
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDye-sensitized photocathodes for oxygen reduction: efficient H2O2 production and aprotic redox reactions†
Jiaonan
Sun
 ,
Yongze
Yu
,
Allison E.
Curtze
,
Xichen
Liang
and
Yiying
Wu
*
,
Yongze
Yu
,
Allison E.
Curtze
,
Xichen
Liang
and
Yiying
Wu
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: wu@chemistry.ohio-state.edu; Fax: +1-614-292-1685; Tel: +1-614-247-7810
First published on 29th April 2019
Abstract
Dye-sensitized photoelectrochemical cells (DSPECs) can be used to store solar energy in the form of chemical bonds. Hydrogen peroxide (H2O2) is a versatile energy carrier and can be produced by reduction of O2 on a dye-sensitized photocathode, in which the design of dye molecules is crucial for the conversion efficiency and electrode stability. Herein, using a hydrophobic donor-double-acceptor dye (denoted as BH4) sensitized NiO photocathode, hydrogen peroxide (H2O2) can be produced efficiently by reducing O2 with current density up to 600 μA cm−2 under 1 sun conditions (Xe lamp as sunlight simulator, λ > 400 nm). The DSPECs maintain currents greater than 200 μA cm−2 at low overpotential (0.42 V vs. RHE) for 18 h with no decrease in the rate of H2O2 production in aqueous electrolyte. Moreover, the BH4 sensitized NiO photocathode was for the first time applied in an aprotic electrolyte for oxygen reduction. In the absence of a proton source, the one-electron reduction of O2 generates stable, nucleophilic superoxide radicals that can then be synthetically utilized in the attack of an available electrophile, such as benzoyl chloride. The corresponding photocurrent generated by this photoelectrosynthesis is up to 1.8 mA cm−2. Transient absorption spectroscopy also proves that there is an effective electron transfer from reduced BH4 to O2 with a rate constant of 1.8 × 106 s−1. This work exhibits superior photocurrent in both aqueous and non-aqueous systems and reveals the oxygen/superoxide redox mediator mechanism in the aprotic chemical synthesis.
Introduction
Hydrogen peroxide (H2O2) is an important commodity chemical that is widely used for cleaning, paper bleaching, and waste water treatment applications.1 Conventional H2O2 production is achieved via the anthraquinone process, which is a large-scale synthesis method capable of producing 1 metric ton H2O2 per day. However, the anthraquinone process is limited by side reactions, the requirement of significant energy input, and contamination by organic impurities. Therefore, it has become of interest to pursue clean, on-site production of H2O2 by electrolysis or photolysis of its elements. The conversion of inexhaustible solar energy into versatile hydrogen peroxide has been proposed as a feasible alternative.2,3 Hydrogen peroxide can be generated by photooxidation of water using BiVO4 or Ge-porphyrin dye sensitized TiO2 photoanodes,4–7 though it suffers from side reactions, namely, the oxidation of water to oxygen. Hydrogen peroxide can also be produced by the two-electron photoreduction of oxygen in aqueous media. Semiconductor metal oxide materials,8,9 graphitic carbon nitride (g-C3N4)10 and organic semiconductors including epindolidione (EPI), the quinacridone (QNC) family, dimethyl perylene tetracarboxylicdiimide (PTCDI), and poly(3-hexylthiophene) (P3HT)11,12 have been used as photocatalysts in the production of H2O2 from oxygen reduction. Observed current densities achieved by these photocathodes ranges from about 3 to 20 μA cm−2 with limited stability under 1 sun or visible light illumination.Dye sensitized photoelectrochemical cells (DSPECs) are promising platforms in targeting solar fuel generation. DSPEC devices are usually composed of mesoporous semiconductors, photosensitizers, and catalysts. Extensive work has been done on dye sensitized photoanodes based on n-type TiO2 semiconductors for water splitting.13 Tandem DSPEC devices, which incorporate both n-type and p-type DSPECs, can operate with greater theoretical efficiency than either n-type or p-type alone, providing motivation for the study of the less developed photocathode. To this end, dye sensitized nickel oxide (NiO) photocathodes for water splitting have been studied with the use of Ru complexes, push–pull dyes and coumarin dyes as light absorbers.14–19 Moreover, dye sensitized photocathodes can be applied not only to water splitting applications, but also to the production of H2O2. Recently, it was noticed that a porphyrin sensitized NiO photocathode enables H2O2 to be produced from oxygen with high faradaic efficiency and decent photocurrent.20 However, it can only achieve a photocurrent of 300 μA cm−2 with a high-power light source at a specific wavelength for excitation of porphyrin dye. Under 1 sun illumination with AM 1.5 filter, the porphyrin sensitized NiO photocathode only achieves an 80 μA cm−2 photocurrent.
Our group has synthesized an organic sensitizer, denoted as BH4, and applied it to p-type DSPECs for hydrogen evolution.21–23 The unique structural design of this photosensitizer allows DSPECs sensitized with BH4 to be operated in extremely acidic conditions. As shown in Scheme 1(a), the BH4 molecule is composed of three moieties: a triphenylamine donor (TPA, in blue), perylenemonoimide acceptors (PMI, in red), and thiophene linkers (in green). Under light illumination, TPA serves as an electron donor to PMI, facilitated by the thiophene linkers connecting PMI and TPA together. Each linker consists of four thiophene units and four hexyl chains that not only aid in the charge separation of electrons and holes, but also provide a hydrophobic environment on electrode surface, providing unprecedented stability to BH4 sensitized devices. Further, BH4 dye absorbs sunlight efficiently with molar extinction coefficient close to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 and provides long-lived charge separated state which enables efficient charge transfer reactions.24
