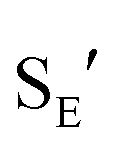DOI:
10.1039/C9SC01574D
(Edge Article)
Chem. Sci., 2019,
10, 6149-6156
Ionization of gold (γ-methoxy)vinyl complexes generates reactive gold vinyl carbene complexes†
Received
31st March 2019
, Accepted 8th May 2019
First published on 10th May 2019
Abstract
Cationic gold vinyl carbene/allylic cation complexes of the form (E)-[(L)AuC(H)C(H)CAr2]+ OTf− {L = IPr, Ar = Ph [(E)-5a], L = IPr, Ar = 4-C6H4OMe [(E)-5b], L = P(t-Bu)2o-biphenyl, Ar = 4-C6H4OMe [(E)-5c]} were generated in solution via Lewis acid-mediated ionization of the corresponding gold (γ-methoxy)vinyl complexes (E)-(L)AuC(H)C(H)C(OMe)Ar2 at or below −95 °C. Complexes (E)-5b and (E)-5c were fully characterized in solution employing multinuclear NMR spectroscopy, which established the predominant contribution of the aurated allylic cation resonance structure and the significant distribution of positive charge into the γ-anisyl rings. Complex (E)-5b reacted rapidly at −95 °C with neutral two-electron, hydride, and oxygen atom donors exclusively at the C1 position of the vinyl carbene moiety and with p-methoxystyrene to form the corresponding vinylcyclopropane. In the absence of nucleophile (E)-5a decomposed predominantly via intermolecular carbene dimerization whereas formation of 1-aryl-5-methoxy indene upon ionization of (Z)-(IPr)AuC(H)C(H)C(OMe)(4-C6H4OMe)2 [(Z)-6b] implicated an intramolecular Friedel–Crafts or electrocyclic Nazarov pathway for the decomposition of the unobserved vinyl carbene complex (Z)-[(IPr)AuC(H)C(H)C(4-C6H4OMe)2]+ OTf− [(Z)-5b].
Introduction
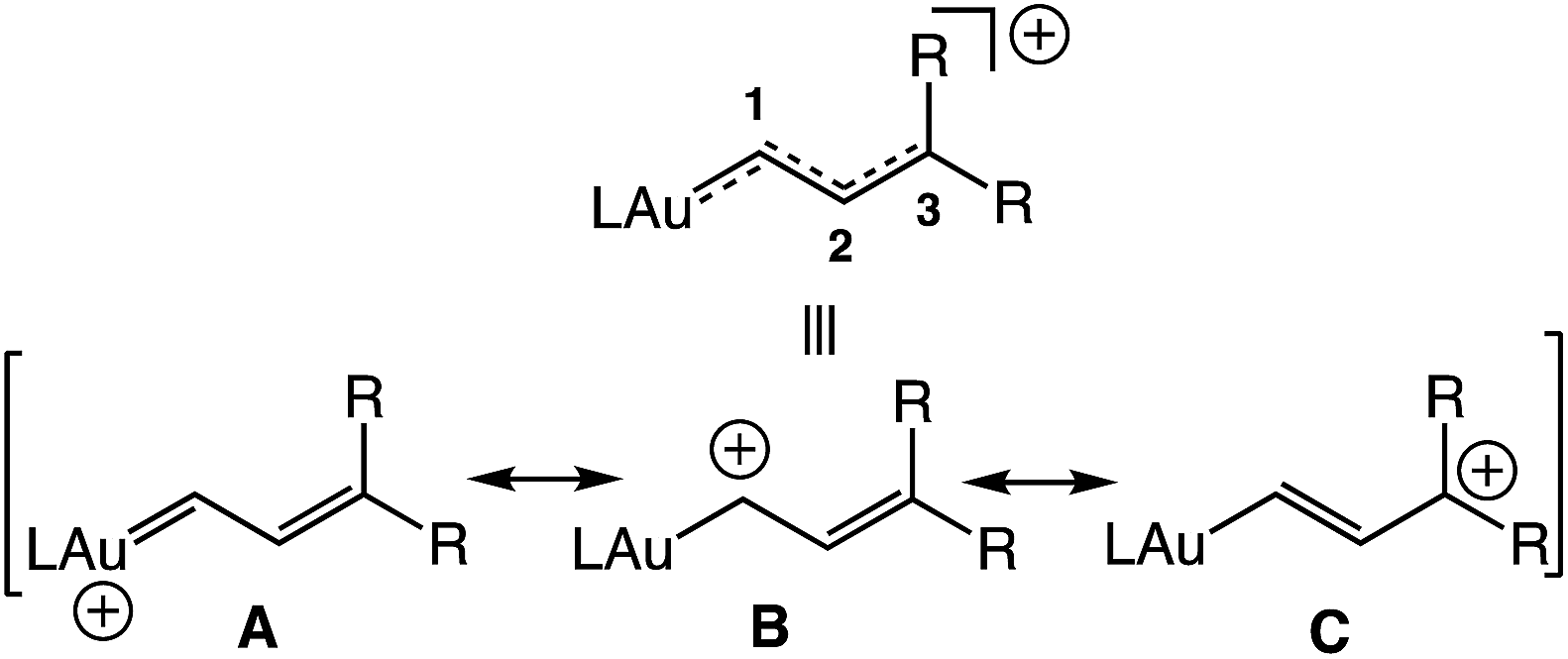
Cationic gold carbene complexes have been invoked as intermediates in a diverse range of gold(I)-catalyzed transformations1–3 and, for this reason, considerable effort has been directed toward understanding the structure and reactivity of these complexes.4 A particularly important subset of the reactive gold carbene complexes is the gold vinyl carbene/allylic cation complexes, with the potential for both the delocalization of positive charge on the Au, C1, and/or C3 atoms and diverse reaction pathways (Fig. 1). Cationic gold vinyl carbene complexes have been invoked as intermediates in range of gold-catalyzed transformations employing cyclopropenes,5–8 propargyl carboxylates,9,10 propargyl ethers,11 enynes,12 ene-allenes,13 vinyl diazo compounds,14 and norcaradienes as vinyl carbene precursors.15
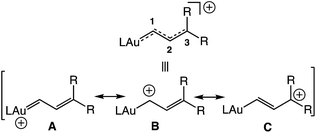 |
| | Fig. 1 Potential resonance contributors and composite representation of an unstabilized gold vinyl carbene/allylic cation complex. | |
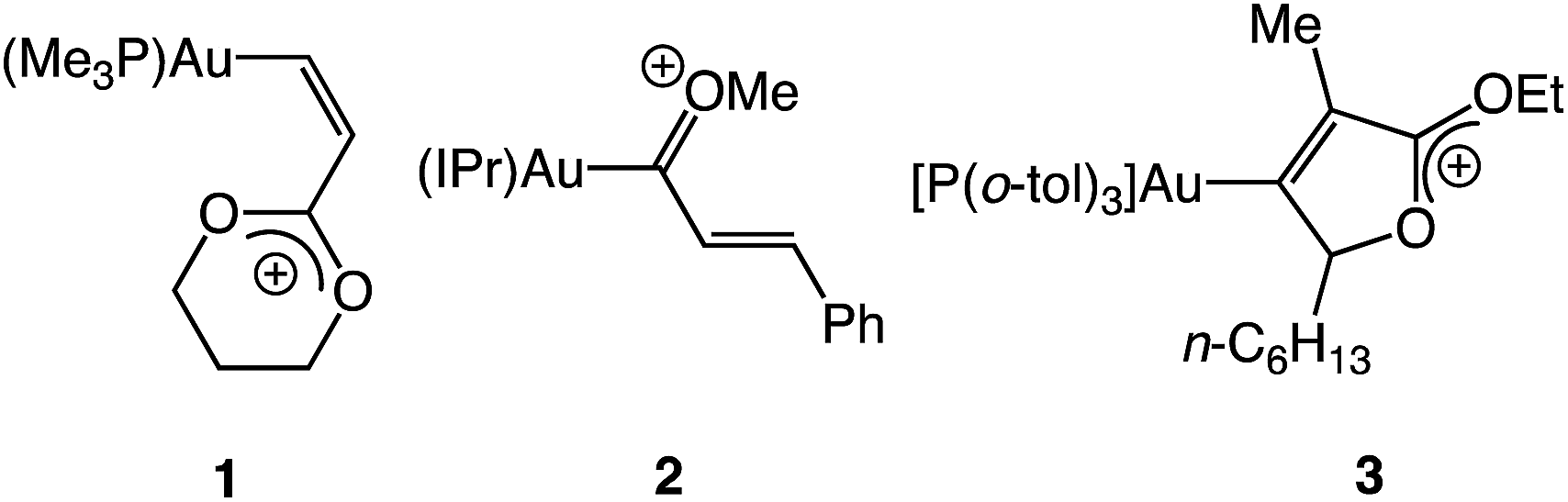
Notwithstanding the insight provided through the computational analysis of Toste and Goddard,16 information regarding the structure and reactivity of cationic gold vinyl carbene complexes is scarce, owing in large part to the absence of suitable model complexes for interrogation.4 Only three cationic gold vinyl carbene/allylic cation complexes (1–3) have been characterized, each of which contains either a C1 or multiple C3 oxygen atoms (Fig. 2).17,18 As such, neither the electronic structure nor reactivity of complexes 1–3 is likely to mimic that of the reactive vinyl carbenes generated under gold(I) catalysis.1–3,5–15 Indeed, well defined, two-coordinate gold carbene complexes that display reactivity analogous to their catalytic counterparts, gold to alkene carbene transfer in particular, are exceedingly rare.19,20 Although weakly-stabilized vinyl carbene complexes are known for a range of transition metals,21 the properties of cationic gold(I), including high d-electron count, electronegativity22 and poor d → π back bonding,4 likely render the electronic structure and reactivity of cationic gold vinyl carbene complexes distinct from these related complexes.
