chem. Phys., 2002, 203, 2540–2550 CrossRef CAS.
- B. Zhang, H. Yu, A. D. Schlüter, A. Halperin and M. Kröger, Synthetic Regimes Due to Packing Constraints in Dendritic Molecules Confirmed by Labelling Experiments, Nat. Commun., 2013, 4, 1993 CrossRef PubMed.
- A. Zhang, L. Wei and a. D. Schlüter, Narrowly Distributed Dendronized Polymethacrylates by Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization, Macromol. Rapid Commun., 2004, 25(7), 799–803 CrossRef CAS.
- O. Bertran, B. Zhang, A. D. Schlüter, M. Kröger and C. Alemán, Computer Simulation of Fifth Generation Dendronized Polymers: Impact of Charge on Internal Organization, J. Phys. Chem. B, 2013, 117(19), 6007–6017 CrossRef CAS PubMed.
- C. Gstrein, B. Zhang, M. A. Abdel-Rahman, O. Bertran, C. Alemán, G. Wegner, A. D. Schlüter, A. Halperin, A. D. Schlüter and Y. Talmon, et al., Solvatochromism of Dye-Labeled Dendronized Polymers of Generation Numbers 1–4: Comparison to Dendrimers, Chem. Sci., 2016, 7(7), 4644–4652 RSC.
- S. Tugulu and A. Klok, Stability and Nonfouling Properties of Poly(Poly(Ethylene Glycol) Methacrylate) Brushes under Cell Culture Conditions, Biomacromolecules, 2008, 9, 906–912 CrossRef CAS PubMed.
- A. Klok and J. Genzer, Expanding the Polymer Mechanochemistry Toolbox through Surface-Initiated Polymerization, ACS Macro Lett., 2015, 4, 636–639 CrossRef.
- B. Lyu, W. Cha, T. Mao, Y. Wu, H. Qian, Y. Zhou, X. Chen, S. Zhang, L. Liu and G. Yang, et al., Surface Confined Retro Diels–Alder Reaction Driven by the Swelling of Weak Polyelectrolytes, ACS Appl. Mater. Interfaces, 2015, 7, 6254–6259 CrossRef CAS PubMed.
- H. Yu, A. D. Schlüter and B. Zhang, Synthesis of Dendronized Polymers by a “n + 2” Approach, Macromolecules, 2012, 45(21), 8555–8560 CrossRef CAS.
- T. Hugel, M. Grosholz, H. Clausen-Schaumann, A. Pfau, H. Gaub and M. Seitz, Elasticity of Single Polyelectrolyte Chains and Their Desorption from Solid Supports Studied by AFM Based Single Molecule Force Spectroscopy, Macromolecules, 2001, 34, 1039–1047 CrossRef CAS.
- I. Park, S. S. Sheiko, A. Nese and K. Matyjaszewski, Molecular Tensile Testing Machines: Breaking a Specific Covalent Bond by Adsorption-Induced Tension in Brushlike Macromolecules, Macromolecules, 2009, 42, 1805–1807 CrossRef CAS.
- S. S. Sheiko, F. C. Sun, A. Randall, D. Shirvanyants, M. Rubinstein, H.-I. Lee and K. Matyjaszewski, Adsorption-Induced Scission of Carbon-Carbon Bonds, Nature, 2006, 440(9), 191–194 CrossRef CAS PubMed.
- S. Panyukov, E. B. Zhulina, S. S. Sheiko, G. C. Randall, J. Brock and M. Rubinstein, Tension Amplification in Molecular Brushes in Solutions and on Substrates, J. Phys. Chem. B, 2009, 113, 3750–3768 CrossRef CAS PubMed.
- N. V. Lebedeva, F. C. Sun, H.-I. Lee, K. Matyjaszewski and S. S. Sheiko, “Fatal Adsorption” of Brushlike Macromolecules: High Sensitivity of C-C Bond Cleavage Rates to Substrate Surface Energy, J. Am. Chem. Soc., 2008, 130, 4228–4229 CrossRef CAS PubMed.
- J. A. Odell, A. Keller and Y. Rabin, Flow-induced Scission of Isolated Macromolecules, J. Chem. Phys., 1988, 88(6), 4022–4028 CrossRef CAS.
- J. A. Odell and A. Keller, Flow-Induced Chain Fracture of Isolated Linear Macromolecules in Solution, J. Polym. Sci., Part B: Polym. Phys., 1986, 24(9), 1889–1916 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and Testing of a General Amber Force Field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- Y. Li and S. S. Sheiko, Molecular Mechanochemistry: Engineering and Implications of Inherently Strained Architectures, in Topics in Current Chemistry, Springer International Publishing, 2015, vol. 369, pp. 1–36 Search PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Mechanochemistry as an Emerging Tool for Molecular Synthesis: What Can It Offer?, Chem. Sci., 2018, 9, 3080–3094 RSC.
- T. Kawashima, S. Shimada, H. Kashiwabara and J. Sohma, ESR Studies on the Molecular Mechanisms of Fracture of Polymers at Low Temperatures, Polym. J., 1973, 5(2), 135–143 CrossRef CAS.