000 M−1 and provides long-lived charge separated state which enables efficient charge transfer reactions.24
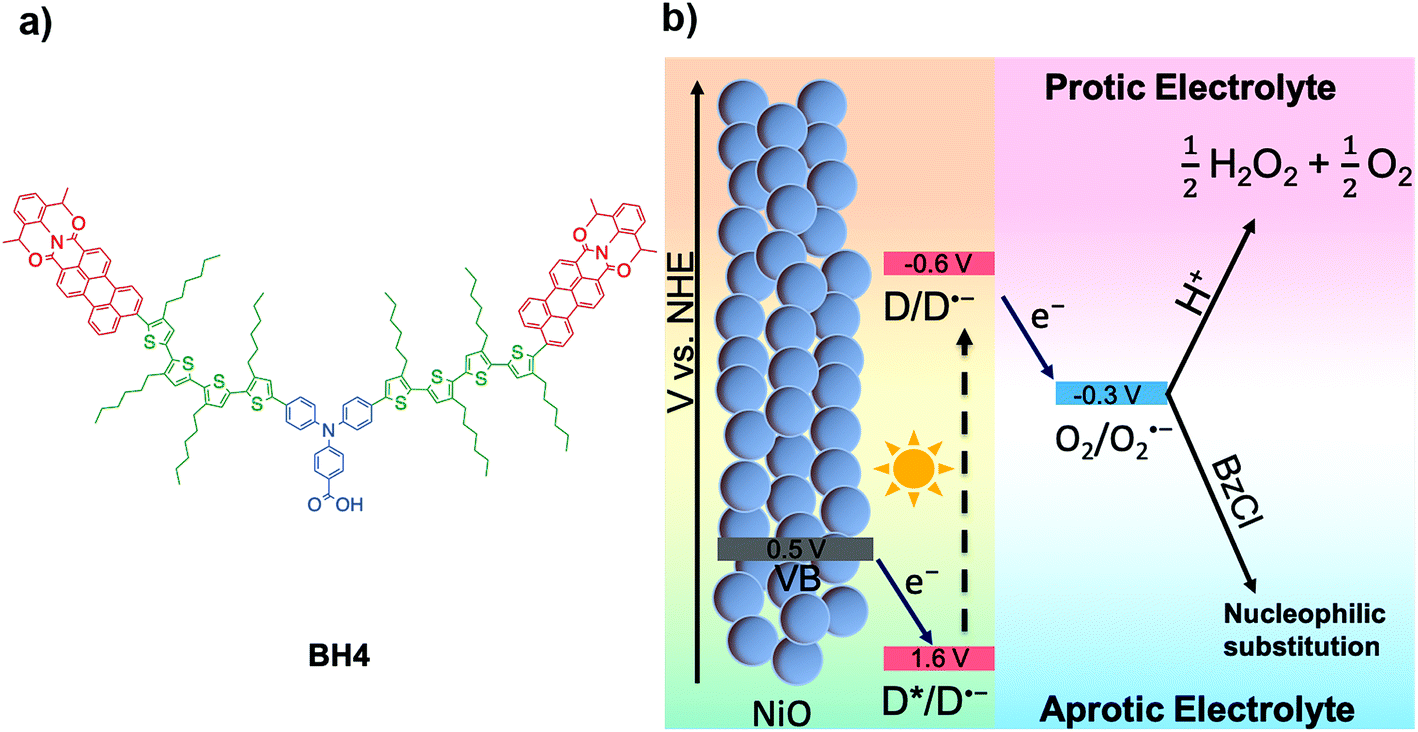 | ||
| Scheme 1 (a) Molecular structure of BH4. (b) Schematic representation of the DSPEC for H2O2 production in protic electrolyte and nucleophilic substitution in aprotic electrolyte. | ||
In this work, the BH4 dye is used as the photosensitizer in DSPECs for the production of H2O2 from O2. We report the highest current density for H2O2 production with a multi-functional organic photosensitizer in aqueous DSPECs and also the highest current density for chemical reactions involving photoelectrochemically generated superoxide radical anions in aprotic solution. The working principle of the BH4 sensitized NiO photocathode in the generation of hydrogen peroxide is demonstrated in Scheme 1b. Under light illumination, BH4 dye molecules are excited, generating a charge separation between the TPA donor and PMI acceptor followed by a fast hole injection into NiO and a one-electron transfer from PMI to dioxygen. Through the one-electron reduction step, dioxygen is reduced to superoxide radical anion, which quickly reacts with a proton and disproportionates into hydrogen peroxide and oxygen in protic electrolytes. The one-electron reduction of O2 to O2˙− and the reduction potential of BH4 are −0.33 and −0.66 V vs. NHE, respectively.21,25 Therefore, the electron transfer between BH4 and O2 is thermodynamically favorable. Employing the BH4 sensitized NiO photocathode, current density up to 600 μA cm−2 was recorded under 1 sun conditions (Xe lamp, λ > 400 nm), and the DSPECs can be operated at a bias of 0.42 V vs. RHE with currents above 200 μA cm−2 for 18 h without decrease in the rate of H2O2 production in aqueous electrolyte.
In addition, we have investigated the performance of a BH4 sensitized photoelectrochemical cell in acetonitrile, which marks the first time that oxygen reduction with a dye sensitized NiO photocathode has been studied in an aprotic electrolyte (Scheme 1b). With the addition of benzoyl chloride (BzCl) as the substrate to react with photoelectrochemically generated superoxide radicals, the observed photocurrent is up to 1.8 mA cm−2. The mechanism of this reaction was probed by ultrafast transient absorption spectroscopy, which reveals effective electron transfer from photoexcited BH4 to O2. Reactions between superoxide radicals and BzCl were also confirmed by cyclic voltammetry. It is worth mentioning that the high current density generated by the reaction of benzoyl chloride with superoxide provides an intriguing strategy in developing in situ organic synthesis from photoelectrochemically generated superoxide in aprotic systems.26
Experimental section
Materials and chemicals
Unless indicated otherwise, chemicals were purchased from Sigma-Aldrich and used without further purification. These chemicals include benzoyl chloride (ACS reagent, 99%), 3-(N-morpholino)-propanesulfonic acid (MOPS) (≥99.5%), ammonium molybdate tetrahydrate (≥99.5%), nickel(II) chloride (98%), potassium iodide (BioUltra, ≥99.5%), 4,4′,4′′,4′′′-(porphine-5,10,15,20-tetrayl)tetrakis(benzoic acid) (porphyrin, 75% dye content), Synperonic F-108 (Fluka Analytical), tetrabutylammonium hexafluorophosphate (≥99.0%), potassium chloride (Fisher Scientific, certified ACS), titanium(IV) oxysulfate solution (15 wt% in dilute sulfuric acid), acetonitrile (Extra dry, Acros Organics), hydrogen peroxide (50% aqueous solution), sulfuric acid (Fisher Scientific, certified ACS Plus), 18-crown-6 (≥99.0%), potassium dioxide. Fluorine-doped tin oxide (FTO) glass (Tech 7, thickness 2.2 mm, 2.5 cm × 2.5 cm) was purchased from Hartford Glass Co. BH4 was synthesized based on our prior report.21 Deionized water was obtained via filtration through a Thermo Scientific Barnstead E-pure Ultrapure Water Purification System until greater than 18 MΩ cm in conductivity.Film preparation
A sol–gel method was used in the preparation of NiO films based on reported procedure.27 A NiCl2 paste solution was first prepared by mixing NiCl2 with F-108 polymer, water, and ethanol by sonication. The green NiCl2 solution was then doctor-bladed onto FTO glass. The films were sintered at 450 °C for 30 minutes with a ramp rate of 2 °C min−1. Thicker films were produced by repeating the doctor-blading and sintering processes. An AlphaStep D-100 profilometer from KLA-Tencor Corporation was used to determine the thickness of films. All films used in this study were ∼0.8 μm thick, with an area of 0.36 cm2. For sensitization, the prepared NiO films were soaked for over 24 hours in 0.025 mM BH4 or 0.25 mM porphyrin dye solutions in DMF. The transmittance of sensitized films was measured by a High-Performance Lambda UV-vis spectrometer from PerkinElmer.Electrochemical setup and tests
The electrochemical setup consisted of glassy carbon as the working electrode and Ag/AgNO3 in 0.1 M TBAPF6 and 0.01 M AgNO3 in acetonitrile as the reference electrode. The counter electrode was a Pt wire. The scan rate was 100 mV s−1. 0.1 M TPAPF6 was used as the supporting electrolyte. The concentration of BzCl and BPO was 6 mM.Photoelectrochemical setup and tests