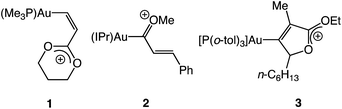 |
| | Fig. 2 Gold vinyl carbene/allylic cation complexes possessing π-conjugated oxygen atoms, depicted in the form of the predominant oxonium resonance contributor. | |
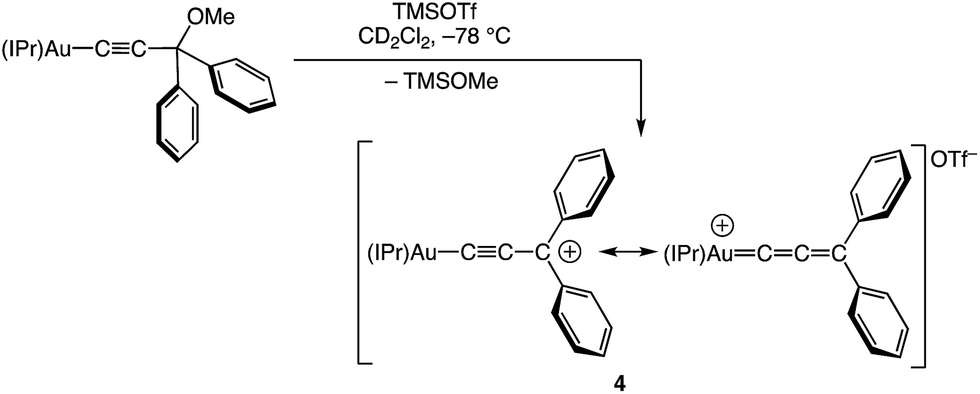
We have recently reported the synthesis of the cationic, two coordinate gold(I) diphenylallenylidene complex 4 and related complexes, generated via the Lewis acid-mediated ionization of the corresponding gold (γ-methoxy)acetylide complexes (Scheme 1),23,24 and which represent rare examples of cationic gold carbene complexes lacking π-conjugated heteroatoms.25,26 The efficiency and facility of the γ-ionization process suggested that a similar approach might provide access to particularly reactive gold vinyl carbene/allylic cation complexes. Indeed, here we report that ionization of gold (γ-methoxy-γ,γ-diaryl)vinyl complexes generates cationic, two-coordinate gold vinyl carbene complexes that react rapidly at −95 °C with a range of nucleophiles, including alkenes, and that decompose in the absence of nucleophile via inter- or intramolecular pathways. The spectroscopy of these complexes points to strong contribution of the aurated allylic cation resonance form and stabilization by the γ-aryl groups.
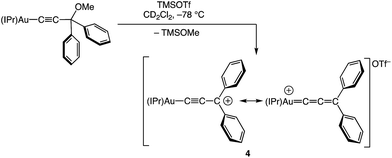 |
| | Scheme 1 Synthesis of gold allenylidene complex 4 [IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]. | |
Results and discussion
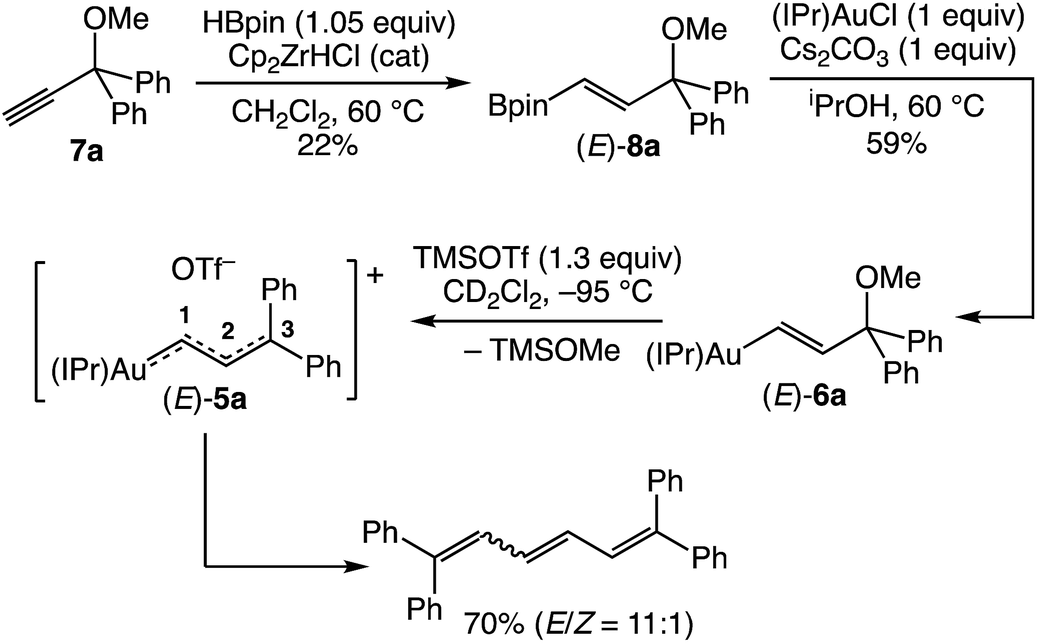
Guided by our investigation of cationic gold allenylidene complexes,23 we targeted the gold γ,γ-(diphenyl)vinylcarbene/allylic cation complex (E)-[(IPr)AuC(H)C(H)CPh2]+ OTf− [(E)-5a] for initial investigation (Scheme 2). The requisite gold (γ,γ-diphenyl)vinyl complex (E)-(IPr)AuC(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)C(OMe)Ph2 [(E)-6a] was synthesized in two steps from propargylic ether 7avia zirconium-catalyzed hydroboration27 followed by transmetallation of the resulting boronic ester (E)-8a with (IPr)AuCl (Scheme 2).28 Treatment of (E)-6a with trimethylsilyl trifluoromethanesulfonate (TMSOTf) in CD2Cl2 at −95 °C led to immediate (<1 min) formation of a bright orange/red solution that became colorless over the course of ∼10 s. 1H NMR analysis of the resulting solution at −95 °C revealed formation of trimethylsilyl methyl ether (TMSOMe) in 92 ± 5% yield, which established that efficient ionization of (E)-6a had occurred, but no resonances were observed that could be attributed to (E)-5a. Rather 1H NMR analysis revealed formation of a ∼11
C(H)C(OMe)Ph2 [(E)-6a] was synthesized in two steps from propargylic ether 7avia zirconium-catalyzed hydroboration27 followed by transmetallation of the resulting boronic ester (E)-8a with (IPr)AuCl (Scheme 2).28 Treatment of (E)-6a with trimethylsilyl trifluoromethanesulfonate (TMSOTf) in CD2Cl2 at −95 °C led to immediate (<1 min) formation of a bright orange/red solution that became colorless over the course of ∼10 s. 1H NMR analysis of the resulting solution at −95 °C revealed formation of trimethylsilyl methyl ether (TMSOMe) in 92 ± 5% yield, which established that efficient ionization of (E)-6a had occurred, but no resonances were observed that could be attributed to (E)-5a. Rather 1H NMR analysis revealed formation of a ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (E)- and (Z)-1,1,6,6-tetraphenylhexa-1,3,5-triene in 70% combined yield, presumably generated via homocoupling of the vinyl carbene groups of (E)-5a (Scheme 2).20,29 In an effort to directly observe vinyl carbene (E)-5a, a solution of (E)-6a was treated with TMSOTf at −110 °C in CDFCl2/CD2Cl2 and analysed immediately by 1H NMR spectroscopy. The resulting spectrum was broadened and largely uninterpretable, but displayed a pair of mutually-coupled doublets at δ 10.83 and 8.18 (J = 17.5 Hz), assigned to the C1 and C2 vinylic protons, respectively, of (E)-5a (Table 1).