- M. Sakaguchi and J. Sohma, ESR Evidence for Main-Chain Scission Produced by Mechanical Fracture of Polymers at Low Temperature, J. Polym. Sci., Polym. Phys. Ed., 1975, 13, 1233–1245 CrossRef CAS.
- M. Tabata, T. Miyazawa, O. Kobayashi and J. Sohma, Direct Evidence of Main-Chain Scissions Induced by Ultrasonic Irradiation of Benzene Solutions of Polymers, Chem. Phys. Lett., 1980, 73(1), 178–180 CrossRef CAS.
- Y. Li, Z. Niu, J. Burdyńska, A. Nese, Y. Zhou, Z. S. Kean, A. V. Dobrynin, K. Matyjaszewski, S. L. Craig and S. S. Sheiko, Sonication-Induced Scission of Molecular Bottlebrushes: Implications of the “Hairy” Architecture, Polymer, 2016, 84, 178–184 CrossRef CAS.
- G. I. Peterson, K.-T. Bang and T.-L. Choi, Mechanochemical Degradation of Denpols: Synthesis and Ultrasound-Induced Chain Scission of Polyphenylene-Based Dendronized Polymers, J. Am. Chem. Soc., 2018, 140(27), 8599–8608 CrossRef CAS PubMed.
- Y. Li, Z. Niu, J. Burdy Nska C, A. Nese, Y. Zhou, Z. S. Kean, A. V. Dobrynin, K. Matyjaszewski, S. L. Craig and S. S. Sheiko, Sonication-Induced Scission of Molecular Bottlebrushes: Implications of the “hairy” Architecture, Polymer, 2016, 84, 178–184 CrossRef CAS.
- N. V. Lebedeva, A. Nese, F. C. Sun, K. Matyjaszewski and S. S. Sheiko, Anti-Arrhenius Cleavage of Covalent Bonds in Bottlebrush Macromolecules on Substrate, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(24), 9276–9280 CrossRef CAS PubMed.
- G. Gooberman, Ultrasonic Degradation of Polystyrene. Part 1. A Proposed Mechanism for Degradation, J. Polym. Sci., 1960, 42(139), 25–33 CrossRef CAS.
- J. Chakraborty, J. Sarkar, R. Kumar and G. Madras, Ultrasonic Degradation of Polybutadiene and Isotactic Polypropylene, Polym. Degrad. Stab., 2004, 85, 555–558 CrossRef CAS.
- C. Schüll and H. Frey, Controlled Synthesis of Linear Polymers with Highly Branched Side Chains by “Hypergrafting”: Poly(4-Hydroxy Styrene)-Graft-Hyperbranched Polyglycerol, ACS Macro Lett., 2012, 1(4), 461–464 CrossRef.
- X. Sun, J.-P. Lindner, B. Bruchmann and A. D. Schlüter, Synthesis of Neutral, Water-Soluble Oligo–Ethylene Glycol-Containing Dendronized Homo- and Copolymers of Generations 1, 1.5, 2, and 3, Macromolecules, 2014, 47(21), 7337–7346 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures; syntheses and characterization of PG5CO5002tBu, PG5CO5002Me and the respective small-molecule precursors; additional GPC, AFM, MALDI-TOF-MS, EPR and NMR spectroscopic data concerning scission experiments and the characterization of scission products; additional MD simulation data and associated discussion. See DOI: 10.1039/c9sc01639b |
| ‡ MeCN is fairly polar, but a very poor solvent for DPs of the type PGgNHBocn. Their mixtures with MeCN remain cloudy even at reflux, while the other solvents listed in Fig. 3a and b afforded clear solutions at 80 °C. |
| § In hindsight, this finding is not surprising: DMPU had been selected as a prospective solvent for the synthesis of PG6NHBoc500 from PG5NHAlloc500 in earlier studies, however, main-chain scission was found to occur; see the ESI for ref. 30, p. 39, freely available at https://pubs.acs.org/doi/10.1021/acs.macromol.8b00891. |
| ¶ Since DPs are polymers with a significant diameter and a rather clear separation between “interior” and “exterior”, some comment should be made on the difference between solvents and swelling agents, which becomes particularly pertinent at high g: many solvents are capable of dispersing DPs, but they are not necessarily good swelling agents, i.e. capable of penetrating deeply into the dendritic branchwork. This is true e.g. for methylene chloride, a very good solvent but a rather poor swelling agent (see ref. 38). A potential example of the opposite case is TFA, which appears to be an excellent swelling agent, but a poor solvent. Polar-aprotics such as DMF and DMPU are both good swelling agents and good solvents. |
| || Protonation of internal amides likely contributes to the swelling in the case of TFA; see main text for further discussions on the influence of charges. |
| ** The impact of inter-dendron interactions such as hydrogen bonds on the basicity of the amides is uncertain. |
| †† For TFA, the shortest fragments observed (n ≈ 40–50) of all main-chain scission reactions were observed, suggesting a possible lower chain length boundary on swelling-induced scission. |
| ‡‡ A discussion of possible mechanisms involved in backbone scission and the generation of macromonomers may be found in the ESI.† |
| This journal is © The Royal Society of Chemistry 2019 |