The photoelectrochemical setup was comprised of our homemade cell, which is designed for three-electrode measurements with one film in one compartment. The working electrode was a BH4 sensitized NiO film on FTO glass as mentioned above. The counter electrode was a Pt wire. Cyclic voltammetry was conducted at a scan rate of 50 mV s−1 in single compartment. The assembled cell was purged with argon for 10–20 minutes for inert conditions or with O2 for 10–20 minutes for oxygen reduction conditions. For solar simulation, a xenon lamp was used as the light source with 400 nm long-path filter. The light intensity is adjusted to 1 sun (100 mW cm−2, Newport optical power meter, model 1830-C) by a silicon photodiode (818-UV).Aqueous photoelectrochemical measurements
The reference electrode for aqueous solution was Ag/AgCl in saturated KCl aqueous solution. The cell was assembled in atmosphere. The electrolyte is a 20 mM MOPS pH 6 buffer, 0.2 M KCl aqueous solution. Chronoamperometry was conducted at a constant potential of 0.22 V vs. RHE. Controlled-potential coulometry was conducted at a constant potential of 0.42 V vs. RHE in a two-compartment homemade cell for separating the counter electrode. Nafion 212 was used as proton exchange membrane between the two compartments of the cell.Non-aqueous photoelectrochemical measurements
The reference electrode was Ag/AgNO3 in 0.1 M TBAPF6 and 0.01 M AgNO3 in acetonitrile. The cell was assembled under an inert atmosphere in a glove box. The electrolyte is a 0.1 M TBAPF6 solution in acetonitrile.H2O2 detection
Iodometric titration28,29 and titanium oxysulfate titration30 were used for the quantification of hydrogen peroxide. High-Performance Lambda 950 UV-vis spectrometer from PerkinElmer was used for product detection.Transient absorption spectroscopy
Nanosecond transient absorption spectroscopy was conducted at the Center for Nanoscale material, Argonne National Lab. An amplified Ti: sapphire laser system (Spectra Physics, Spitfire-Pro) and an automated data acquisition system (EOS Ultrafast Systems, 0–100 μs) were applied. The amplifier with 120 fs output from oscillator (Spectra Physics, Tsunami) was operated at 1.0 kHz. An optical parametric amplifier (Topas) was pumped by 90% of the beam from the oscillator and provided the pump beam. Another supercontinuum light source (EOS Ultrafast System) which was generated from the 10% of the beam was used as the probe. The excitation light was set at 500 nm with a power of 0.3 μJ per pulse. The size of the pump beam is 0.3 mm in diameter. BH4/NiO film sample was loaded on a glass slide instead of an FTO glass and was inserted in a 2 mm sealed quartz cuvette. The cuvette was degassed by argon or purged through O2 for more than 20 minutes prior to the experiments. A moving stage was used during the measurements to avoid destroying the samples. All the transient absorption spectra were subtracted by background, scattering light and corrected for t0 before analysis.Results and discussion
The transmittance of a bare NiO film and a BH4 sensitized NiO film were both measured (Fig. S1†). Compared with the bare NiO film, the BH4 sensitized sample shows a broad peak at around 500 nm with 13% transmittance, which indicates the successful surface modification of the NiO film by BH4 dye. The amount of BH4 dye molecules on NiO film surface was quantified via a Langmuir adsorption isotherm. The absorbance of BH4 dye solutions at various concentrations was measured before and after NiO film soaking (Fig. S2 and S3†). The obtained adsorption isotherm of BH4 dye on NiO surface agrees well with the Langmuir adsorption model (Fig. S4†).31,32 Both the direct method of measuring film transmittance and the indirect method of measuring adsorption isotherm demonstrate that the sensitization of NiO films by BH4 molecules has been achieved, thus the sensitized films are suitable for further photoelectrochemical analysis.With the BH4 sensitized NiO film as the working electrode, electrochemical performance was tested in our homemade cell by cyclic voltammetry (Fig. 1a). Under the argon-purged condition, two typical reversible redox features from oxidation of Ni(II) to Ni(III) and Ni(IV) are observed at potentials of 0.90 V and 1.3 V, respectively.33 No light response was observed. Upon introduction of oxygen gas into the electrolyte, current density starts to increase at applied voltages more negative than 0.25 V vs. RHE under dark conditions. This increase in current density may be due to electrochemical oxygen reduction at the NiO films, which is similar to the result of bulk NiO on a glassy carbon electrode.34 To simulate sunlight illumination, a Xe lamp was used as the solar simulator, and the light intensity was adjusted to 1 sun condition. To avoid the decomposition of H2O2 by UV light irradiation,10,35 a glass filter was used to filter off light with λ < 400 nm. Under this light condition at 1 atm O2, the current density of the DSPEC increases to 600 μA cm−2, which reveals superior efficiency of the BH4 sensitized photocathode in oxygen reduction (Fig. 1a and d). Light-chopping chronoamperometry on the BH4 sensitized NiO photocathode is shown in Fig. 1b at an applied bias of 0.22 V vs. RHE. The dark current density is around 100 μA cm−2, which can be attributed to the reduction on the NiO films. The photocurrent density is stable at 600 μA cm−2 for more than 10 min and reveals rapid photoresponse with the chopping of light.