:1 mixture of (E)- and (Z)-1,1,6,6-tetraphenylhexa-1,3,5-triene in 70% combined yield, presumably generated via homocoupling of the vinyl carbene groups of (E)-5a (Scheme 2).20,29 In an effort to directly observe vinyl carbene (E)-5a, a solution of (E)-6a was treated with TMSOTf at −110 °C in CDFCl2/CD2Cl2 and analysed immediately by 1H NMR spectroscopy. The resulting spectrum was broadened and largely uninterpretable, but displayed a pair of mutually-coupled doublets at δ 10.83 and 8.18 (J = 17.5 Hz), assigned to the C1 and C2 vinylic protons, respectively, of (E)-5a (Table 1).
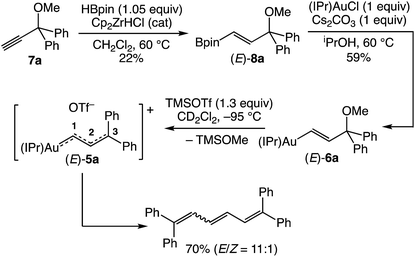 |
| | Scheme 2 Synthesis and thermal decomposition of gold vinyl carbene/allylic cation complex (E)-5a. | |
Table 1 Selected NMR data for gold vinyl carbene complexes (E)-5. δ Values reported in ppm; J values reported in Hz
|
|
(E)-5aa |
(E)-5bb |
(E)-5cc |
|
−110 °C in CDFCl2/CD2Cl2.
−95 °C in CD2Cl2.
−90 °C in CD2Cl2.
|
|
δH1 |
10.83 |
9.51 |
8.89 |
|
δH2 |
8.18 |
7.71 |
7.83 |
|
δC1 |
— |
243.7 |
252.3 |
|
δC2 |
— |
146.9 |
148.2 |
|
δC3 |
— |
185.7 |
185.6 |
|
J
H1,H2
|
17.5 |
17.8 |
17.4 |
|
J
C1,C2
|
— |
48 |
— |
|
J
C2,C3
|
— |
48 |
— |
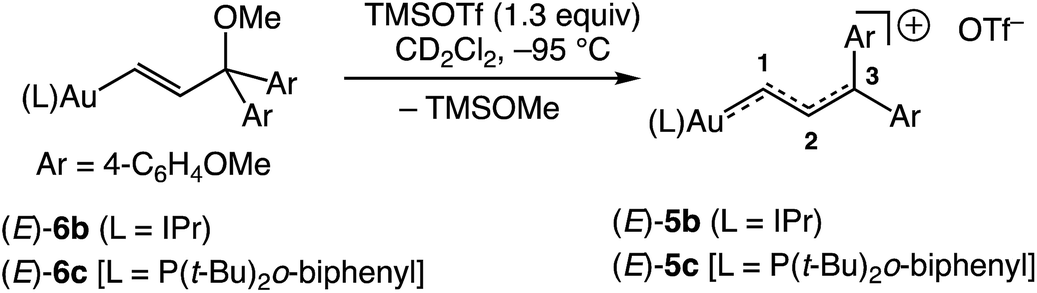
In an effort to enhance the stability of the gold vinyl carbene/allylic cation complex to allow more rigorous spectroscopic interrogation and reactivity analysis, we turned our attention to the gold γ,γ-bis(anisyl)vinyl carbene/allylic cation complex (E)-[(IPr)AuC(H)C(H)C(4-C6H4OMe)2]+ OTf− [(E)-5b]. To this end, ionization of gold vinyl complex (E)-6b in CD2Cl2 at −95 °C led to immediate (<1 min) formation of the bright red-orange solution of (E)-5b in 96 ± 5% yield (1H NMR), which persisted indefinitely at this temperature (Scheme 3). In a similar manner, ionization of the phosphine-supported gold vinyl complex (E)-6c at −95 °C led to formation of (E)-5c in 95% yield (1H NMR; Scheme 3), which darkened noticeably over the course of several hours at −95 °C.
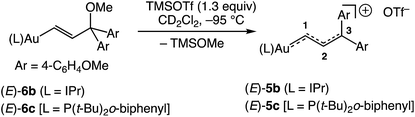 |
| | Scheme 3 Syntheses of gold vinyl carbene/allylic cation complexes (E)-5b and (E)-5c. | |
Complexes (E)-5b and (E)-5c were fully characterized by NMR spectroscopy at or below −85 °C (Table 1). For example, the 13C NMR spectrum of (E)-5b displayed downfield resonances at δ 243.7, 146.9, and 185.7 assigned to the C1, C2, and C3 carbon atoms, respectively, of the vinyl carbene/allylic cation moiety. These resonances as well as 1JC1,C2 and 1JC2,C3 were unambiguously assigned via13C NMR analysis of the corresponding 13C-labelled isotopomer (E)-[(IPr)Au13C(H)13C(H)C(4-C6H4OMe)2]+ OTf− [(E)-5b-13C2]. The 13C NMR spectrum of (E)-5b also displayed a pair of downfield resonances at δ 168.3 and 167.6 assigned to the chemically inequivalent methoxy-bound aromatic carbon atoms. These resonances were shifted significantly downfield relative to those of neutral (E)-6b (δ 140), which points to the significant delocalization of positive charge onto the γ-anisyl rings of (E)-5b.30
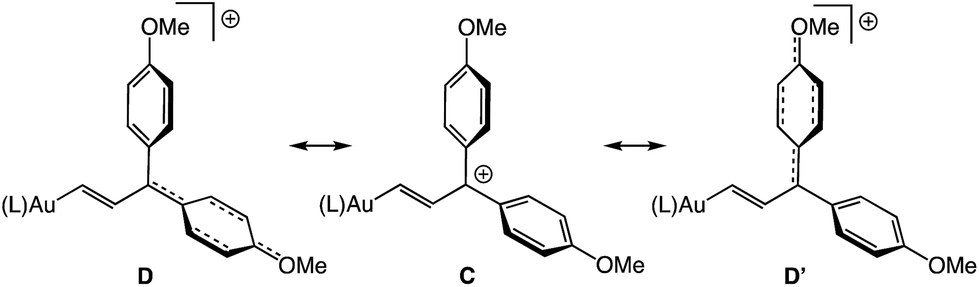
The 1H NMR spectrum of (E)-5b at −95 °C displayed two mutually-coupled doublets at δ 9.51 and 7.71 (J = 17.8 Hz) assigned to the C1 and C2 protons, respectively, and a pair of singlets at δ 3.89 and 3.96 assigned to the chemically inequivalent aryl methoxy protons. As the temperature was raised, the methoxy resonances broadened and coalesced at −42 °C, albeit with significant decomposition, which provided an energy barrier of ΔG‡231K = 10.5 kcal mol−1 for interconversion of the anisyl groups of (E)-5b (Fig. 3). Both the facile rotation about the C2–C3 bond of (E)-5b and the significant delocalization of positive charge onto the γ-anisyl rings of (E)-5b point to the predominant contribution resonance structures D to the electronic structure of (E)-5b (Fig. 4), which is likewise consistent with the significantly greater thermal stability of (E)-5b relative to (E)-5a.
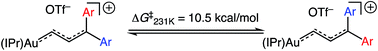 |
| | Fig. 3 Fluxional behaviour of (E)-5b. | |
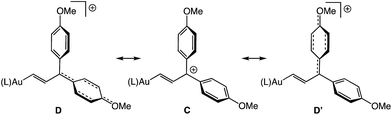 |
| | Fig. 4 Resonance structures D representing delocalization of positive charge onto the γ-anisyl rings of (E)-5b. | |
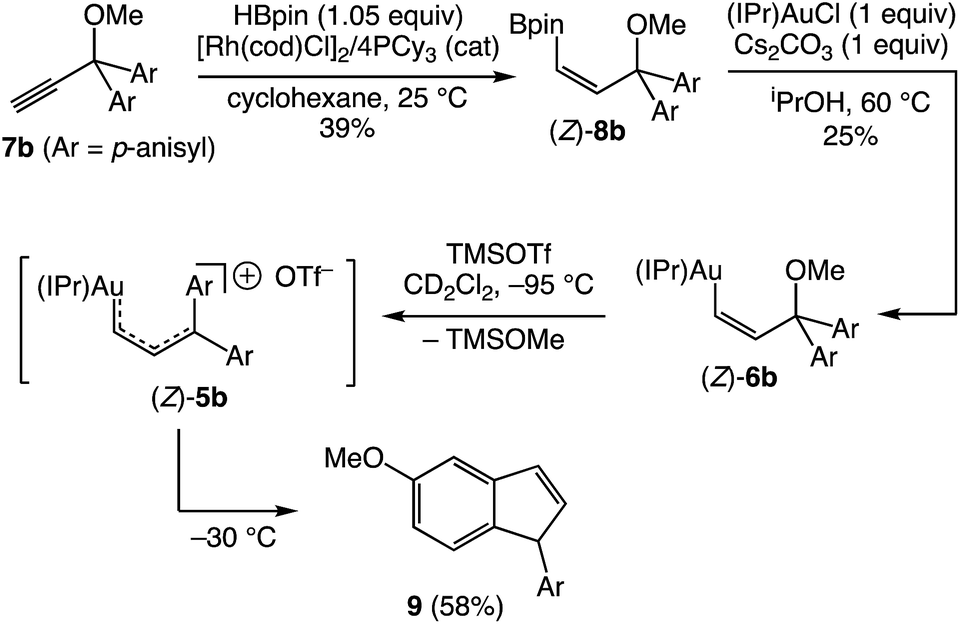
Warming a solution of (E)-5b at −40 °C (t1/2 = 35 min) or (E)-5c at −60 °C (t1/2 ≈ 10 min) led to decomposition to form unidentified mixtures of products that included neither the products of intramolecular Friedel–Crafts alkylation nor carbene dimerization as determined by GCMS and LCMS analysis. We were somewhat surprised by the absence of Friedel–Crafts/Nazarov reactivity for complexes (E)-5, which is well documented for 1-aryl allylic cations31,32 and which is directly implicated in the gold(I)-catalyzed ring-opening of aryl cyclopropenes.5,6 We reasoned that the less stable gold Z-vinyl carbene complexes, which are generated as the kinetic products of cyclopropene ring opening,33 might be more susceptible toward intramolecular cyclization processes than are the E-vinyl carbene complexes. To evaluate this hypothesis, we sought the independent synthesis of vinyl carbene complex (Z)-5b.‡ To this end, the gold vinyl complex (Z)-6b was synthesized in two steps from propargyl ether 7bvia rhodium-catalyzed, anti-selective hydroboration34 followed by transmetallation of boronic ester (Z)-8b (Scheme 4).
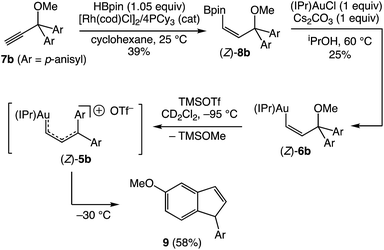 |
| | Scheme 4 Generation and decomposition of gold vinyl carbene/allylic cation complex (Z)-5b. | |
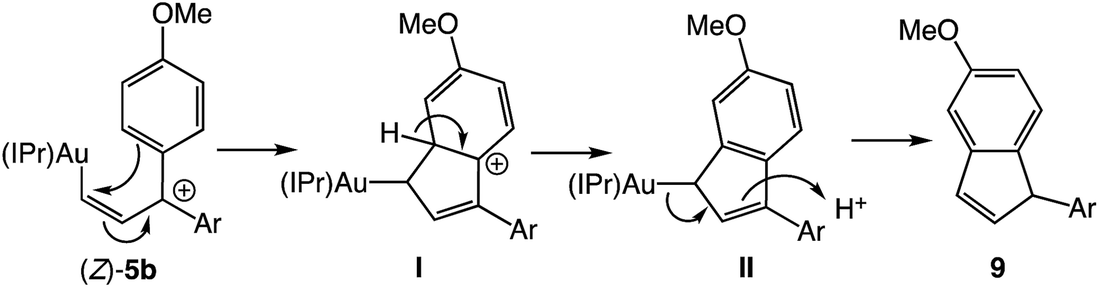
Ionization of (Z)-6b with TMSOTf at −95 °C formed a pale orange solution that displayed no resonances in the 1H NMR spectrum that could be attributed to vinyl carbene complex (Z)-5b.§ Rather, warming this solution above −50 °C led to formation of the 1-aryl-5-methoxy indene 9 in 58% yield (1H NMR; Scheme 4). Because 1-aryl indenes are less stable than are the isomeric 3-aryl indenes,359 must be formed kinetically, which points to a mechanism for the conversion of (Z)-5b to 9 involving intramolecular Friedel–Crafts alkylation or electrocyclic Nazarov-type cyclization followed by selective protonation of the γ-position of gold allyl intermediate II through an  pathway (Scheme 5). Interestingly, (3-phenyl-1H-inden-1-yl)gold(I) complexes very similar to intermediate II have been recently isolated via thermal rearrangement of neutral (3,3-diphenylcycloprop-1-en-1-yl)gold(I)complexes and were shown to undergo protodeauration with HCl predominantly through an
pathway (Scheme 5). Interestingly, (3-phenyl-1H-inden-1-yl)gold(I) complexes very similar to intermediate II have been recently isolated via thermal rearrangement of neutral (3,3-diphenylcycloprop-1-en-1-yl)gold(I)complexes and were shown to undergo protodeauration with HCl predominantly through an 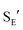 pathway.36,37 The facility of the intramolecular Friedel–Crafts/Nazarov reaction of (Z)-5b and the failure to form 9 in the decomposition of (E)-5b therefore suggests that the energy barrier for E/Z isomerization of (E)-5b exceeds that of the lowest energy decomposition pathway available to (E)-5b (ΔG‡ ≈ 16 kcal mol−1).
pathway.36,37 The facility of the intramolecular Friedel–Crafts/Nazarov reaction of (Z)-5b and the failure to form 9 in the decomposition of (E)-5b therefore suggests that the energy barrier for E/Z isomerization of (E)-5b exceeds that of the lowest energy decomposition pathway available to (E)-5b (ΔG‡ ≈ 16 kcal mol−1).
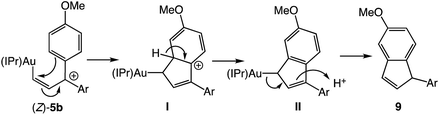 |
| | Scheme 5 Proposed mechanism of the conversion of (Z)-5b to 9. | |
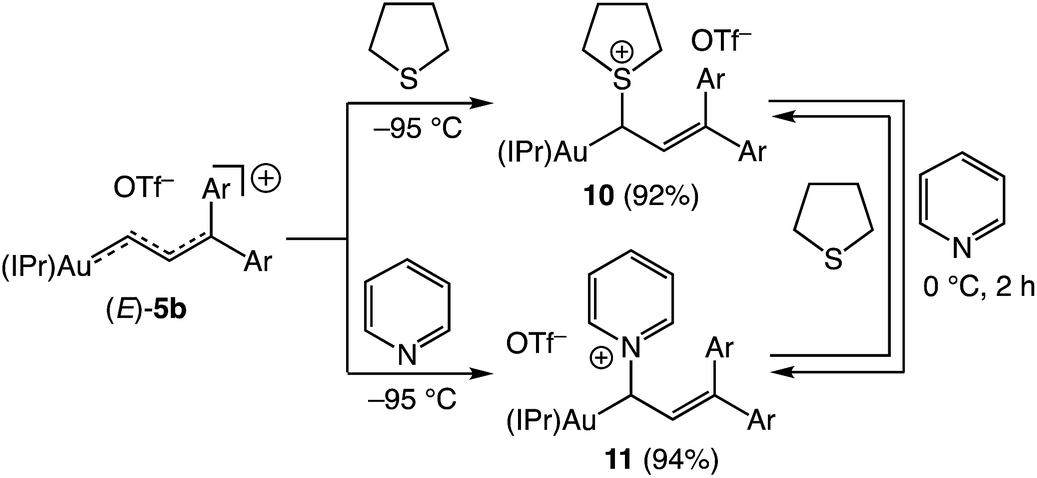
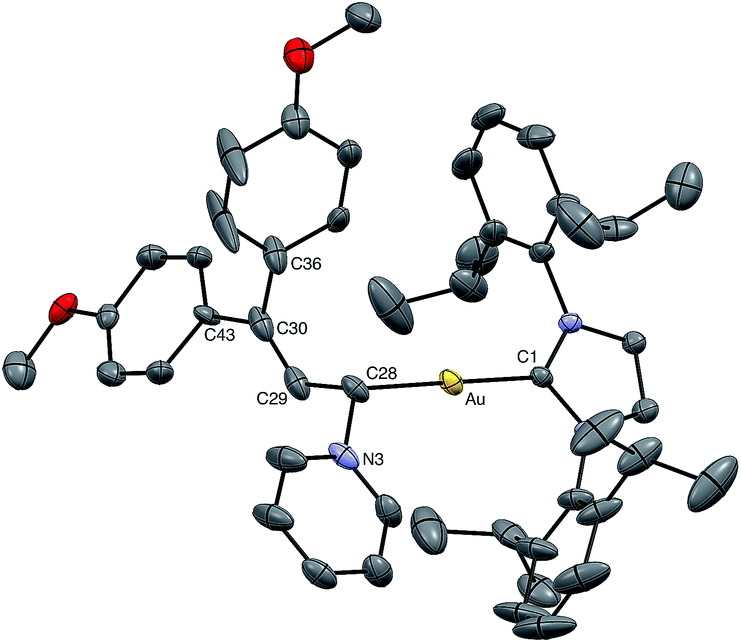
Nucleophilic trapping of a cationic gold vinyl carbene complex represents one of the most common terminating steps of catalytic transformations involving these intermediates.5,7,8 In a similar manner, treatment of (E)-5b with either tetrahydrothiophene (THT) or pyridine at −95 °C in CD2Cl2 led to immediate (<1 min) disappearance of the distinctive red color to form pale yellow solutions of the gold allyl carbenoid complexes 10 and 11, respectively, in >90% yield (1H NMR; Scheme 6). These complexes were characterized in solution, and in the case of 11, by X-ray crystallography (Fig. 5). For example, selective attack of THT at the C1 vinyl carbene carbon atom of (E)-5b was established by a pair of mutually-coupled doublets at δ 3.22 and 5.60 (J = 11.5 Hz) assigned to the aliphatic C1 and vinylic C2 protons, respectively of 10. In the solid state, 11 adopts a slightly distorted linear conformation (C1–Au–C28 = 177°) with the C1 allylic carbon atom adopting a distorted sp3 geometry with a larger Au–C28–C29 angle (115°) and smaller Au–C28–N3 and N3–C28–C29 angles (108°) (Fig. 5). Treatment of 10 (15 mM) with pyridine (1 equiv.) at 0 °C for 2 h led to formation of an equilibrium 1.25:1 mixture of 11:10 (Keq = 1.8 ± 0.1; Scheme 6). Kinetic analysis of the conversion of 10 to 11 established first-order approach to equilibrium through 2.5 half-lives with forward and reverse rate constants kf = 4.1 ± 0.9 × 10−4 s−1 and kr = 2.3 ± 0.5 × 10−4 s−1. The first-order kinetic behaviour points to a dissociative mechanism for the interconversion of 10 and 11 involving free (E)-5b.