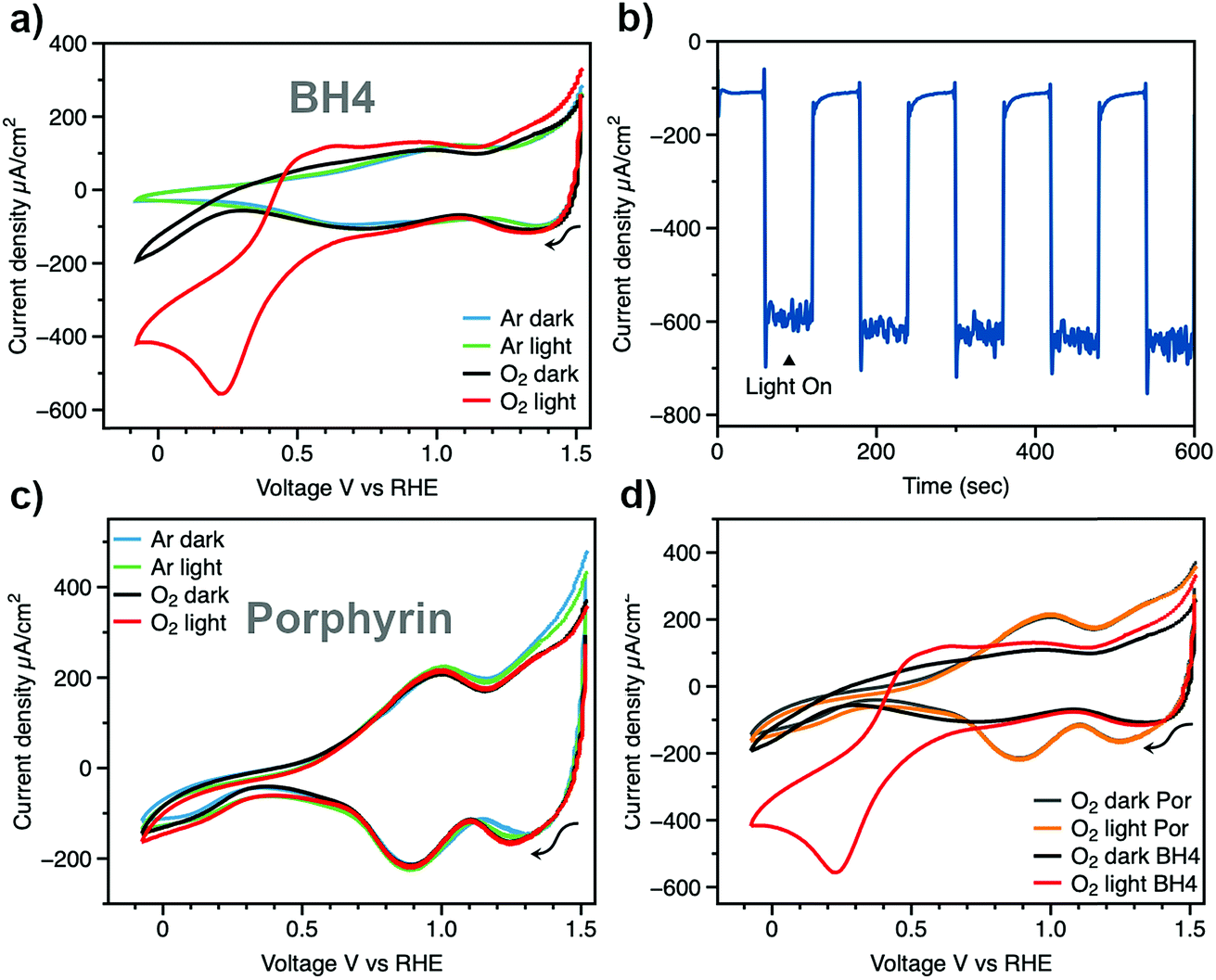 | ||
| Fig. 1 Photoelectrochemical tests: (a) cyclic voltammograms of BH4 sensitized NiO films under 1 atm Ar environment in the dark (blue line) and under illumination (green line), and under 1 atm O2 environment in the dark (black line) and under illumination (red line). (b) Chronoamperometry of a BH4 sensitized NiO film under 1 atm O2 at an applied bias of 0.22 V vs. RHE with light chopping. (c) Cyclic voltammograms of porphyrin sensitized NiO films under inert conditions (Ar dark, Ar light) and under 1 atm O2 (O2 dark, O2 light). (d) Cyclic voltammograms of porphyrin and BH4 sensitized NiO films in O2 environment. Light source: Xe lamp, 400 nm long path filter, 1 sun intensity. Electrolyte: 20 mM MOPS pH 6 buffer, 0.2 M KCl aqueous solution. | ||
Jung et al. have recently used porphyrin dye (4,4′,4′′,4′′′-(porphine-5,10,15,20-tetrayl)tetrakis(benzoic acid)) sensitized NiO films as photocathodes for the production of H2O2 and reported 300 μA cm−2 current density under high intensity 405 nm and 623 nm LED light illumination.20 However, under 1 sun conditions with AM 1.5 filter, it was mentioned that the photocurrent density decreased to 80 μA cm−2. In our present study, 1 sun conditions were created using a Xe lamp equipped with a 400 nm filter, thus, it was necessary to compare photocathodes sensitized with BH4 dye and porphyrin dye under the same conditions. Following the same procedure as BH4/NiO photocathode preparation, porphyrin sensitized NiO photocathodes were prepared. The 17% transmittance at 420 nm indicates the successful sensitization of NiO film by porphyrin dye (Fig. S5†). Control experiments using porphyrin dye sensitized NiO films as the working electrode are shown in Fig. 1c and d. In an argon-purged electrolyte with the porphyrin sensitized NiO electrode, only the distinctive redox features of Ni(II) and Ni(III) transitions were observed, which is similar to that of BH4 sensitized films. The greater magnitude of the anodic and cathodic peak currents observed for the Ni(II) and Ni(III) transitions on porphyrin sensitized NiO as compared to BH4 sensitized NiO suggest a lesser degree of surface protection provided by porphyrin sensitization. The BH4 dye's high degree of hydrophobicity provides more protection for the NiO films, effectively decreasing redox reactions at the NiO surface. Thus, the stability of BH4 sensitized electrode is superior in solar-simulated conditions. Under 1 sun conditions in the oxygen-saturated solution, the improvement in current density with porphyrin dye is only 22 μA cm−2, which is marginal in comparison with dark conditions (Fig. 1d, yellow and gray traces, respectively). By replacing porphyrin dye with BH4 dye, the photocurrent density increases by 500 μA cm−2 (Fig. 1d, red trace).
Controlled-potential coulometry was done for the quantification of hydrogen peroxide as well as to test the long-term stability of the BH4 sensitized photocathode under 1 sun condition. The electrolysis lasted for 24 hours with applied constant voltage of 0.42 V vs. RHE (Fig. 2 and S6†) and accumulated 7.94 C of charge. During this 24 h electrolysis, the photocurrent density decreased from 300 μA cm−2 to 155 μA cm−2 due to the detachment of BH4 dye molecules from the NiO surface (see below). An iodometric titration was used for the quantification of produced H2O2.28,29 Hydrogen peroxide can oxidize potassium iodide (KI) with catalytic (NH4)6Mo7O24 to produce triiodide, which absorbs light at 352 nm (Fig. 2a). Throughout the course of this 24 hour electrolysis experiment, aliquots of electrolyte were collected, to which KI and (NH4)6Mo7O24 were added, and the concentration of H2O2 in bulk electrolyte was calculated based on a calibration curve obtained from a standard H2O2 solution (Fig. S7†). The concentration of H2O2 increased linearly with time and reached 0.97 mM at 18 h, after which the production of H2O2 almost ceases with the final concentration decreasing to 0.96 mM at 24 h (Fig. 2b). The decrease in H2O2 concentration after 18 h of illumination may stem from the decay of the photocathode or decomposition of H2O2 under light exposure. To confirm the result of iodometric titration, titanium oxysulfate (TiO(SO4)) titration was also used for quantifying the final concentration of H2O2 in the electrolyte after the 24 hour electrolysis.30 The final H2O2 concentration was measured to be 0.963 mM (Fig. S8†), in agreement with the iodometric titration results. The turnover number (TON) is calculated to be 2700 based on the final concentration of H2O2 and spectroscopic measurements of dyes (Fig. S4†), which reveals a good stability of BH4/NiO electrodes. The faradaic efficiency for H2O2 production increased from 51% to 60% within the first 2 h and then decreased to 49% after 18 h. The loss of faradaic efficiency is possibly a result of the decomposition of H2O2 under high-intensity light illumination or the generation of side products, such as H2O. H2 was not detected by GC as a possible side product in headspace. Electrolysis at 0.32 V vs. RHE and in citrate buffer solution (pH = 5) was also performed with similar current density and faradaic efficiency (Fig. S9 and S10†).