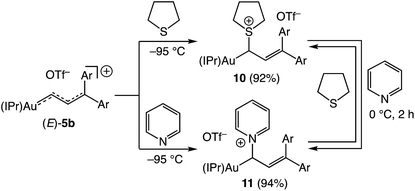 |
| | Scheme 6 Reactions of (E)-5b with THT and pyridine and interconversion of 10 and 11. Yields determined by 1H NMR spectroscopy versus internal standard. | |
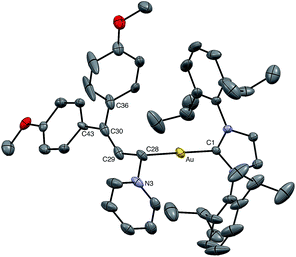 |
| | Fig. 5 ORTEP diagram of 11. One of two crystallographically independent molecules is depicted with ellipsoids shown at the 50% probability level with counterion and hydrogen atoms omitted for clarity. Selected bond distances (Å) and bond angles (deg) for 11: Au–C1 = 2.005(4), Au–C28 = 2.067(5), C28–C29 = 1.495(5), C29–C30 = 1.343(7), C28–N3 = 1.515(6), C30–C36 = 1.501(8), C30–C43 = 1.55(1), C1–Au–C28 = 177.4(2), Au–C28–C29 = 115.6(3), Au–C28–N3 = 107.8(3), N3–C28–C29 = 108.3(4), C28–C29–C30 = 125.5(5), C29–C30–C43 = 126.1(9), C29–C30–C36 = 121.6(5), C36–C30–C43 = 112.0(9). | |
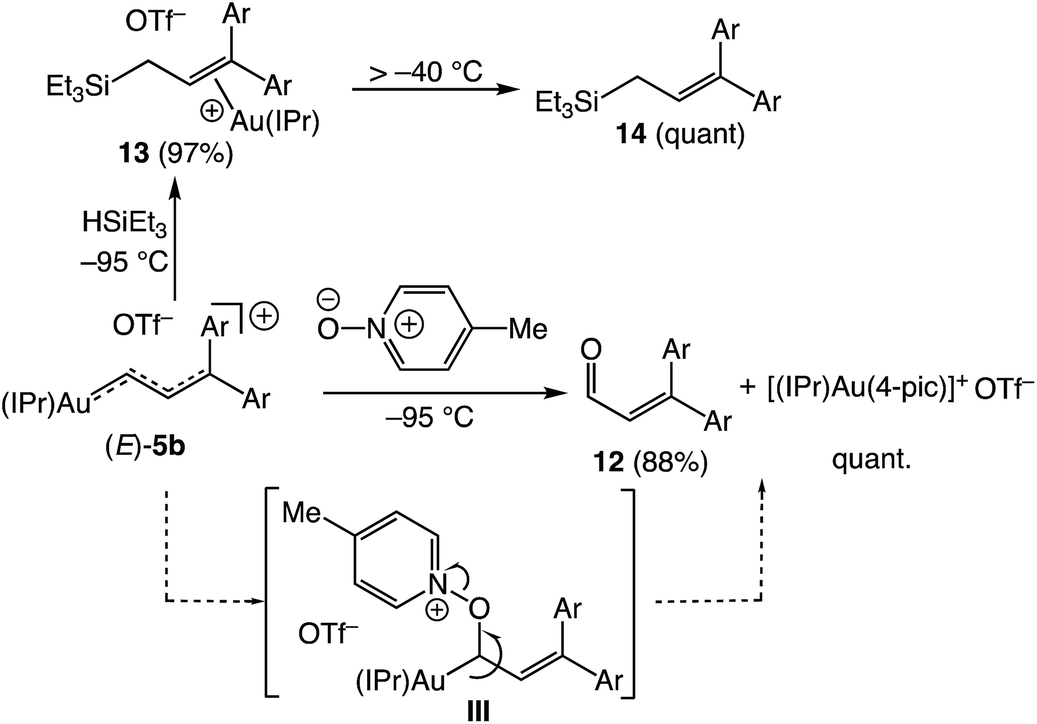
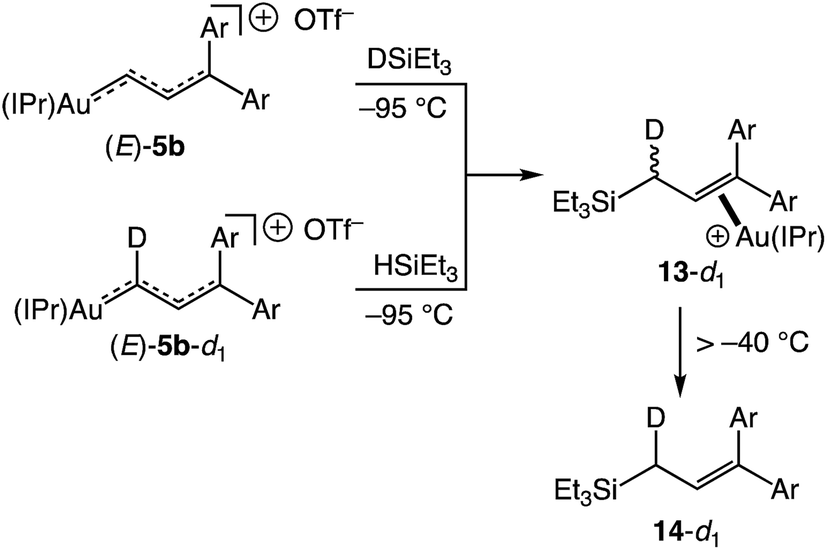
Oxygen atom transfer to the C1 position of a gold vinyl carbene intermediate has been invoked in the gold-catalyzed reactions of cyclopropenes with diphenylsulfoxide.7,38,39 In a similar manner, treatment of (E)-5b with 4-picoline N-oxide (4-PNO) (1.9 equiv.) in CD2Cl2 at −95 °C for ∼1 min led to formation of 3-arylcinnamaldehyde 12 in 88% yield and the 4-picoline gold complex [(IPr)Au(4-pic)]+ OTf− in quantitative yield (Scheme 7). This outcome is consistent with selective oxygen atom transfer to the C1 carbon atom of the vinyl carbene moiety of (E)-5b followed by elimination from the unobserved α-oxopyridinium intermediate III. Related to this, treatment of (E)-5b with triethylsilane at −95 °C led to immediate (<1 min) and selective formation of the cationic gold π-allyl silane complex 13, presumably via hydride addition to the C1 position of (E)-5b followed by silyldeauration of the resulting gold allyl intermediate through an SE pathway.40 Warming this solution above −40 °C led to decomplexation and formation of allylic silane 14 in quantitative yield. Both the reaction of (E)-5b with DSiEt3 and the reaction of the deuterated isotopomer (E)-5b-d1 with HSiEt3 at −95 °C led selective incorporation of deuterium into the allylic position to form 13-d1 as a ∼1:1 mixture of diastereomeric isotopomers, pointing to a stepwise deauration/complexation process (Scheme 8).