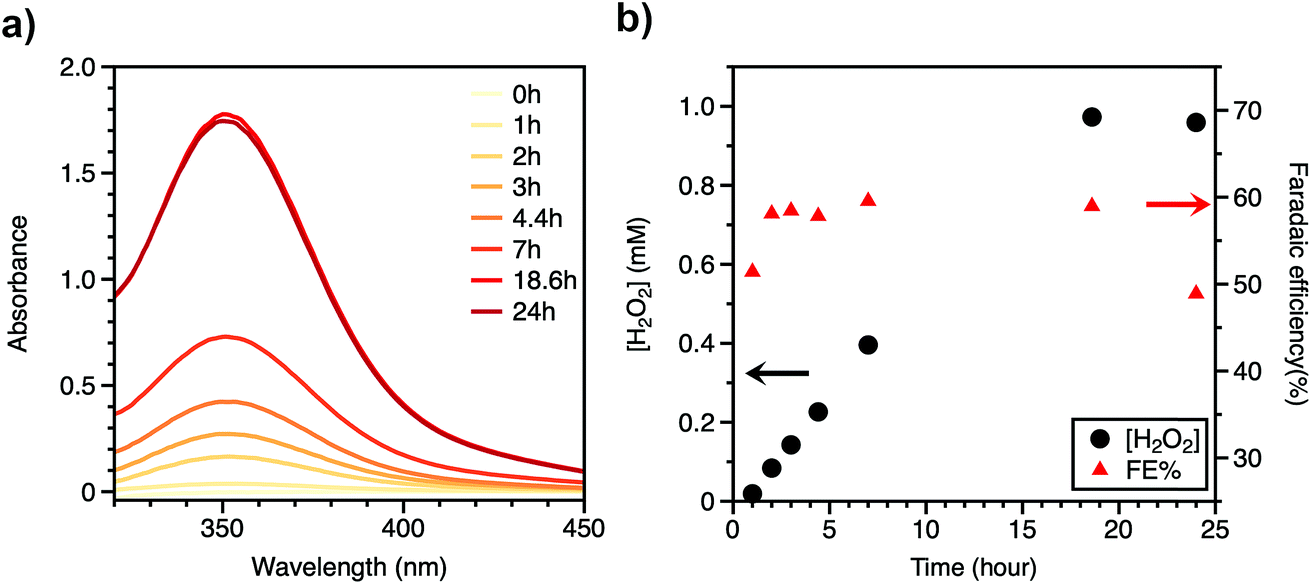 | ||
| Fig. 2 (a) Absorbance of triiodide (generated from reaction with H2O2) at different time (0 h, 1 h, 2 h, 3 h, 4.4 h, 7 h, 18.6 h, 24 h). (b) Concentration of H2O2 from titrations and corresponding faradaic efficiency during bulk electrolysis with applied bias at 0.42 V vs. RHE under 1 sun light illumination. | ||
UV-vis spectroscopy was used to determine the transmittance of BH4/NiO film before and after electrochemical tests. The BH4/NiO film shows almost no change in transmittance following CV scans. However, at 520 nm, the transmittance increases from 13% to 32% and the corresponding absorbance decreases from 0.88 to 0.50 after 24 h long-term electrolysis (Fig. S1†). With the porphyrin-sensitized electrode, the transmittance increases from 17% to 35% at 420 nm after only 20 minutes of CV scans (Fig. S5†). These results reveal the enhanced stability of BH4/NiO electrodes as compared to porphyrin/NiO, due to electrode surface protection by hydrophobic BH4 sensitization.
In the mechanism of photoelectrochemical production of hydrogen peroxide, the superoxide radical anion is produced via outer sphere electron transfer and detected using electron paramagnetic resonance.20 In aqueous solution, superoxide radical anions are quenched by protons and disproportionate into H2O2 and O2. Research on the reaction of photo-generated superoxide radical anions in non-aqueous solution, however, has been rare. The high reactivity and nucleophilicity of superoxide ions inspired us to investigate reactions involving this photoelectrochemically generated active species in aprotic solvent with intentionally added substrate. Alkyl and acyl halides are known to undergo nucleophilic substitution reactions with superoxide,36 therefore, benzoyl chloride was chosen as a model compound to react with photoelectrochemically generated superoxide. The proposed reaction scheme between benzoyl chloride and superoxide radical anions and the subsequent reactions which ultimately yield benzoate as the product are shown in Scheme 2.37 The series of reactions begins with nucleophilic attack by a superoxide radical anion on benzoyl chloride to give a peroxy radical (1). The peroxy radical is further reduced to the peroxy anion via a one-electron reduction by superoxide (2), and subsequently attacks another benzoyl chloride to give dibenzoyl peroxide (BPO) (3). Finally, BPO is reduced by superoxide, yielding benzoate (4).38 Electrochemical tests were conducted in order to validate the proposed reaction scheme (Fig. 3a). In acetonitrile solution, with O2 as the only reactant, a symmetrical trace of the O2/O2˙− redox reaction with E1/2 = −0.66 V vs. NHE is observed. After the addition of benzoyl chloride, the oxidation peak of superoxide was diminished due to the consumption of the electrogenerated superoxide radical anion via its reaction with BzCl. The peak at −1.2 V vs. NHE can be assigned to the reduction of BzCl itself. The reduction of BPO by superoxide was also probed in this manner. Accompanying the slight shift of the O2 reduction peak, the oxidation peak of superoxide decreased, which confirms the reaction between benzoyl peroxide and superoxide. The peak at 0.1 V vs. NHE can be assigned to BPO itself.