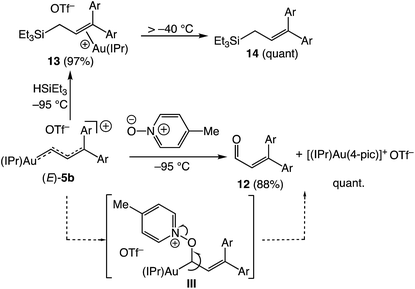 |
| | Scheme 7 Reactions of (E)-5b with 4-picoline-N-oxide and HSiEt3. Yields determined by 1H NMR spectroscopy versus internal standard. | |
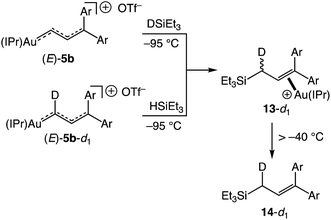 |
| | Scheme 8 Reaction of (E)-5b with DSiEt3 and reaction (E)-5b-d1 with HSiEt3. | |
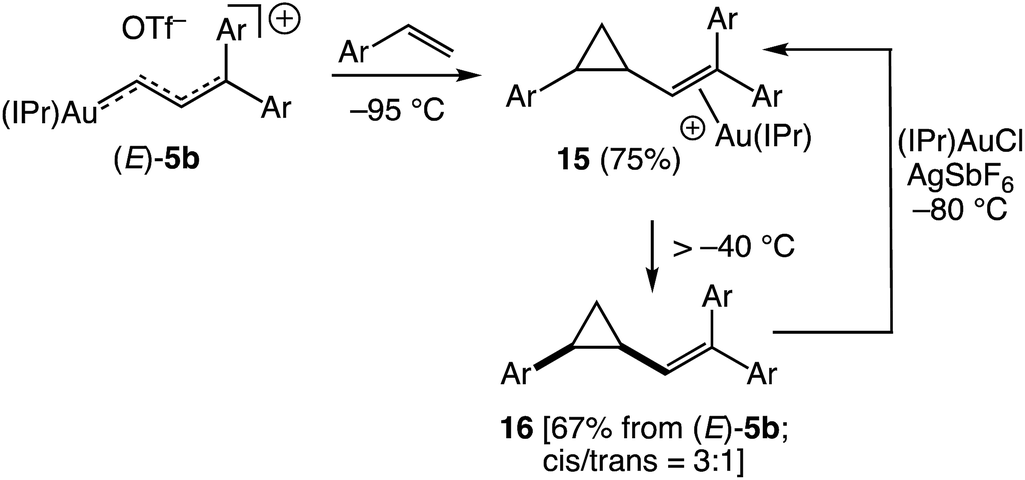
Despite the dearth of examples of stoichiometric gold to alkene carbene transfer,19,20 perhaps no process is more closely associated with the reactive gold carbene complexes, including vinyl carbene complexes, generated as catalytic intermediates.1,2 It is therefore significant that vinyl carbene complex (E)-5b underwent facile gold to alkene carbene transfer with p-methoxystyrene. For example, treatment of (E)-5b with p-methoxystyrene (3 equiv.) at −95 °C led to immediate (<3 min) consumption of (E)-5b to form the gold π-vinyl cyclopropane complex 15 (Scheme 9). Although 1H NMR analysis of this solution at −95 °C was complicated by the presence of free p-methoxystyrene and byproducts, a doublet was observed at δ 4.99 (J = 10.2 Hz), assigned to the vinyl proton of 15, which corresponded to a 75% yield of 15 from (E)-5b (Scheme 9). Warming this solution of 15 above −40 °C led to decomplexation to form free vinyl cyclopropane 16 in 67% NMR yield from (E)-5b as a ∼3:1 mixture of cis/trans isomers (Scheme 9). Assignment of the δ 4.99 resonance to the vinyl proton of 15 was confirmed by independent synthesis of 15 from reaction of 16 (cis/trans = 3:1) with a mixture of (IPr)AuCl and AgSbF6 in CD2Cl2 at −80 °C.¶
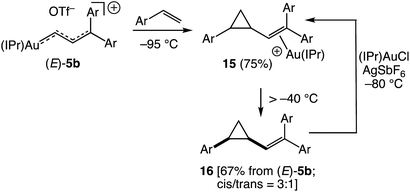 |
| | Scheme 9 Reaction of (E)-5b with p-methoxystyrene and independent synthesis of π-(vinyl cyclopropane) complex 15. | |
Conclusions
We have generated highly reactive gold vinyl carbene/allylic cation complexes via low-temperature ionization of gold γ-methoxy-(γ,γ-diaryl)vinyl complexes. Spectroscopic analysis of vinyl carbene/allylic cation complex (E)-5b established the predominant contribution of the aurated allylic cation canonical form to the electronic structure of the vinyl carbene complex (C; Fig. 1) with significant delocalization of positive charge into the γ-anisyl groups. Indeed, the significantly greater stability of the γ,γ-bis(anisyl) vinyl carbene complex (E)-5b relative to the γ,γ-diphenyl vinyl carbene complex (E)-5a points to the importance of the p-methoxy groups in the stabilization of (E)-5b.41 Complex (E)-5b reacted rapidly at −95 °C with neutral two-electron, hydride, and oxygen atom donors exclusively at the C1 position of the vinyl carbene/allylic cation moiety and with p-methoxystyrene to form vinyl cyclopropane. These transformations are close or direct analogues to processes attributed to gold vinyl carbene intermediates generated under catalytic conditions.1–3,5–15 Two distinct pathways for decomposition of these gold vinyl carbene complexes in the absence of nucleophile were identified; complex (E)-5a decomposed predominantly via intermolecular carbene homocoupling whereas formation of indene 9 upon ionization of (Z)-6b directly implicated an intramolecular Friedel–Crafts or electrocyclic Nazarov pathway for the decomposition of the unobserved vinyl carbene complex (Z)-5b.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the NSF (CHE-1800273) for support of this research. X-ray crystallography measurements were made by Dr Roger Sommer in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University.
Notes and references
- D. Qian and J. Zhang, Chem. Soc. Rev., 2015, 44, 677–698 RSC.
-
(a) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506–516 RSC;
(b) M. R. Fructos, M. M. Diáz-Requejo and P. J. Pérez, Chem. Commun., 2016, 52, 7326–7335 RSC.
-
(a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed;
(b) H. Yeom and S. Shin, Acc. Chem. Res., 2014, 47, 966–977 CrossRef CAS PubMed;
(c) L. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed.
-
(a) R. J. Harris and R. A. Widenhoefer, Chem. Soc. Rev., 2016, 45, 4533–4551 RSC;
(b) Y. Wang, M. E. Muratore and A. M. Echavarren, Chem.–Eur. J., 2015, 21, 7332–7339 CrossRef CAS PubMed.
-
(a) F. Miege, C. Meyer and J. Cossy, Beilstein J. Org. Chem., 2011, 7, 717–734 CrossRef CAS PubMed;
(b) A. Archambeau, F. Miege, C. Meyer and J. Cossy, Acc. Chem. Res., 2015, 48, 1021–1031 CrossRef CAS PubMed.
-
(a) C. Li, Y. Zeng and J. Wang, Tetrahedron Lett., 2009, 50, 2956–2959 CrossRef CAS;
(b) Z.-B. Zhu and M. Shi, Chem.–Eur. J., 2008, 14, 10219–10222 CrossRef CAS PubMed.
-
(a) J. T. Bauer, M. S. Hadfield and A.-L. Lee, Chem. Commun., 2008, 6405–6407 RSC;
(b) M. S. Hadfield, J. T. Bauer, P. E. Glen and A.-L. Lee, Org. Biomol. Chem., 2010, 8, 4090–4095 RSC.