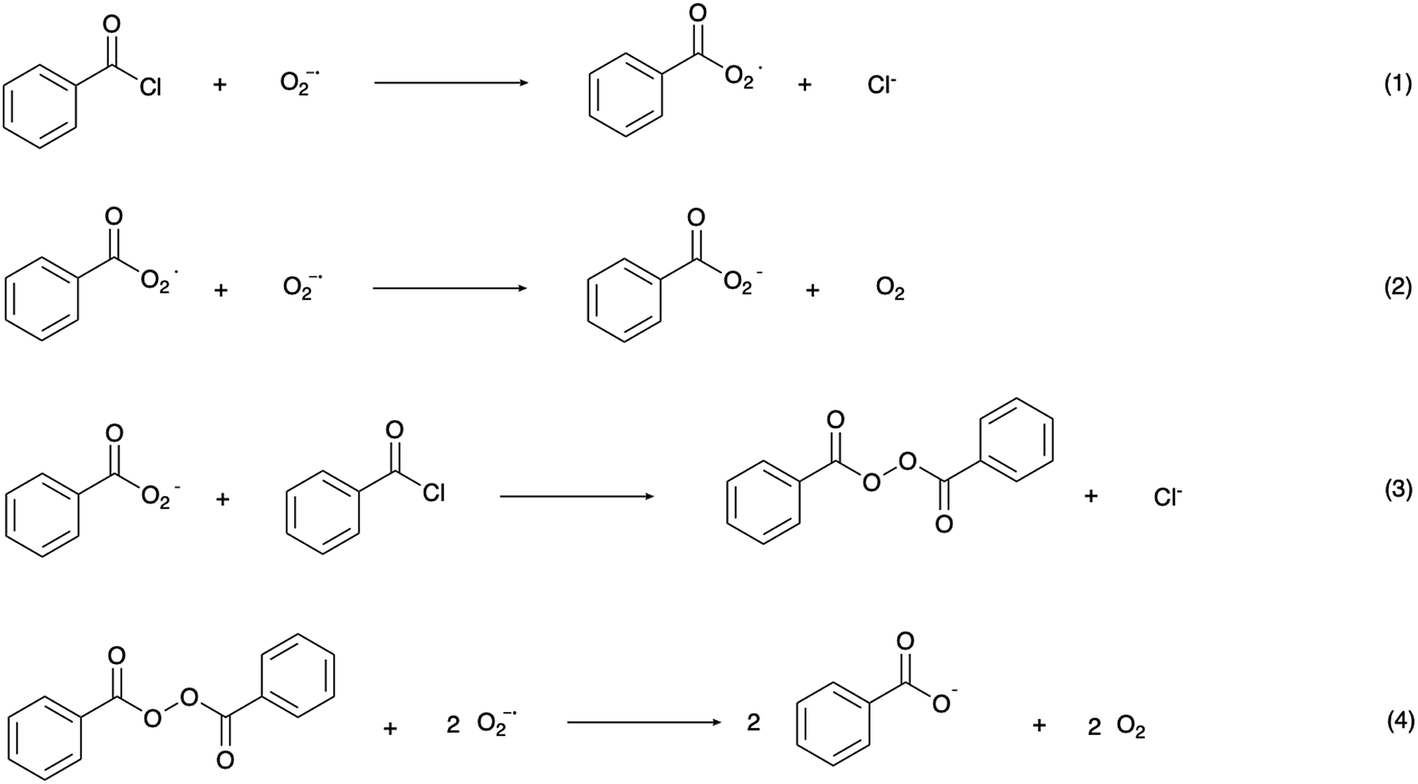 | ||
| Scheme 2 Proposed reactions between benzoyl chloride and superoxide radical anions.37,38 | ||
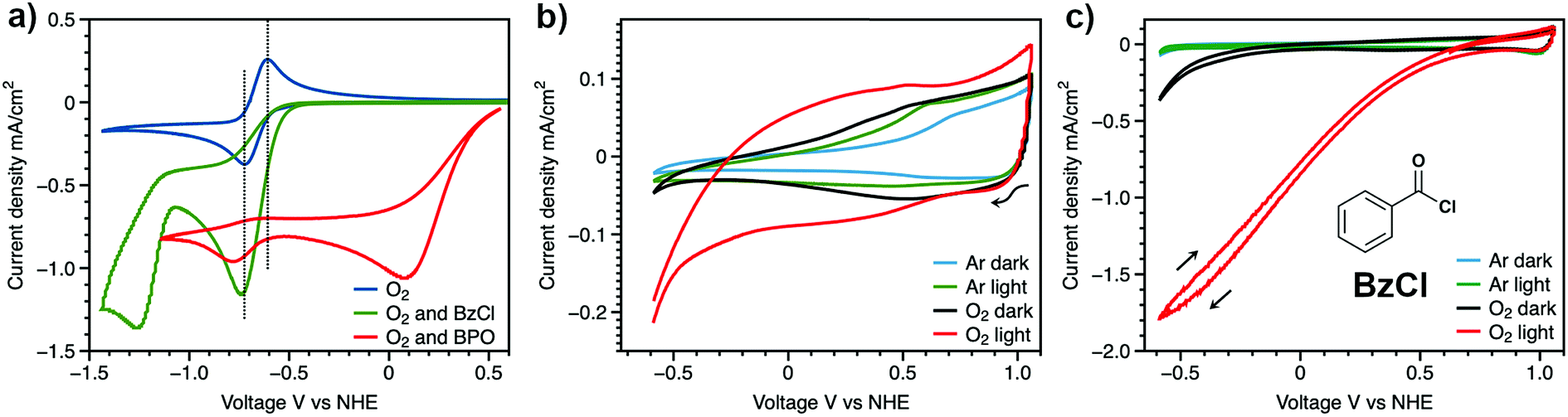 | ||
| Fig. 3 (a) Electrochemical setup: cyclic voltammograms of electrogenerated superoxide ion, oxygen (blue line), oxygen and BzCl (green line), oxygen and BPO (red line). Working electrode was glassy carbon and the reference electrode was Ag/AgNO3. The counter electrode was a Pt wire. The scan rate was 100 mV s−1. 0.1 M TPAPF6 was used as the supporting electrolyte. The concentration of BzCl and BPO was 6 mM. Photoelectrochemical setup: cyclic voltammograms of BH4 sensitized NiO film (b) in 0.1 M TBAPF6 acetonitrile solution (c) in 0.1 M BzCl, 0.1 M TBAPF6 acetonitrile solution (structure of BzCl is shown in the inset). Ar and dark (blue line), Ar and light (green line), O2 and dark (black line), O2 and light (red line). Light: Xe lamp, 400 nm long path filter, 1 sun intensity. | ||
With the reactivity of electrogenerated superoxide with BzCl confirmed, we further explored the use of BzCl in a DSPEC device equipped with a BH4/NiO photocathode. A control group of this DSPEC device in pure aprotic solvent was first investigated (Fig. 3b). Without the addition of BzCl, only 0.2 mA cm−2 of current density is achieved accompanying the degradation of the BH4/NiO photocathode under light illumination at 1 atm O2 (Fig. 3b, red trace). The result is as expected – in the absence of a proton source or other quencher, the generated superoxide on the electrode surface may attack the attached dye, causing desorption, and resulting in the decay of the photocathode. The UV-vis spectrum of a BH4 sensitized NiO film before and after the CV scans confirms the detachment of BH4 (Fig. S11†).