-
(a) P.-L. Zhu, X.-Y. Tang and M. Shi, Chem. Commun., 2016, 52, 7245–7248 RSC;
(b) X. Cheng, L. Zhu, M. Lin, J. Chen and X. Huang, Chem. Commun., 2017, 53, 3745–3748 RSC;
(c) R. J. Mudd, P. C. Young, J. A. Jordan-Hore, G. M. Rosair and A.-L. Lee, J. Org. Chem., 2012, 77, 7633–7639 CrossRef CAS PubMed;
(d) E. Seraya, E. Slack, A. Ariafard, B. F. Yates and C. J. T. Hyland, Org. Lett., 2010, 12, 4768–4771 CrossRef CAS PubMed;
(e) P. C. Young, M. S. Hadfield, L. Arrowsmith, K. M. Macleod, R. J. Mudd, J. A. Jordan-Hore and A.-L. Lee, Org. Lett., 2012, 14, 898–901 CrossRef CAS PubMed;
(f) M. S. Hadfield and A.-L. Lee, Chem. Commun., 2011, 47, 1333–1335 RSC.
-
(a) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed;
(b) R. K. Shiroodi and V. Gevorgyan, Chem. Soc. Rev., 2013, 42, 4991–5001 RSC;
(c) X.-Z. Shu, D. Shu, C. M. Schienebeck and W. Tang, Chem. Soc. Rev., 2012, 41, 7698–7711 RSC.
-
(a) M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS PubMed;
(b) M. Petrović, D. Scarpi, B. Fiser, E. Gómez-Bengoa and E. G. Occhiato, Eur. J. Org. Chem., 2015, 3943–3956 CrossRef;
(c) W. Zi, H. Wu and F. D. Toste, J. Am.
Chem. Soc., 2015, 137, 3225–3228 CrossRef CAS PubMed;
(d) W. Rao and P. W. H. Chan, Chem.–Eur. J., 2014, 20, 713–718 CrossRef CAS PubMed;
(e) T. Lauterbach, W. Ganschow, M. W. Hussong, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 680–686 CrossRef CAS;
(f) C. H. Oh, J. H. Kim, L. Piao, J. Yu and S. Y. Kim, Chem.–Eur. J., 2013, 19, 10501–10505 CrossRef CAS PubMed;
(g) W. Rao, M. J. Koh, D. Li, H. Hirao and P. W. H. Chan, J. Am. Chem. Soc., 2013, 135, 7926–7932 CrossRef CAS PubMed;
(h) T. Lauterbach, S. Gatzweiler, P. Nösel, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2013, 355, 2481–2487 CrossRef CAS;
(i) J. Liu, M. Chen, L. Zhang and Y. Liu, Chem.–Eur. J., 2015, 21, 1009–1013 CrossRef CAS PubMed.
-
(a) E. Jiménez-Núñez, M. Raducan, T. Lauterbach, K. Molawi, C. R. Solorio and A. M. Echavarren, Angew. Chem., Int. Ed., 2009, 48, 6152–6155 CrossRef PubMed;
(b) A. Rinaldi, M. Petrovic, S. Magnolfi, D. Scarpi and E. G. Occhiato, Org. Lett., 2018, 20, 4713–4717 CrossRef CAS PubMed.
-
(a) M. Gaydou, R. E. Miller, N. Delpont, J. Ceccon and A. M. Echavarren, Angew. Chem., Int. Ed., 2013, 52, 6396–6399 CrossRef CAS PubMed;
(b) J. Carreras, M. Livendahl, P. R. McGonigal and A. M. Echavarren, Angew. Chem., Int. Ed., 2014, 53, 4896–4899 CrossRef CAS PubMed;
(c) P. Calleja, Ó. Pablo, B. Ranieri, M. Gaydou, A. Pitaval, M. Moreno, M. Raducan and A. M. Echavarren, Chem.–Eur. J., 2016, 22, 13613–13618 CrossRef CAS PubMed.
-
(a) J. H. Lee and F. D. Toste, Angew. Chem., Int. Ed., 2007, 46, 912–914 CrossRef CAS PubMed;
(b) B. D. Mokar, P. D. Jadhav, Y. B. Pandit and R.-S. Liu, Chem. Sci., 2018, 9, 4488–4492 RSC.
-
(a) J. Barluenga, G. Lonzi, M. Tomás and L. A. López, Chem.–Eur. J., 2013, 19, 1573–1576 CrossRef CAS PubMed;
(b) E. López, G. Lonzi and L. A. López, Organometallics, 2014, 33, 5924–5927 CrossRef;
(c) D. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070–11074 CrossRef CAS PubMed;
(d) G. Xu, C. Zhu, W. Gu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 883–887 CrossRef CAS PubMed;
(e) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 13314–13317 CrossRef CAS PubMed;
(f) G. Lonzi and L. A. López, Adv. Synth. Catal., 2013, 355, 1948–1954 CrossRef CAS;
(g) V. V. Pagar, A. M. Jadhav and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 20728–20731 CrossRef CAS PubMed;
(h) A. M. Jadhav, V. V. Pagar and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 11809–11813 CrossRef CAS PubMed;
(i) V. V. Pagar and R.-S. Liu, Angew. Chem., Int. Ed., 2015, 54, 4923–4926 CrossRef CAS PubMed;
(j) V. V. Pagar, A. M. Jadhav and R.-S. Liu, J. Org. Chem., 2013, 78, 5711–5716 CrossRef CAS PubMed;
(k) S. N. Karad and R.-S. Liu, Angew. Chem., Int. Ed., 2014, 53, 5444–5448 CrossRef CAS PubMed.
-
(a) B. Herlé, P. M. Holstein and A. M. Echavarren, ACS Catal., 2017, 7, 3668–3675 CrossRef PubMed;
(b) X. Yin, M. Mato and A. M. Echavarren, Angew. Chem., Int. Ed., 2017, 56, 14591–14595 CrossRef CAS PubMed.
- D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III and F. D. Toste, Nat. Chem., 2009, 1, 482–486 CrossRef CAS PubMed.
-
(a) M. Fañanás-Mastral and F. Aznar, Organometallics, 2009, 28, 666–668 CrossRef;
(b) L.-P. Liu, B. Xu, M. S. Mashuta and G. B. Hammond, J. Am. Chem. Soc., 2008, 130, 17642–17643 CrossRef CAS PubMed.
- G. Seidel, R. Mynott and A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 2510–2513 CrossRef CAS PubMed.
-
(a) G. Seidel and A. Fürstner, Angew. Chem., Int. Ed., 2014, 53, 4807–4811 CrossRef CAS PubMed;
(b) C. Werlé, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 15452–15456 CrossRef PubMed.
- C. García-Morales, X.-L. Pei, J. M. Sarria Toro and A. M. Echavarren, Angew. Chem., Int. Ed., 2019, 58, 3957–3961 CrossRef PubMed.
-
(a) P. Binger, P. Müller, R. Benn and R. Mynott, Angew. Chem., Int. Ed., 1989, 28, 610–611 CrossRef;
(b) S. G. Feng, A. S. Gamble and J. L. Templeton, Organometallics, 1989, 8, 2024–2031 CrossRef CAS;
(c) S. T. Nguyen, L. K. Johnson, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1992, 114, 3974–3975 CrossRef CAS;
(d) L. K. Johnson, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1993, 115, 8130–8145 CrossRef CAS;
(e) C. S. Yi, G. L. Geoffroy, C. A. White and A. L. Rheingold, J. Am. Chem. Soc., 1993, 115, 3806–3807 CrossRef CAS;
(f) A. Mayr, M. F. Asaro, T. J. Glines, D. Van Engen and G. M. Tripp, J. Am. Chem. Soc., 1993, 115, 8187–8196 CrossRef CAS;
(g) B. T. Flatt, R. H. Grubbs, R. L. Blanski, J. C. Calabrese and J. Feldman, Organometallics, 1994, 13, 2728–2732 CrossRef CAS;
(h) M. A. Esteruelas, F. J. Lahoz, E. Oñate, L. A. Oro and B. Zeier, Organometallics, 1994, 13, 1662–1668 CrossRef CAS;
(i) M. A. Esteruelas, F. J. Lahoz, E. Oñate, L. A. Oro, C. Valero and B. Zeier, J. Am. Chem. Soc., 1995, 117, 7935–7942 CrossRef CAS;
(j) C. Gauss, D. Veghini, O. Orama and H. Berke, J. Organomet. Chem., 1997, 541, 19–38 CrossRef CAS;
(k) J. Foerstner, A. Kakoschke, D. Stellfeldt, H. Butenschön and R. Wartchow, Organometallics, 1998, 17, 893–896 CrossRef CAS.
-
(a) S. G. Bratsch, J. Chem. Educ., 1988, 65, 34 CrossRef CAS;
(b)
J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, HarperCollins, New York, USA, 4th edn, 1993 Search PubMed.
- N. Kim and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2018, 57, 4722–4726 CrossRef CAS PubMed.
- For examples of heteroatom-stabilized gold allenylidene complexes see:
(a) M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 4, 1552–1559 RSC;
(b) X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem.–Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed;
(c) L. Jin, M. Melaimi, A. Kostenko, M. Karni, Y. Apeloig, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Sci., 2016, 7, 150–154 RSC;
(d) X.-S. Xiao, C. Zou, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem. Commun., 2016, 52, 4983–4986 RSC.
-
(a) M. W. Hussong, F. Rominger, P. Krämer and B. F. Straub, Angew. Chem., Int. Ed., 2014, 53, 9372–9375 CrossRef CAS PubMed;
(b) R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015, 54, 6867–6869 CrossRef CAS PubMed.
- R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2014, 53, 9369–9371 CrossRef CAS PubMed.
- C. G. Frost, H. J. Edwards, S. D. Penrose and R. Gleave, Synthesis, 2010, 3243 CrossRef CAS.
-
(a) D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2006, 45, 8188–8191 CrossRef CAS PubMed;
(b) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592–597 CrossRef CAS.
- For related homocoupling processes of gold carbene complexes see ref. 20 and J. M. Sarria Toro, C. García-Morales, M. Raducan, E. S. Smirnova and A. M. Echavarren, Angew. Chem., Int. Ed., 2017, 56, 1859–1863 CrossRef CAS PubMed.
-
(a) H.-U. Siehl, F.-P. Kaufmann and K. Hori, J. Am. Chem. Soc., 1992, 114, 9343–9349 CrossRef CAS;
(b) R. Jost, J. Sommer, C. Engdahl and P. Ahlberg, J. Am. Chem. Soc., 1980, 102, 7663–7667 CrossRef CAS.
-
(a) C. U. Pittman Jr and W. G. Miller, J. Am. Chem. Soc., 1973, 95, 2947–2956 CrossRef;
(b) N. C. Deno, C. U. Pittman Jr and J. O. Turner, J. Am. Chem. Soc., 1965, 87, 2153–2157 CrossRef CAS;
(c) W. G. Miller and C. U. Pittman Jr, J. Org. Chem., 1974, 39, 1955–1956 CrossRef CAS;
(d) D. M. Dytnerski, K. Ranganaykublu, P. B. Singh and T. S. Sorensen, Can. J. Chem., 1982, 60, 2993–3004 CrossRef CAS;
(e) G. A. Olah, G. Asensio and H. Mayr, J. Org. Chem., 1978, 43, 1518–1520 CrossRef CAS;
(f) J. Wang, L. Zhang, Y. Jing, W. Huang and X. Zhou, Tetrahedron Lett., 2009, 50, 4978–4982 CrossRef CAS.
- H. Mayr, W. Förner and P. von Ragué Schleyer, J. Am. Chem. Soc., 1979, 101, 6032–6040 CrossRef CAS.
-
(a) M. S. Hadfield, L. J. L. Häller, A.-L. Lee, S. A. Macgregor, J. A. T. O'Neill and A. M. Watson, Org. Biomol. Chem., 2012, 10, 4433–4440 RSC;
(b) N. A. Rajabi, M. J. Atashgah, R. BabaAhmadi, C. Hyland and A. Ariafard, J. Org. Chem., 2013, 78, 9553–9559 CrossRef CAS PubMed.
- T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990–4991 CrossRef CAS.
- E. C. Friedrich and D. B. Taggart, J. Org. Chem., 1975, 40, 720–723 CrossRef CAS.
- F. F. Mulks, P. W. Antoni, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 1810–1821 CrossRef CAS.
- For the protodeauration of gold σ-allyl complexes through an
 pathway see ref. 8e and:
(a) A. S. K. Hashmi, A. M. Schuster, S. Litters, F. Rominger and M. Pernpointner, Chem.–Eur. J., 2011, 17, 5661–5667 CrossRef CAS PubMed;
(b) R. BabaAhmadi, P. Ghanbari, N. A. Rajabi, A. S. K. Hashmi, B. F. Yates and A. Ariafard, Organometallics, 2015, 34, 3186–3195 CrossRef CAS.
pathway see ref. 8e and:
(a) A. S. K. Hashmi, A. M. Schuster, S. Litters, F. Rominger and M. Pernpointner, Chem.–Eur. J., 2011, 17, 5661–5667 CrossRef CAS PubMed;
(b) R. BabaAhmadi, P. Ghanbari, N. A. Rajabi, A. S. K. Hashmi, B. F. Yates and A. Ariafard, Organometallics, 2015, 34, 3186–3195 CrossRef CAS.
- Analogous oxidations of gold carbene moieties have been observed under catalytic conditions39 and from well defined gold carbene complexes.20,26.
- C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS PubMed.
- For an example of silyldeauration implicated in a catalytic transformation see: I. Nakamura, T. Sato, M. Terada and Y. Yamamoto, Org. Lett., 2007, 9, 4081–4083 CrossRef CAS PubMed.
- A similar conclusion was reached by Fürstner regarding the gold bis(anisyl) carbene complex (PCy3)AuC(p-C6H4OMe)2.19.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and scans of NMR spectra. CCDC 1897597. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01574d |
| ‡ Fürstner has previously reported that attempted synthesis of the PCy3-analog of (Z)-5avia reaction of 1,1-diphenylcyclopropene with (PCy3)PAuNTf2 led instead to rapid oligomerization of the cyclopropene.19 In our hands, attempted synthesis of (Z)-5bvia reaction of 1,1-(bisanisyl)cyclopropene with either (IPr)AuOTf or (IPr)AuCl/AgSbF6 produced a similar outcome. |
| § Two discrete intermediates were detected by 1H NMR spectroscopy over the temperature range −95 to −30 °C. However, broadening, overlapping resonances, and the dearth of potentially diagnostic resonances precluded extraction of any meaningful information from these data. |
| ¶ Reaction of 16 (cis/trans = 3:1, 1 equiv.) with a 1:1 mixture of (IPr)AuCl and AgSbF6 in CD2Cl2 at −80 °C generated a single resonance [δ 4.99 (br d, J = 10.2 Hz)] that could be attributed to 15 and which constituted ∼25% of the reaction mixture, the remainder being free 16. This suggests that either (1) the vinylic 1H resonances for the various diastereomers of 15 are coincidental or (2) only a single diastereomer of 15 is formed in detectable concentrations from this reaction. Therefore, the 75% yield determined for the conversion of (E)-5b to 15 (determined by integrating the δ 4.99 resonance relative to internal standard) represents a lower limit for the yield of 15 formed from (E)-5b and the 67% yield determined for 16 (determined by integrating the vinylic resonances of cis- and trans-16 relative to internal standard after warming to room temperature and filtering through silica gel) points to decomposition of 16 under reaction conditions, which was confirmed experimentally. Treatment of (E)-5b with 1-(p-methoxyphenyl)-1,3-butadiene at −95 °C led to immediate consumption of (E)-5b but produced no products that could be attributed to either cyclopropanation or [4 + 3] cycloaddition. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
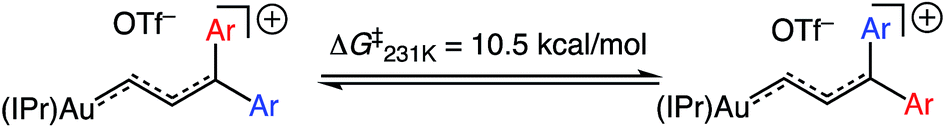
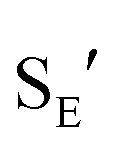 pathway (Scheme 5). Interestingly, (3-phenyl-1H-inden-1-yl)gold(I) complexes very similar to intermediate II have been recently isolated via thermal rearrangement of neutral (3,3-diphenylcycloprop-1-en-1-yl)gold(I)complexes and were shown to undergo protodeauration with HCl predominantly through an
pathway (Scheme 5). Interestingly, (3-phenyl-1H-inden-1-yl)gold(I) complexes very similar to intermediate II have been recently isolated via thermal rearrangement of neutral (3,3-diphenylcycloprop-1-en-1-yl)gold(I)complexes and were shown to undergo protodeauration with HCl predominantly through an 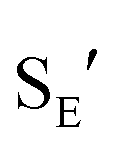 pathway.36,37 The facility of the intramolecular Friedel–Crafts/Nazarov reaction of (Z)-5b and the failure to form 9 in the decomposition of (E)-5b therefore suggests that the energy barrier for E/Z isomerization of (E)-5b exceeds that of the lowest energy decomposition pathway available to (E)-5b (ΔG‡ ≈ 16 kcal mol−1).
pathway.36,37 The facility of the intramolecular Friedel–Crafts/Nazarov reaction of (Z)-5b and the failure to form 9 in the decomposition of (E)-5b therefore suggests that the energy barrier for E/Z isomerization of (E)-5b exceeds that of the lowest energy decomposition pathway available to (E)-5b (ΔG‡ ≈ 16 kcal mol−1).