After the addition of benzoyl chloride, the photocurrent reaches 1.8 mA cm−2 (Fig. 3c, red line). This enhanced current observed upon the addition of BzCl is thought to be the result of the reaction between benzoyl chloride and photoelectrochemically generated superoxide radicals. An increased local concentration of O2 at the electrode surface is generated by reactions 2 and 4 (Scheme 2), which in turn, drives superoxide generation, and ultimately leads to higher current densities. However, the current was observed to degrade within five CV scans, and photocathode decomposition was also observed via UV-vis analysis (Fig. S11†). The reason for the fast decay could be caused by reactions between extra superoxide radicals and BH4/NiO photocathode. As a final confirmation of photocathode degradation being due to the reaction between superoxide and BH4/NiO, a BH4 sensitized NiO film was exposed to superoxide in the form of potassium superoxide (Fig. S12†). Within three hours, the color of the BH4/NiO film changed from red to colorless, which indicates dye desorption from the NiO electrode under the attack of superoxide. Superoxide radicals can either attack the bonds between the carboxylic anchoring groups of BH4 and NiO or break the bonds of BH4 molecules. The weakest bond is thought to be between Ni2+ and a carboxylic anchoring group, thus, it is very likely that the photocathode degradation is from dye detachment. In an effort to protect against dye detachment, an Al2O3 overlayer was added on the surface of BH4/NiO films by atomic layer deposition.39,40 However, the Al2O3 treatment was unable to protect the surface of BH4/NiO from the attack of superoxide radicals, and it caused a decrease in conductivity (Fig. S15 and S16†).
Transient absorption spectroscopy was performed in order to dissect the electron transfer process of a BH4 dye sensitized system for oxygen reduction. Based on our prior reports, a rapid hole transfer from BH4 to NiO occurs within 65 ps following the excitation of BH4 dye molecules and generates a long-lived BH4˙−/NiO+ species (Fig. 4a).24 The BH4˙− species has a characteristic absorption at 615 nm, which was monitored under Ar and O2 conditions (Fig. S17†). In Ar, charge recombination is the only pathway of dye decay. However, in O2, both charge recombination and a one-electron transfer from BH4˙− to O2 can occur, resulting in a shorter lifetime of the BH4˙−/NiO+ species. Nanosecond transient absorption spectroscopy allows us to obtain the lifetime of BH4˙−/NiO+ from an absorption decay of BH4˙− at around 615 nm in Ar and in O2 (Fig. 4b). After fitting the spectra by a stretched exponential decay model, the lifetime of BH4˙− was found to be 1.3 μs in Ar and is 0.38 μs in O2 (Fig. 4b, inset and more details in Table S1†). From these data, the rate constants for charge recombination and for electron transfer were determined to be 7.7 × 105 s−1 and 1.8 × 106 s−1, respectively (details in ESI†). The result of the transient study reveals the effective electron transfer from BH4˙− to O2 and provides concrete evidence of the production of superoxide radicals, which play an important role in the formation of new chemicals. It is noted that the potential applied to the film strongly affects the charge recombination kinetics.41,42 A kinetic analysis of TA spectra with the same potential applied to the film as the one applied for the photoelectrochemical measurements would directly reveal pertinent recombination kinetics.
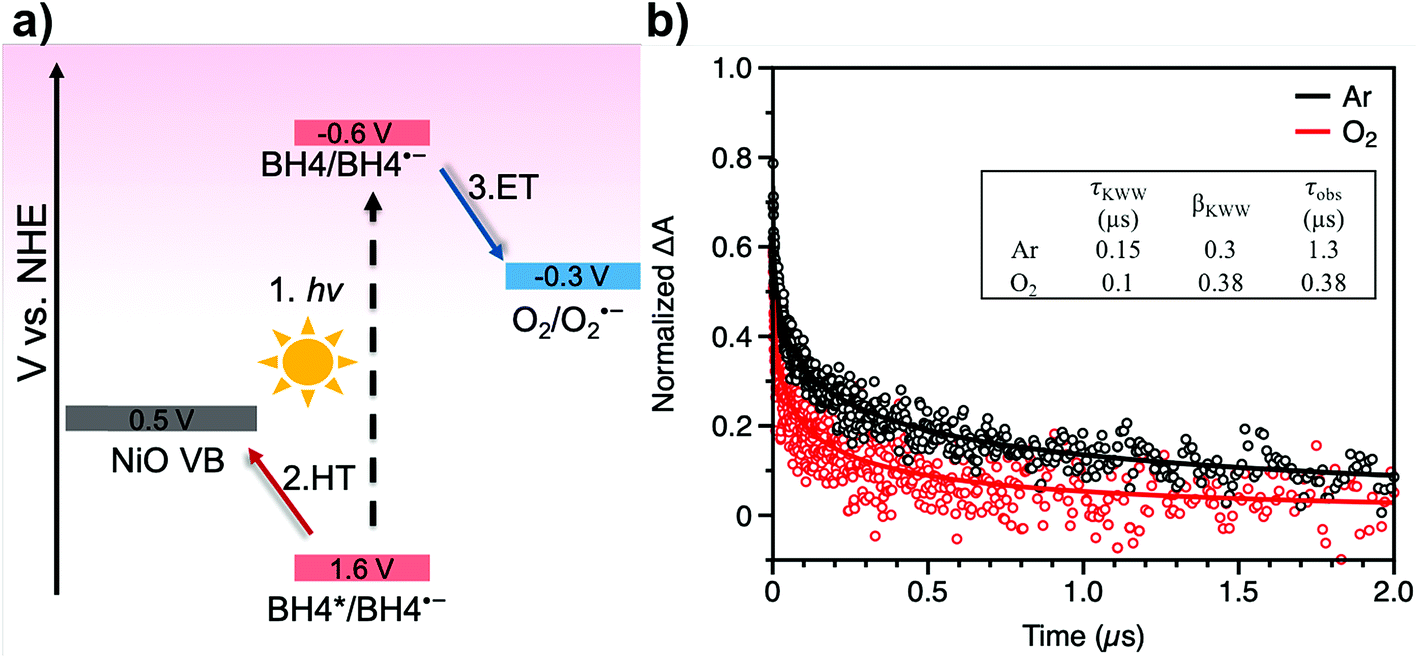 | ||
| Fig. 4 (a) Energy diagram for charge transfer kinetics of BH4-sensitized NiO film. (b) Nanosecond TA spectra of BH4-sensitized NiO film at 615 nm in 0.1 M LiClO4 inert electrolyte in acetonitrile, Ar (black trace) and O2 (red trace). Excitation: 500 nm. | ||
Bare NiO film electrodes without dye sensitization were also applied as a control in the aprotic electrolyte with the benzoyl chloride substrate. Under light illumination, the photocurrent density only reached 0.1 mA cm−2, which demonstrates that the unsensitized NiO film does not react with benzoyl chloride (Fig. S14†). To sum up, this is the first time that a dye sensitized photocathode for oxygen reduction has been investigated in an aprotic electrolyte. With the addition of an electrophilic substrate, namely benzoyl chloride, current densities up to 1.8 mA cm−2 are observed. Further work needs to be done in order to stabilize the photocathode and elucidate the mechanism among superoxide, benzoyl chloride and photocathode.
Conclusions
This study first applies a novel push-double-pull BH4 dye sensitized mesoporous NiO photocathode in DSPECs for hydrogen peroxide production. In aqueous electrolyte, the photocurrent density can achieve 600 μA cm−2 under 1 sun illumination conditions, and the photocathode is stable up to 18 h in generating H2O2 on a millimolar level. The bulky, hydrophobic BH4 sensitizer plays an important role in realizing efficient light absorption, charge separation, and surface protection. Further work can be done in further improving the stability of photocathode and faradaic efficiency of H2O2 production. In addition, with the improved efficiency, dye sensitized photocathode may be integrated with water oxidation photoanode in tandem DSPECs, which could produce hydrogen peroxide in both compartments under light illumination. In non-aqueous electrolyte, reactions involving in situ photoelectrochemically generated superoxide and benzoyl chloride were investigated, which exhibit an unprecedented current density of 1.8 mA cm−2. This finding paves the way for the potential creation of numerous novel high-value added chemicals and organic synthesis strategies involving in situ generated superoxide radical anions. Mechanistic studies of the reactions between superoxide, benzoyl chloride and the photocathode are also of interest for future research, which could in turn provide possible solutions for the improvement of the stability of the photocathode in the presence of superoxide in non-aqueous solvents. Overall, this study brings to light exciting advances toward the application of tandem DSPECs for clean energy conversion and alternative liquid fuel production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the U.S. Department of Energy (Award No. DE-FG02-07ER46427). We thank Dr Kevin Click for providing part of the BH4 sample, TPA sample and the synthesis strategy. We also acknowledge the technical support from Dr David J. Gosztola on nanosecond TA experiments in Argonne National Lab. Use of the Center for Nanoscale Materials, an Office of Science user facility, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.References
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- K. Sayama, ACS Energy Lett., 2018, 3, 1093–1101 CrossRef CAS.
- K. Fuku, Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, RSC Adv., 2017, 7, 47619–47623 RSC.
- T. Shiragami, H. Nakamura, J. Matsumoto, M. Yasuda, Y. Suzuri, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2015, 313, 131–136 CrossRef CAS.
- K. Fuku, Y. Miyase, Y. Miseki, T. Funaki, T. Gunji and K. Sayama, Chem.–Asian J., 2017, 12, 1111–1119 CrossRef CAS PubMed.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406–5409 RSC.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- M. Teranishi, S. I. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
- M. Gryszel, M. Sytnyk, M. Jakešová, G. Romanazzi, R. Gabrielsson, W. Heiss and E. D. Głowacki, ACS Appl. Mater. Interfaces, 2018, 10, 13253–13257 CrossRef CAS PubMed.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- V. Nikolaou, A. Charisiadis, G. Charalambidis, A. G. Coutsolelos and F. Odobel, J. Mater. Chem. A, 2017, 5, 21077–21113 RSC.
- L. J. Antila, P. Ghamgosar, S. Maji, H. Tian, S. Ott and L. Hammarström, ACS Energy Lett., 2016, 1, 1106–1111 CrossRef CAS.
- P. B. Pati, L. Zhang, B. Philippe, R. Fernández-Terán, S. Ahmadi, L. Tian, H. Rensmo, L. Hammarström and H. Tian, ChemSusChem, 2017, 10, 2480–2495 CrossRef CAS PubMed.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- O. Jung, M. L. Pegis, Z. Wang, G. Banerjee, C. T. Nemes, W. L. Hoffeditz, J. T. Hupp, C. A. Schmuttenmaer, G. W. Brudvig and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 4079–4084 CrossRef CAS PubMed.
- K. A. Click, D. R. Beauchamp, B. R. Garrett, Z. Huang, C. M. Hadad and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 26103–26111 RSC.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- K. A. Click, B. M. Schockman, J. T. Dilenschneider, W. D. Mcculloch, B. R. Garrett, Y. Yu, M. He, A. E. Curtze and Y. Wu, J. Phys. Chem. C, 2017, 121, 8787–8795 CrossRef CAS.
- Y. Yu, K. A. Click, S. M. Polen, M. He, C. M. Hadad and Y. Wu, J. Phys. Chem. C, 2017, 121, 20720–20728 CrossRef CAS.
- P. M. Wood, FEBS Lett., 1974, 44, 22–24 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. Alnashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS.
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199, 1–7 CrossRef CAS.
- W. A. Patrick and H. B. Wagner, Anal. Chem., 1949, 21, 1279–1280 CrossRef CAS.
- J. Brode, J. Phys. Chem., 1901, 37, 257–307 Search PubMed.
- N. Xiao, R. T. Rooney, A. A. Gewirth and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 1227–1231 CrossRef CAS PubMed.
- K.-J. Hwang, W.-G. Shim, S.-H. Jung, S.-J. Yoo, J.-W. Lee and J.-W. Lee, Appl. Surf. Sci., 2010, 256, 5428–5433 CrossRef CAS.
- G. Natu, Z. Huang, Z. Ji and Y. Wu, Langmuir, 2012, 28, 950–956 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3029–3044 CrossRef.
- S. Ci, J. Zou, G. Zeng, Q. Peng, S. Luo and Z. Wen, RSC Adv., 2012, 2, 5185–5192 RSC.
- S. Goldstein, D. Aschengrau, Y. Diamant and J. Rabani, Environ. Sci. Technol., 2007, 41, 7486–7490 CrossRef CAS PubMed.
- S. Donald T, N. Edward J and R. Julian L, Electrochem. Spectrochem. Stud. Biol. Redox Components, 2014, vol. 201, pp. 585–600 Search PubMed.
- R. Dietz, A. E. J. Forno, B. E. Larcombe and M. E. Peover, J. Chem. Soc. B, 1970, 816–820 RSC.
- M. J. Gibian, D. T. Sawyer, T. Ungermann, R. Tangpoonpholvivat and M. M. Morrison, J. Am. Chem. Soc., 1979, 101, 640–644 CrossRef CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- R. J. Kamire, K. L. Materna, W. L. Hoffeditz, B. T. Phelan, J. M. Thomsen, O. K. Farha, J. T. Hupp, G. W. Brudvig and M. R. Wasielewski, J. Phys. Chem. C, 2017, 121, 3752–3764 CrossRef CAS.
- L. D'Amario, L. J. Antila, B. Pettersson Rimgard, G. Boschloo and L. Hammarström, J. Phys. Chem. Lett., 2015, 6, 779–783 CrossRef PubMed.
- R. J. Dillon, L. Alibabaei, T. J. Meyer and J. M. Papanikolas, ACS Appl. Mater. Interfaces, 2017, 9, 26786–26796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01626k |
| This journal is © The Royal Society of Chemistry 2019 |